Design, Synthesis and in Vivo Anti-inflammatory Activities of 2,4-Diaryl-5-4H-imidazolone Derivatives


 ,
,
Abstract
:1. Introduction

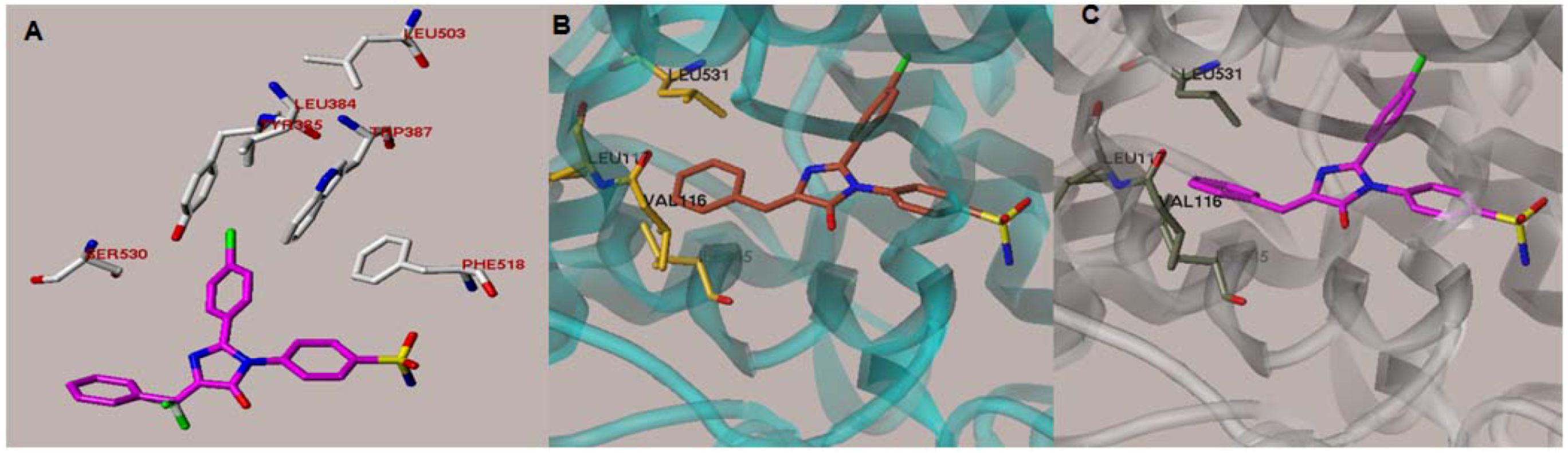
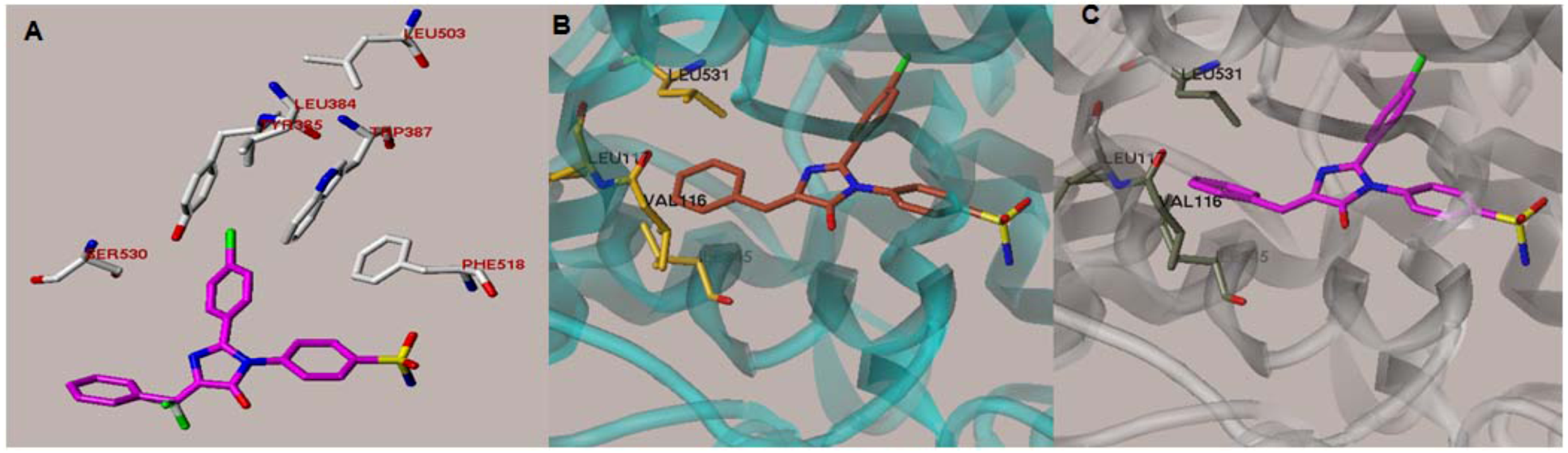{kind=link}
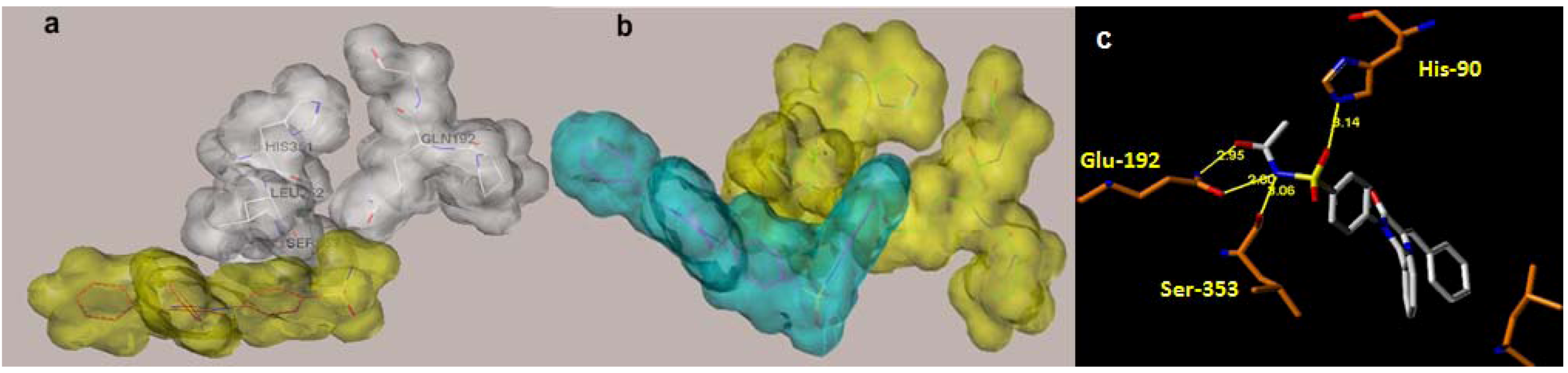{kind=link}
{kind=link}

| Approval | 1999 | 1999 | 2001 | 2006 | 2005 |
| Withdrawal | ------- | 2004 | 2005 | 2007 | ------- |
| Main AEs | ------- | Cardiovascular | Cardiovascular | Hepatotoxicity | ------- |
