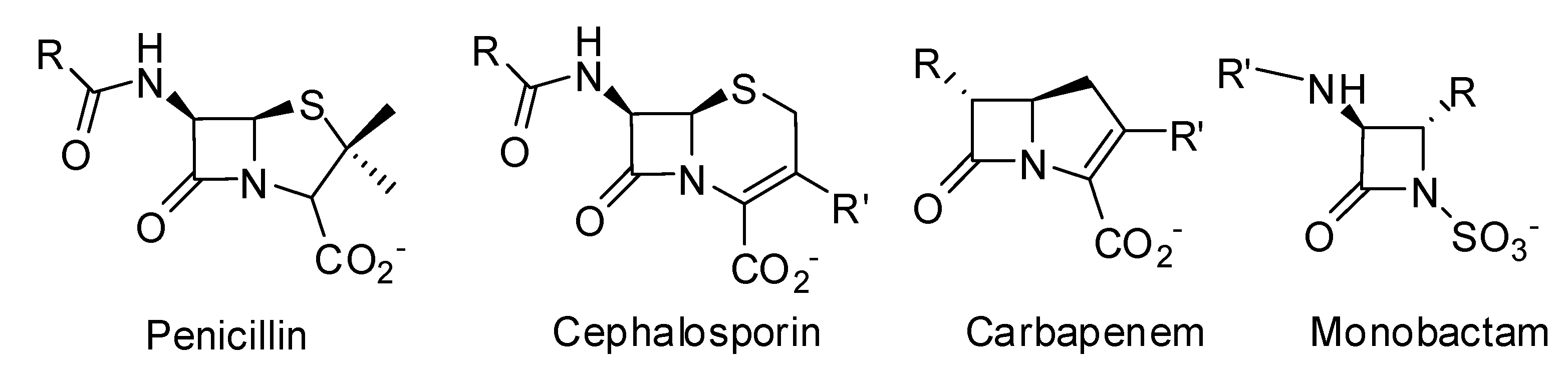
Development of New Drugs for an Old Target — The Penicillin Binding Proteins


Abstract
:Abbreviations
| PBP: | penicillin binding protein |
| ESBL: | extended spectrum β-lactamase |
| MRSA: | methicillin-resistant strains of Staphylococcus aureus |
1. Introduction
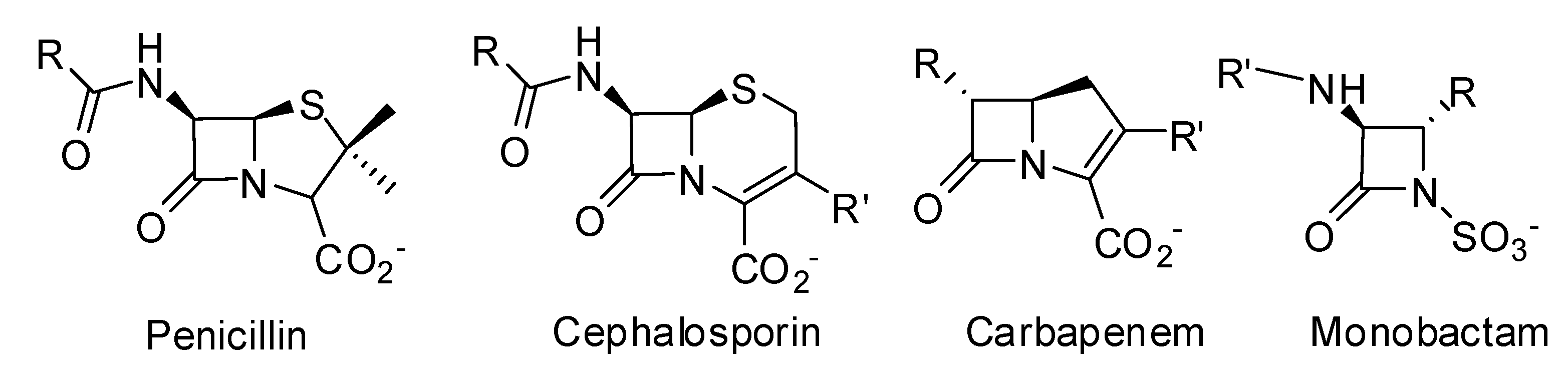
- The production of β-lactamases, which catalyze the hydrolysis of the β-lactam cycle, is the most important mechanism of resistance in Gram-negative bacteria. Transfer of plasmid encoded β-lactamases rapidly disseminates resistance over a broad range of bacteria [7].
- The production of low-affinity PBPs which catalyze the transpeptidation reaction even in the presence of high concentrations of β-lactam antibiotics is an important mechanism of resistance in some Gram-positive bacteria [e.g., methicillin resistant S. aureus (MRSA)]. Mutation of residues surrounding the active sites of these PBPs enhances the resistance of these microorganisms to β-lactam antibiotics. Mutation of residues lowering the affinity of PBPs to β-lactams is also frequently observed in non β-lactamase producing Gram-negative bacteria and in some Gram-positive bacteria like Streptococcus pneumoniae. The mechanism generally affects the class-B PBPs involved in cell division (homologous to Escherichia coli PBP3), which is one of the main targets of β-lactams in these organisms. For example mutations are encountered in S. pneumoniae PBP2x [8], Neisseria gonorrhoeae PBP2 [9] and Haemophilus influenzae PBP3 [10]. Furthermore, horizontal gene transfer allows dissemination of resistance. For example, in Streptococci, resistance is disseminated via natural transformation [11], and resistance in MRSA probably originates from transduction of the mecA gene, coding for a methicillin-resistant PBP2a protein, into the chromosome of S. aureus [7,12].
- A decrease of the production of outer membrane proteins (OMPs), which allow the transfer of β-lactams through the outer membrane, lowers the effective concentration of antibiotics in the periplasm and increases MIC-values. Resistant phenotypes are observed if this mechanism is combined with another resistance mechanism such as the expression of a β-lactamase [13,14].
- In Gram-negative bacteria efflux pumps, which can export β-lactams outside the cells through the outer membrane, can also decrease the effective concentration of drugs in the periplasm [14].