| Authority | FDA | FDA | FDA | EMA, TGA | UK |
2. Results and Discussion
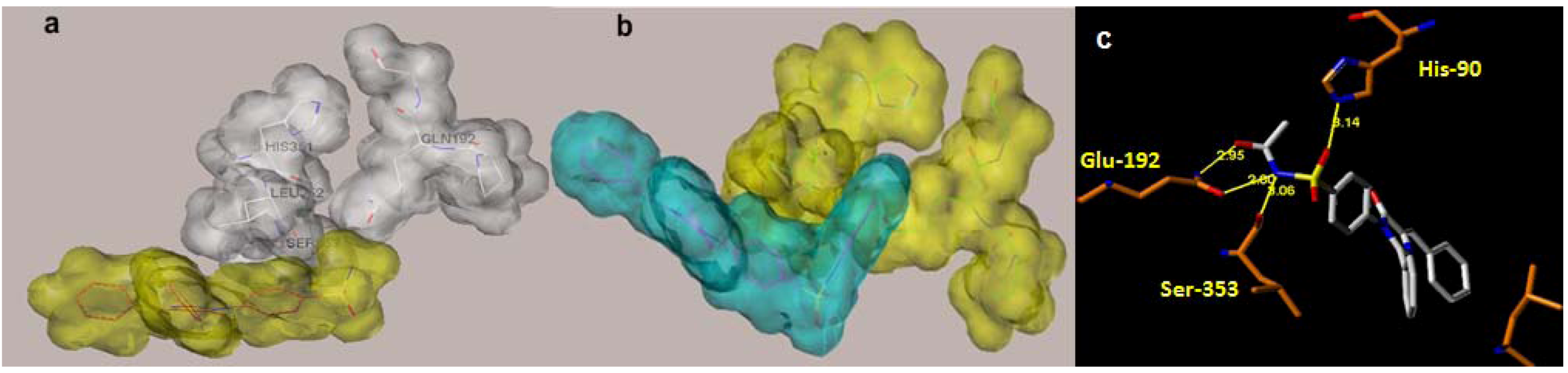
2.1. Rationale and Structure-Based Design
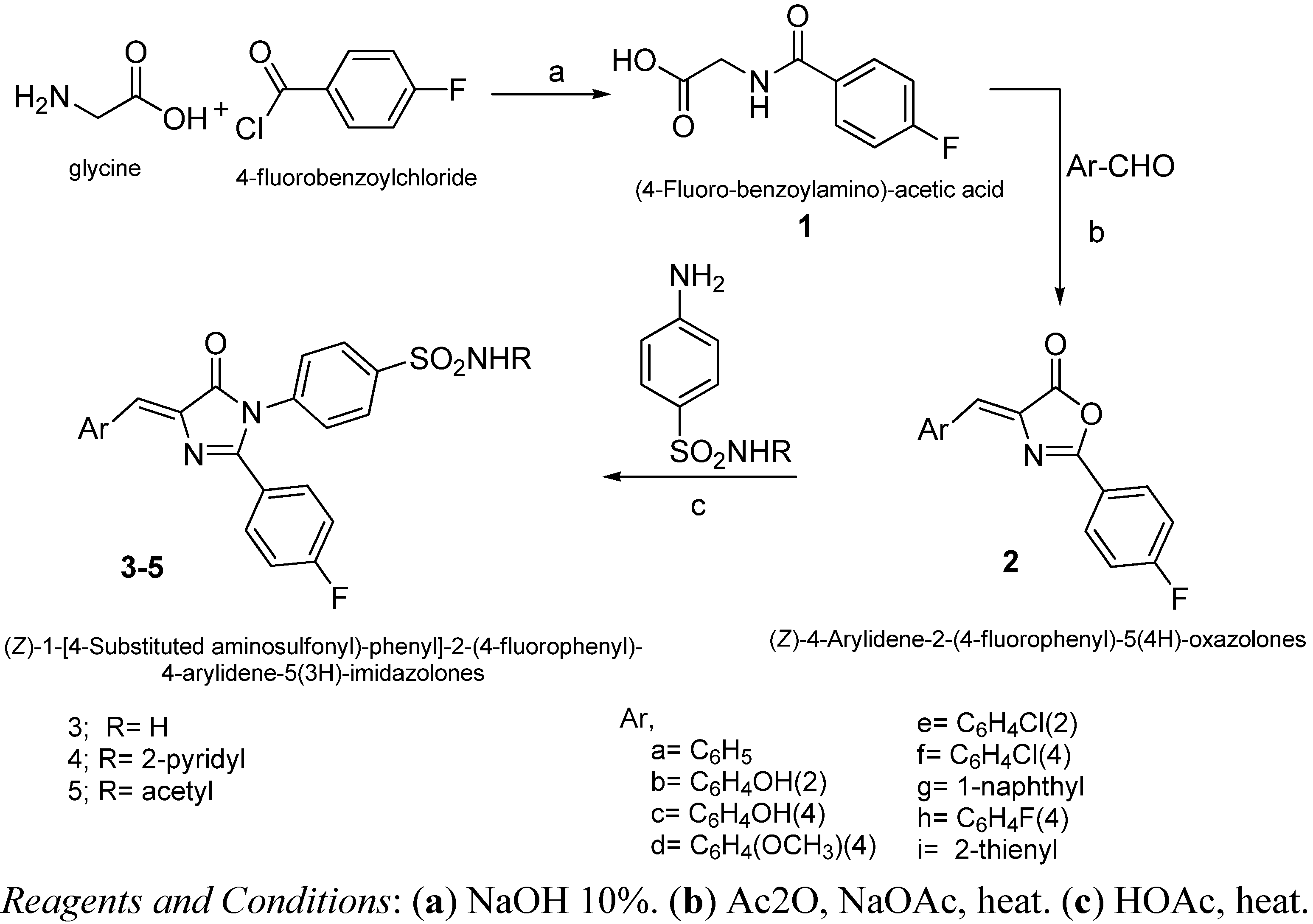
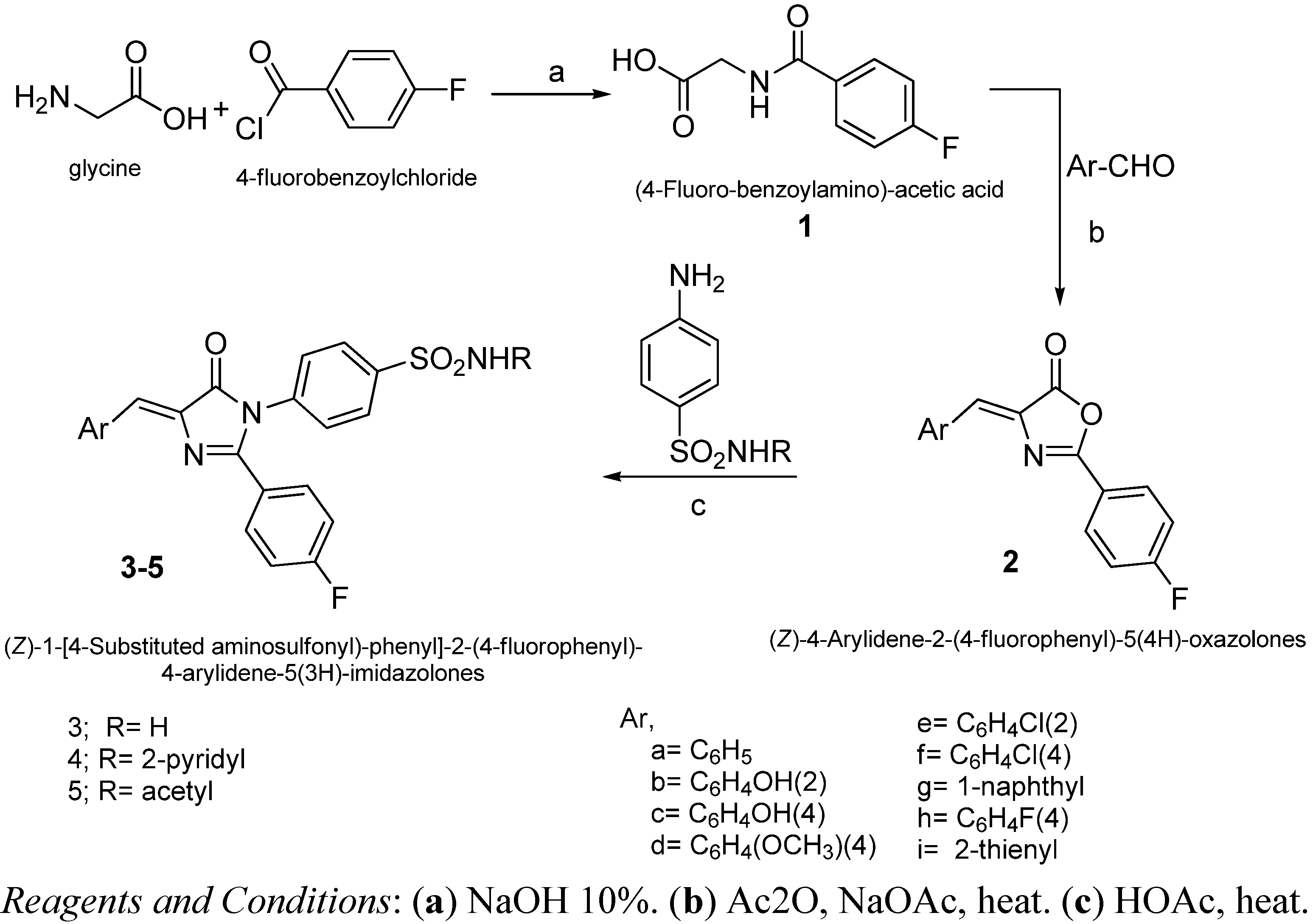
2.2. Chemistry
2.3. Biological Screening
2.3.1. Anti-inflammatory Activity
| Tested Compounds | Increase in paw edema (mL) ± SEM a,b | % Protection | Activity relative to indomethacin |
|---|---|---|---|
| Control | 0.96 ± 0.026 | 0.0 | 0.0 |
| Indomethacin | 0.25 ± 0.024 | 74.0 | 100 |
| Meloxicam | 0.23 ± 0.019 | 76.0 | 103 |
| 3-a | 0.32 ± 0.028 | 66.7 | 90 |
| 3-c | 0.42 ± 0.032 | 56.3 | 76 |
| 3-d | 0.36 ± 0.016 | 62.5 | 84 |
| 3-g | 0.31 ± 0.027 | 67.7 | 91 |
| 3-h | 0.28 ± 0.022 | 70.8 | 96 |
| 4-a | 0.64 ± 0.026 | 33.3 | 45 |
| 4-c | 0.73 ± 0.022 | 24.0 | 32 |
| 4-d | 0.71 ± 0.028 | 26.0 | 35 |
| 4-g | 0.68 ± 0.032 | 29.2 | 39 |
| 4-h | 0.49 ± 0.024 | 49.0 | 66 |
| 5-a | 0.17 ± 0.016 | 82.3 | 111 |
| 5-c | 0.27 ± 0.023 | 71.9 | 97 |
| 5-d | 0.20 ± 0.024 | 79.2 | 107 |
| 5-g | 0.19 ±0.029 | 80.2 | 108 |
| 5-h | 0.08 ± 0.032 | 91.7 | 124 |
2.3.2. Ulcerogenic Effects
| Compound | ED50 (μM/kg) | % Ulceration | Compound | ED50 (μM/kg) | % Ulceration |
|---|---|---|---|---|---|
| Indomethacin | 9.7 | 100 | 4-d | 29 | 20 |
| Meloxicam | 12 | 0 | 4-g | 28 | 20 |
| 3-a | 23 | 10 | 4-h | 27 | NT |
| 3-c | 25 | NT | 5-a | 17.9 | 5 |
| 3-d | 24 | NT | 5-c | 18 | 10 |
| 3-g | 22 | NT | 5-d | 16.5 | 5 |
| 3-h | 19.4 | 10 | 5-g | 14.4 | NT |
| 4-a | 28.7 | 25 | 5-h | 12 | 5 |
| 4-c | 31 | NT |
2.4. Discussion
3. Experimental
3.1. General
3.2. General Procedure for Preparation 3-[4-Substituted aminosulfonyl)-phenyl]-2-(4-fluorophenyl)-5-arylidene-5(4H)-imidazolones 3–5
3.3. Anti-inflammatory Test
3.4. Ulcerogenicity Test
4. Conclusions
Acknowledgments
- Sample Availability: Contact the authors.