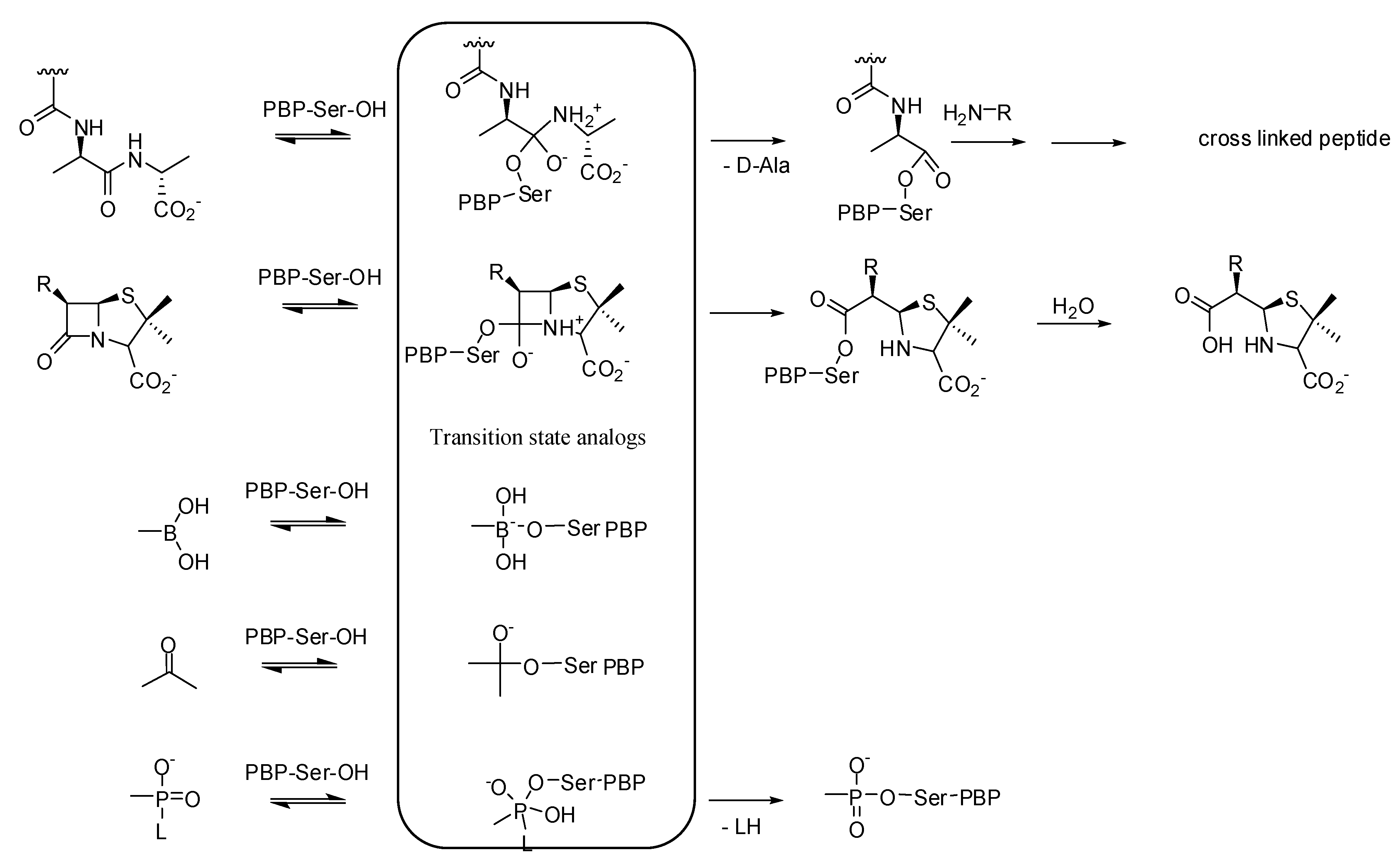
2. Milestones on the Way to Discover Non-β-lactam Inhibitors
2.1. Availability of the Target
2.2. Assay Development

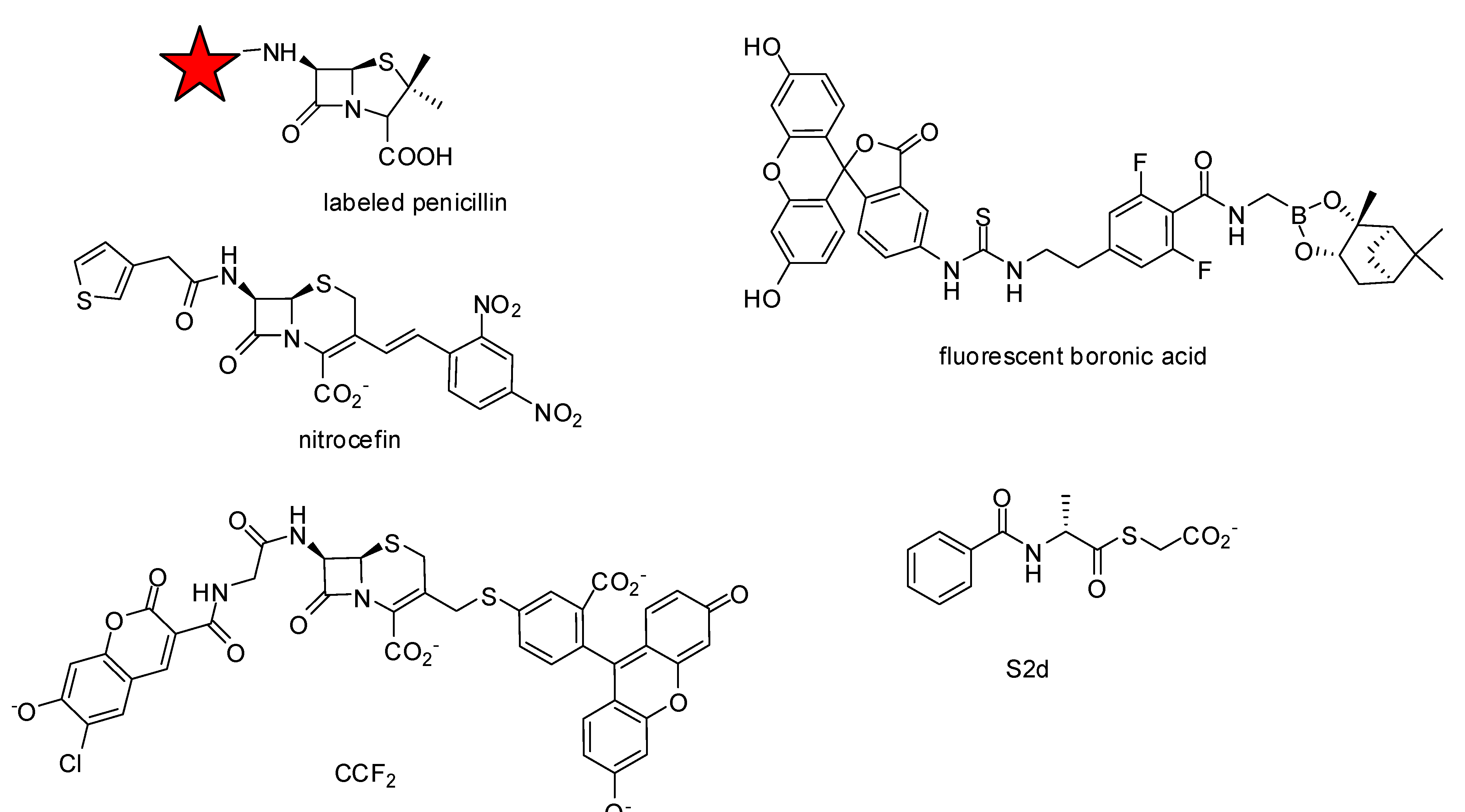