References
- Bazan, N.G.; Flower, R.J. Medicine: Lipid signals in pain control. Nature 2002, 420, 135–138. [Google Scholar] [CrossRef]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F.; et al. Gastrointestinal toxicity with celecoxib vs. nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA 2000, 284, 1247–1255. [Google Scholar]
- Zhang, J.; Ding, E.L.; Song, Y. Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: Meta-analysis of randomized trials. JAMA 2006, 296, 1619–1632. [Google Scholar]
- Lenzer, J. FDA advisers warn: COX 2 inhibitors increase risk of heart attack and stroke. BMJ 2005, 330, 440. [Google Scholar] [CrossRef]
- Mason, R.P.; Walter, M.F.; Day, C.A.; Jacob, R.F. A biological rationale for the cardiotoxic effects of rofecoxib: Comparative analysis with other COX-2 selective agents and NSAids. Subcell. Biochem. 2007, 42, 175–190. [Google Scholar] [CrossRef]
- Corey, E.J.; Reddy, L.R. Facile air oxidation of the conjugate base of rofecoxib. Tetrahedron Lett. 2005, 46, 927–929. [Google Scholar]
- Walter, M.F.; Jacob, R.F.; Day, C.A.; Dahlborg, R.; Weng, Y.; Mason, R.P. Sulfone COX-2 inhibitors increase susceptibility of human LDL and plasma to oxidative modification: Comparison to sulfonamide COX-2 inhibitors and NSAIDs. Atherosclerosis 2004, 177, 235–243. [Google Scholar]
- Liu, J.Y.; Li, N.; Yang, J.; Qiu, H.; Ai, D.; Chiamvimonvat, N.; Zhu, Y.; Hammock, B.D. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proc. Natl. Acad. Sci. USA 2010, 107, 17017–17022. [Google Scholar]
- Burton, B. Australian drugs regulator cancels registration of COX 2 inhibitor. BMJ 2007, 335, 363. [Google Scholar] [CrossRef]
- Singer, J.B.; Lewitzky, S.; Leroy, E.; Yang, F.; Zhao, X.; Klickstein, L.; Wright, T.M.; Meyer, J.; Paulding, C.A. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat. Genet. 2010, 42, 711–714. [Google Scholar]
- Hayashi, S.; Ueno, N.; Murase, A.; Nakagawa, Y.; Takada, J. Novel acid-type cyclooxygenase-2 inhibitors: Design, synthesis, and structure-activity relationship for anti-inflammatory drug. Eur. J. Med. Chem. 2012, 50, 179–195. [Google Scholar] [CrossRef]
- Biava, M.; Porretta, G.C.; Poce, G.; Battilocchio, C.; Manetti, F.; Botta, M.; Sautebin, L.; Rossi, A.; Pergola, C.; Ghelardini, C.; et al. Novel ester and acid derivatives of the 1,5-diarylpyrrole scaffold as anti-inflammatory and analgesic agents. J. Med. Chem. 2010, 53, 723–733. [Google Scholar]
- Wallace, J.L.; Ferraz, J.G. New pharmacologic therapies in gastrointestinal disease. Gastroenterol. Clin. North Am. 2010, 39, 709–720. [Google Scholar] [CrossRef]
- Yamakawa, N.; Suemasu, S.; Okamoto, Y.; Tanaka, K.I.; Ishihara, T.; Asano, T.; Miyata, K.; Otsuka, M.; Mizushima, T. Synthesis and Biological Evaluation of Derivatives of 2-{2-Fluoro-4-[(2-oxocyclopentyl)methyl]phenyl}propanoic Acid: Nonsteroidal Anti-Inflammatory Drugs with Low Gastric Ulcerogenic Activity. J. Med. Chem. 2012, 55, 5143–5150. [Google Scholar]
- SYBYL-X Suite. Tripos Associates Inc.: St. Louis, MO, USA, 2010; Version SYBYL-X 1.1.