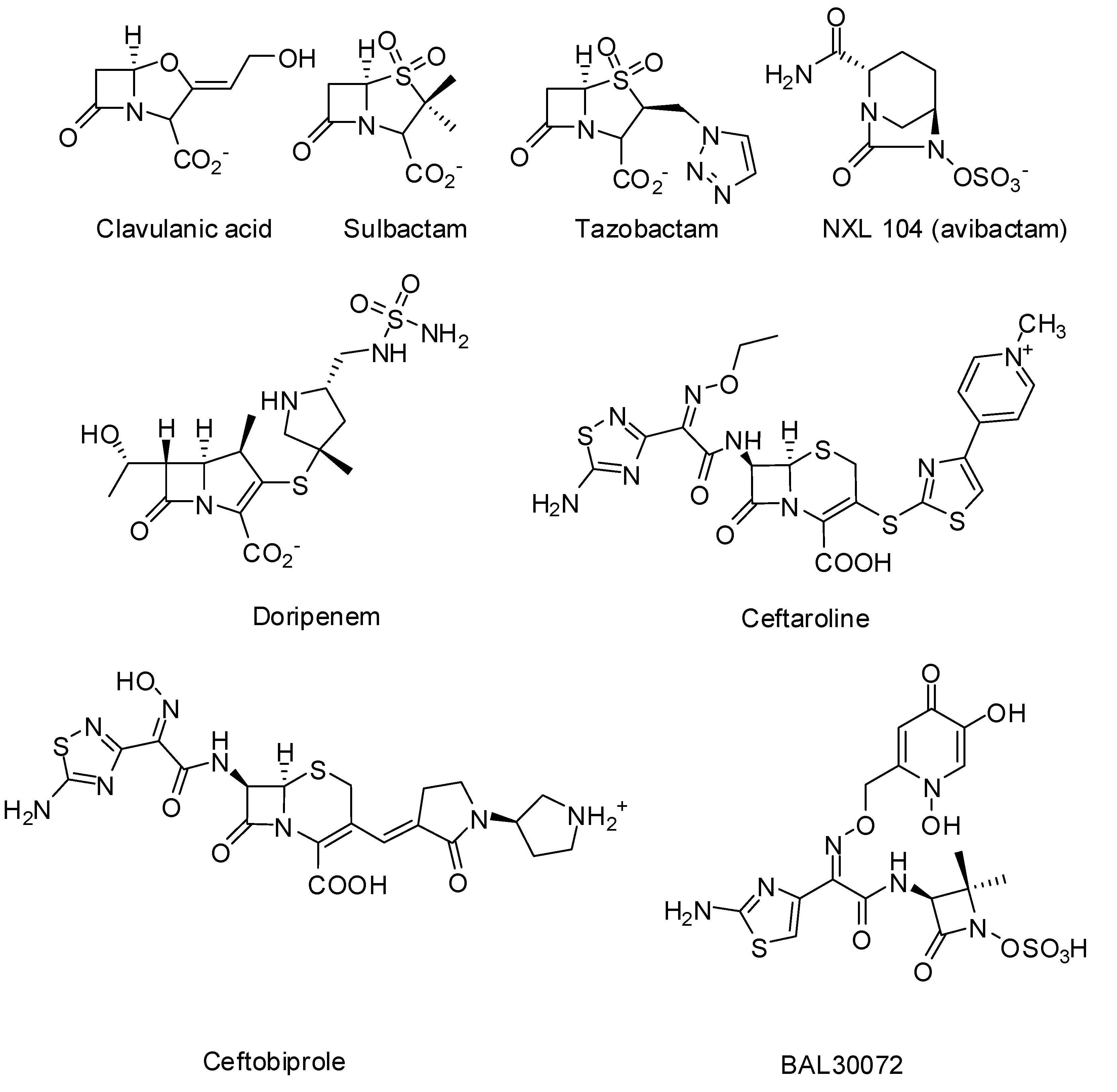
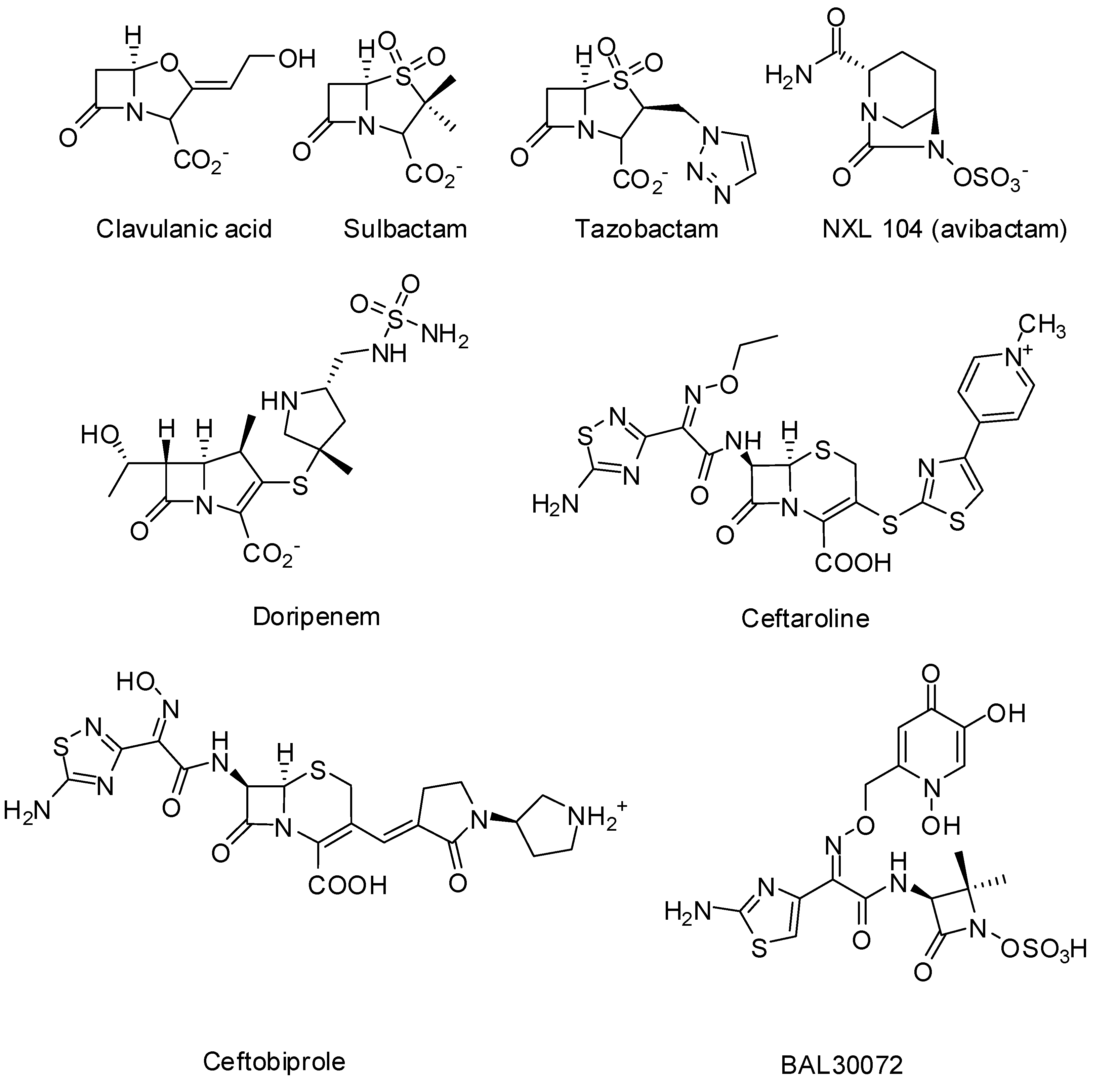
3. Non-beta-lactams
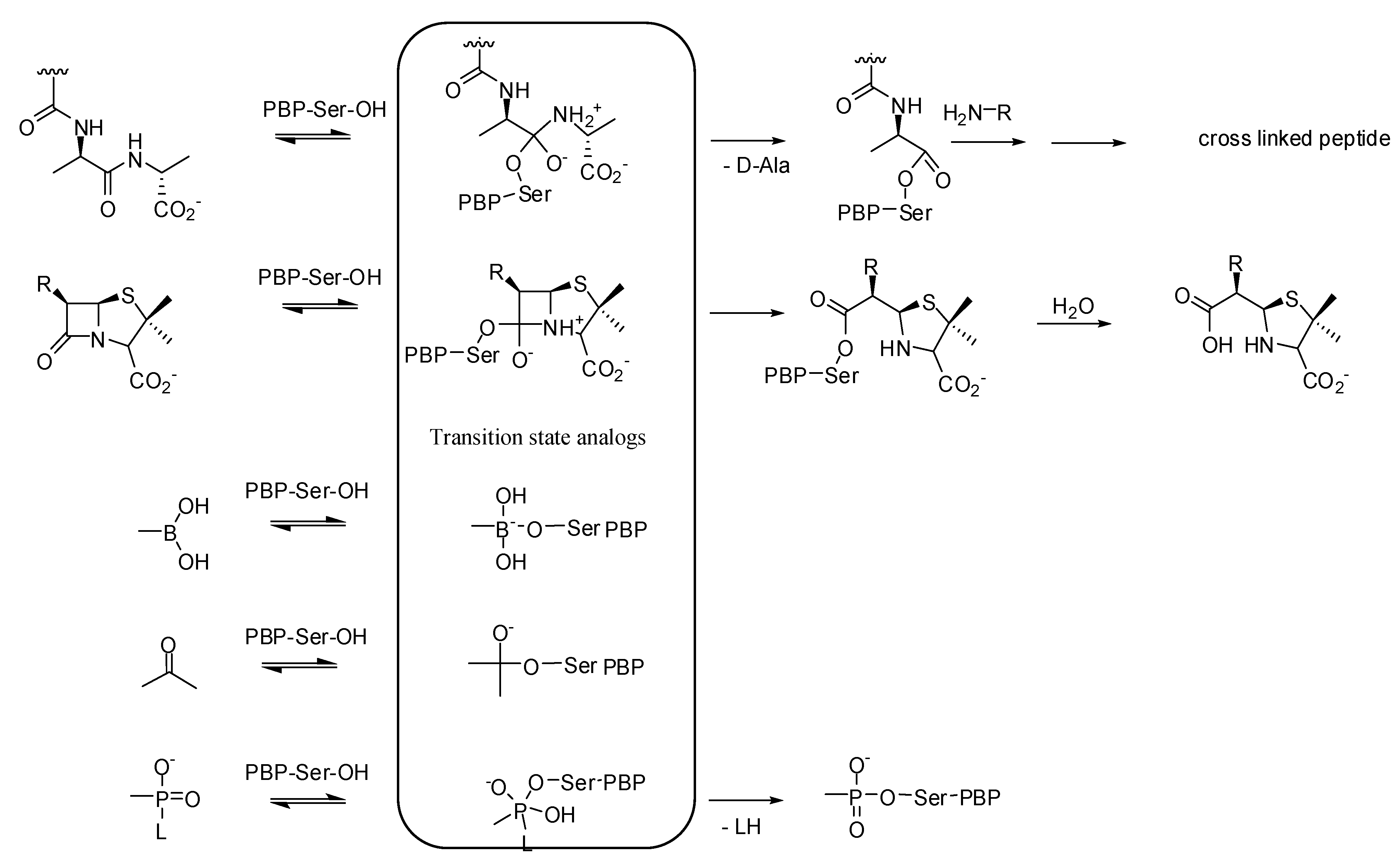
3.1. Transition State Analogs