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Miyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar]
- Crawford, M.; Little, W.T. The Erlenmeyer Reaction with aliphatic aldehydes, 2-phenyloxazol-5-one being used instead of hippuric acid. J. Chem. Soc. 1959, 729–731. [Google Scholar] [CrossRef]
- Joshi, H.; Upadhyay, P.; Karia, D.; Baxi, A.J. Synthesis of some novel imidazolinones as potent anticonvulsant agents. Eur. J. Med. Chem. 2003, 38, 837–840. [Google Scholar] [CrossRef]
- Winter, C.A.; Risley, E.A.; Nuss, G.W. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar]
- Meshali, M.; El-Sabbagh, H.; Foda, A. Effect of encapsulation of flufenamic acid with acrylic resins on its bioavailability and gastric ulcerogenic activity in rats. Acta Pharm. 1983, 29, 217–219. [Google Scholar]
- Mauser, H.; Guba, W. Recent developments in de novo design and scaffold hopping. Curr. Opin. Drug Discov. Dev. 2008, 11, 365–374. [Google Scholar]
- Cheer, S.M.; Goa, K.L. Parecoxib (parecoxib sodium). Drugs 2001, 61, 1133–1141. [Google Scholar] [CrossRef]
- Khalifa, M.; Abdelbaky, N.A. Synthesis of new imidazolyl acetic acid derivatives with anti-inflammatory and analgesic activities. Arch. Pharm. Res. 2008, 31, 419–423. [Google Scholar] [CrossRef]
- Wang, P.; Naduthambi, D.; Mosley, R.T.; Niu, C.; Furman, P.A.; Otto, M.J.; Sofia, M.J. Phenylpropenamide derivatives: Anti-hepatitis B virus activity of the Z isomer, SAR and the search for novel analogs. Bioorg. Med. Chem. Lett. 2011, 21, 4642–4647. [Google Scholar]
- Chavez, F.; Kennedy, N.; Rawalpally, T.; Williamson, R.T.; Cleary, T. Substituents Effect on the Erlenmeyer-Plochl Reaction: Understanding an Observed Process Reaction Time. Org. Process Res. Dev. 2010, 14, 579–584. [Google Scholar] [CrossRef]
- Isobe, T.; Ishikawa, T. 2-Chloro-1,3-dimethylimidazolinium Chloride. 2. Its Application to the Construction of Heterocycles through Dehydration Reactions. J. Org. Chem. 1999, 64, 6989–6992. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
El-Araby, M.; Omar, A.; Hassanein, H.H.; El-Helby, A.-G.H.; Abdel-Rahman, A.A. Design, Synthesis and in Vivo Anti-inflammatory Activities of 2,4-Diaryl-5-4H-imidazolone Derivatives. Molecules 2012, 17, 12262-12275. https://doi.org/10.3390/molecules171012262
El-Araby M, Omar A, Hassanein HH, El-Helby A-GH, Abdel-Rahman AA. Design, Synthesis and in Vivo Anti-inflammatory Activities of 2,4-Diaryl-5-4H-imidazolone Derivatives. Molecules. 2012; 17(10):12262-12275. https://doi.org/10.3390/molecules171012262
Chicago/Turabian StyleEl-Araby, Moustafa, Abdelsattar Omar, Hassanein H. Hassanein, Abdel-Ghany H. El-Helby, and Asharf A. Abdel-Rahman. 2012. "Design, Synthesis and in Vivo Anti-inflammatory Activities of 2,4-Diaryl-5-4H-imidazolone Derivatives" Molecules 17, no. 10: 12262-12275. https://doi.org/10.3390/molecules171012262