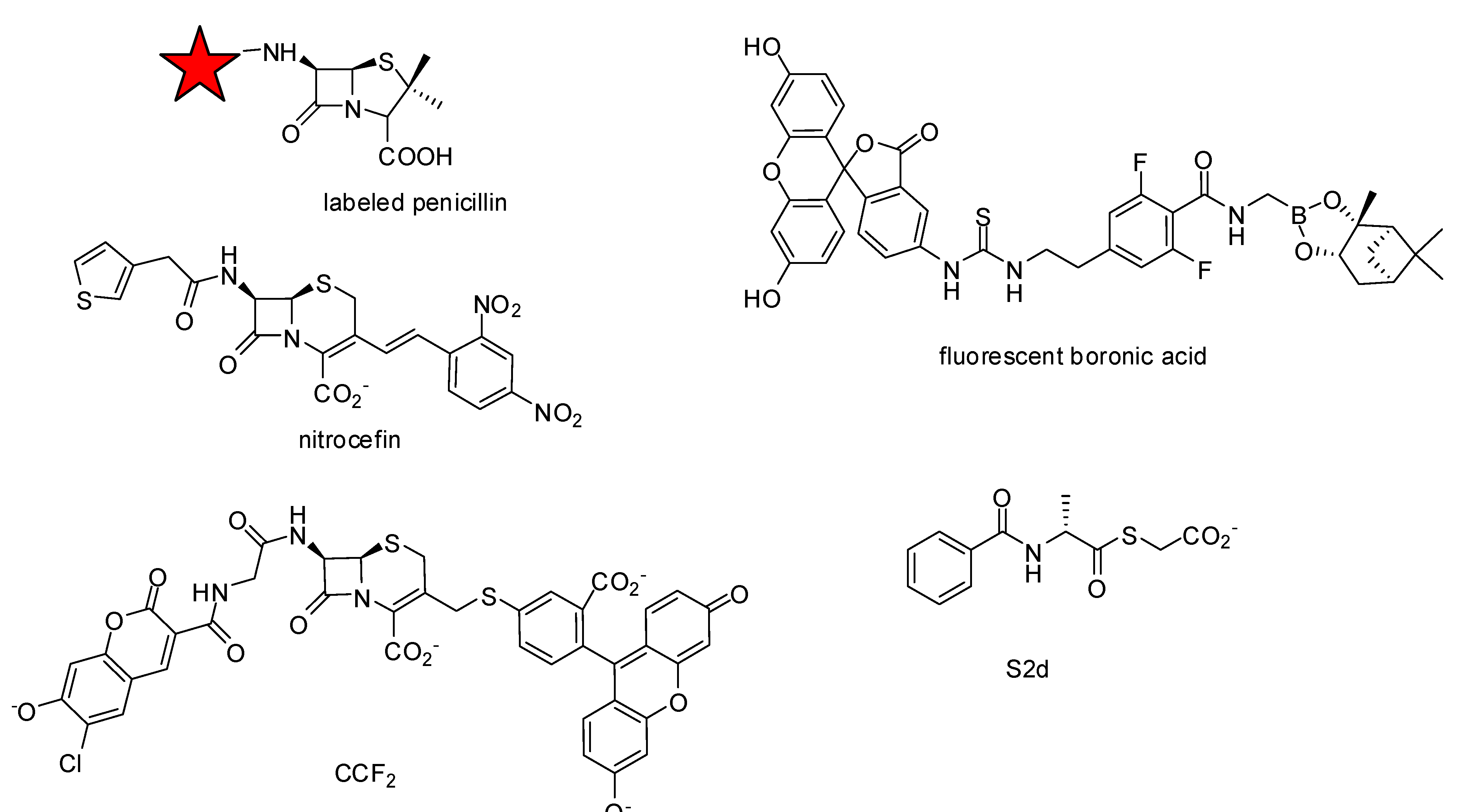
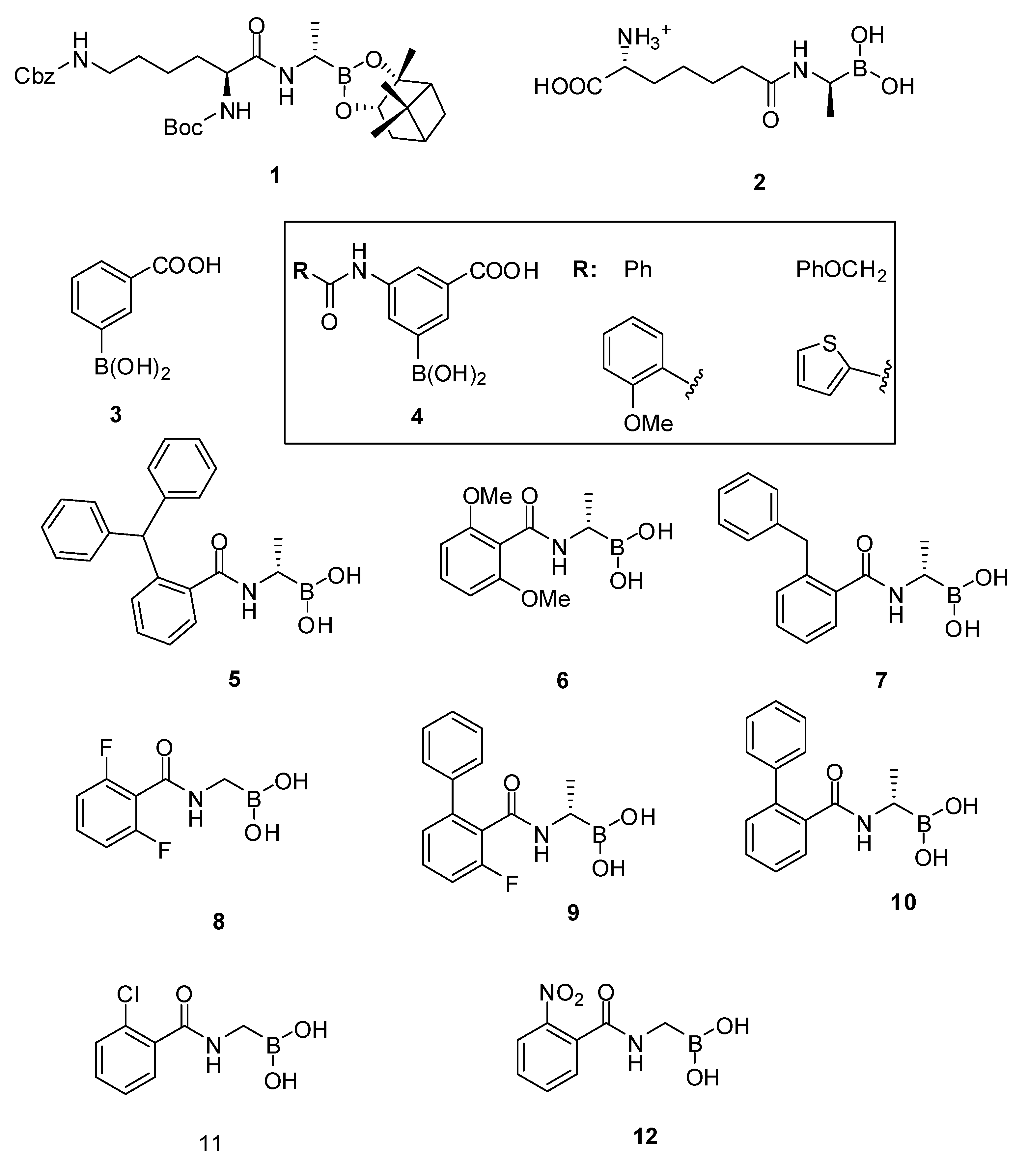
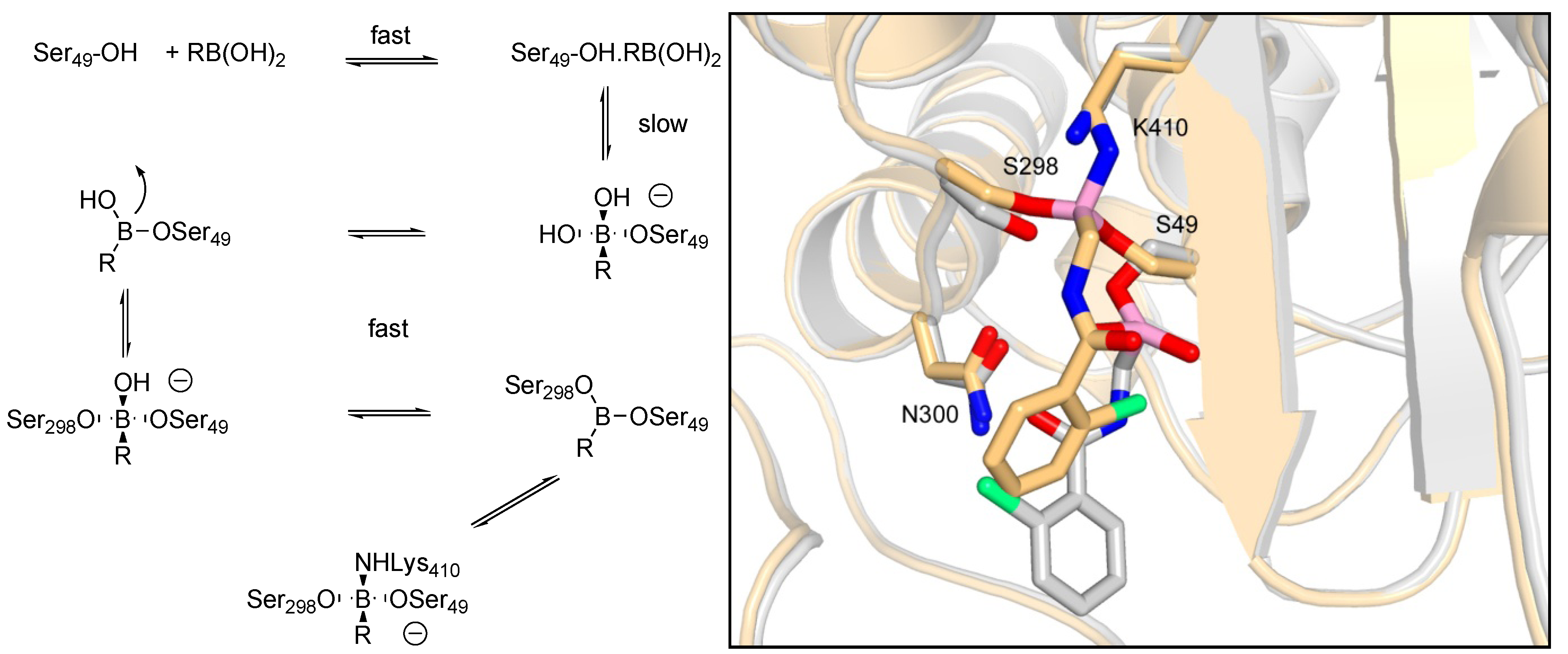
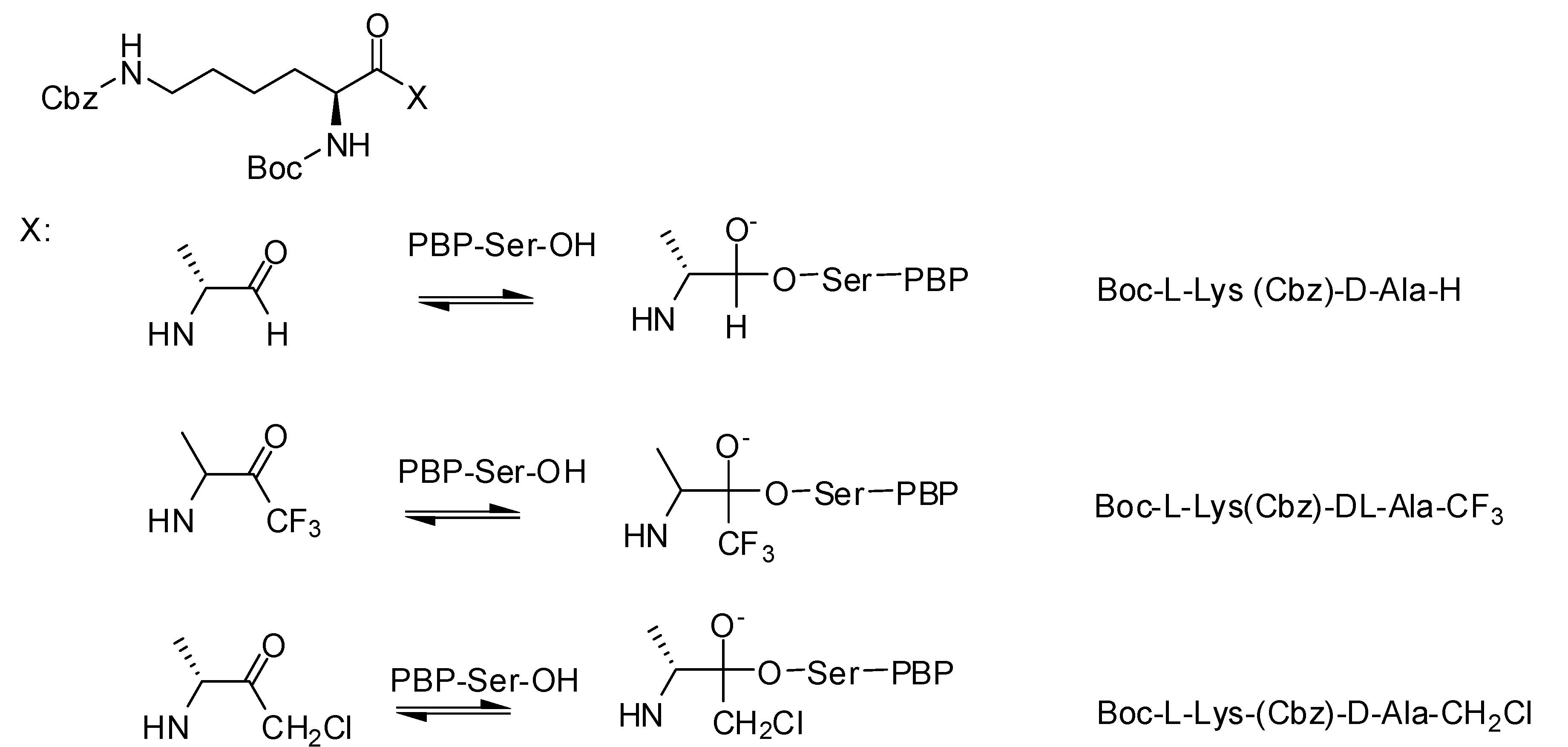
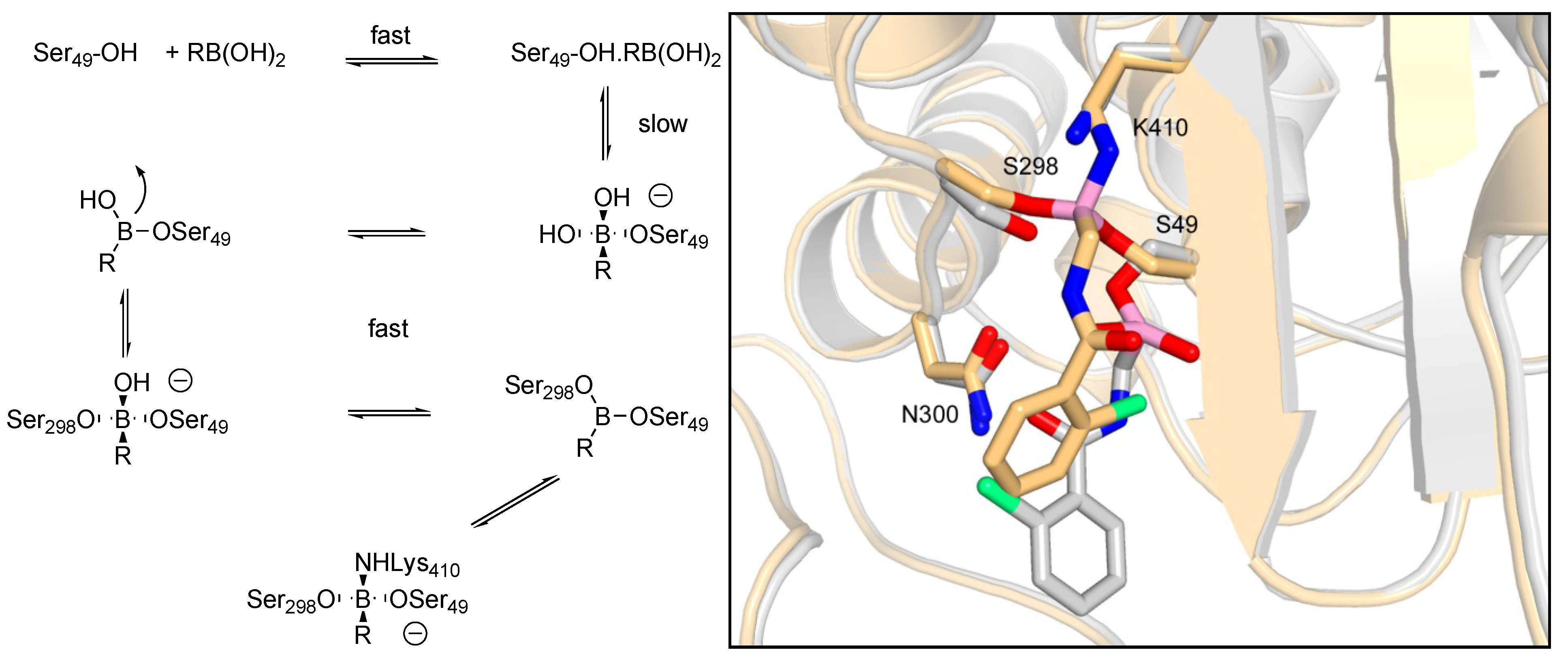
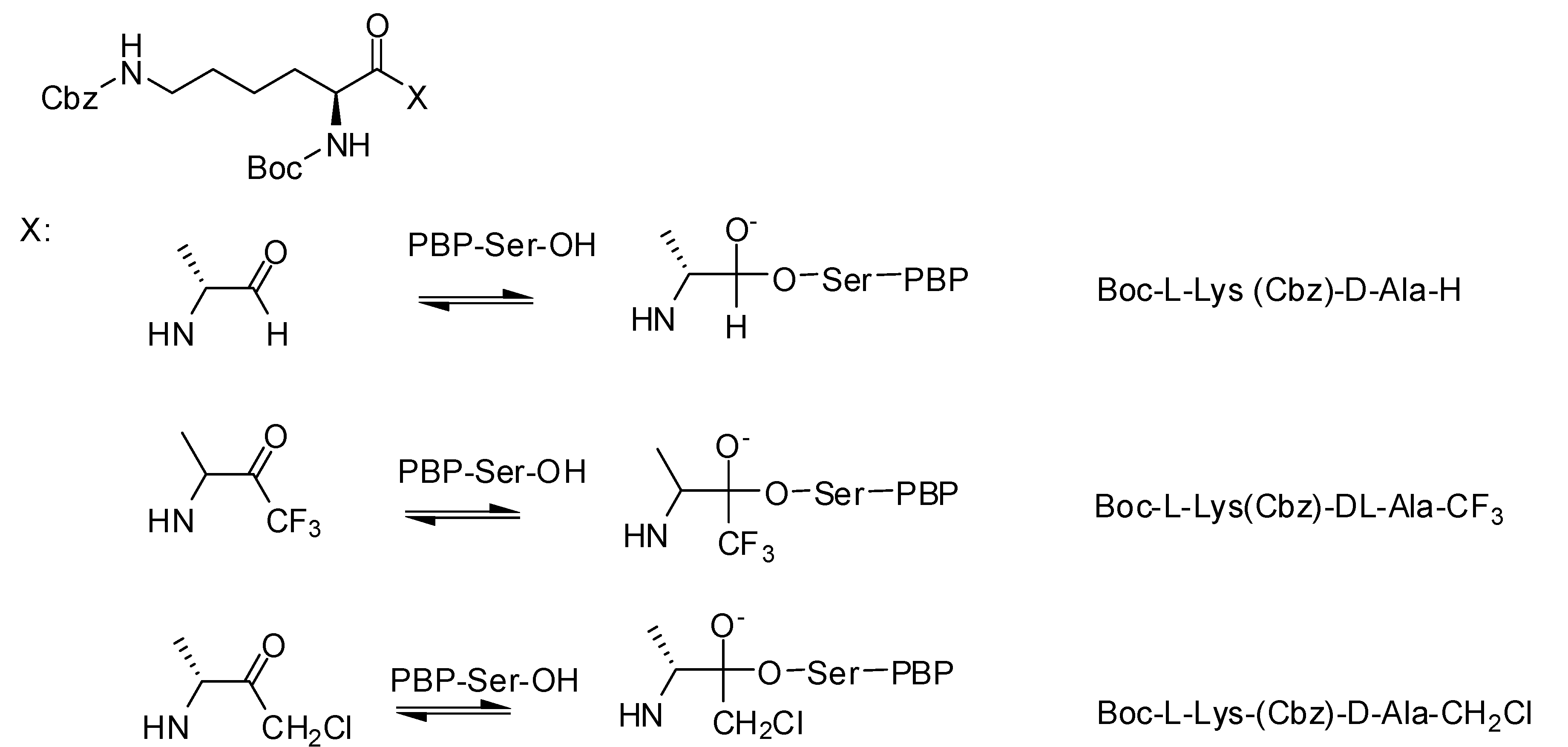
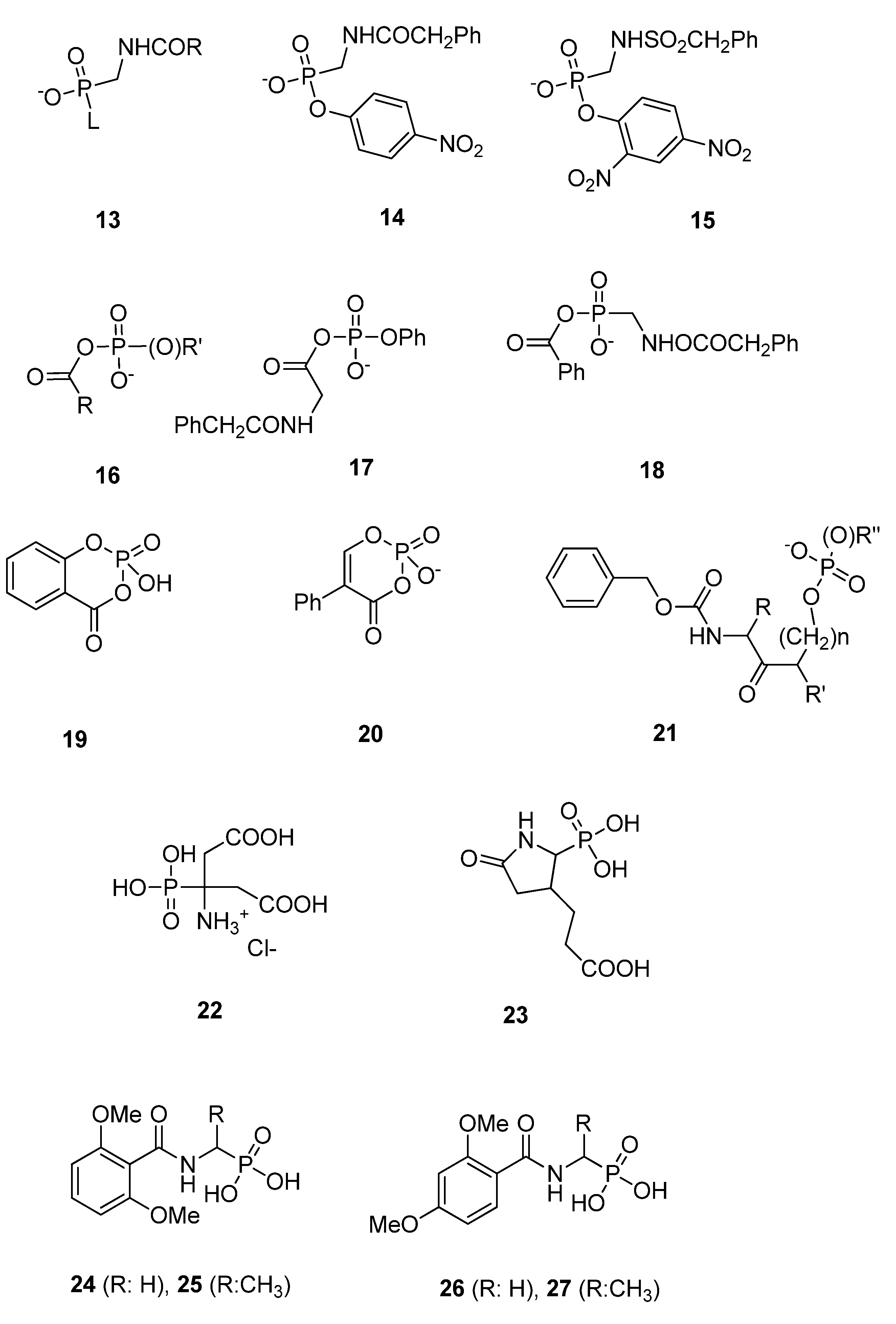
3.1.1. Boronic Acids
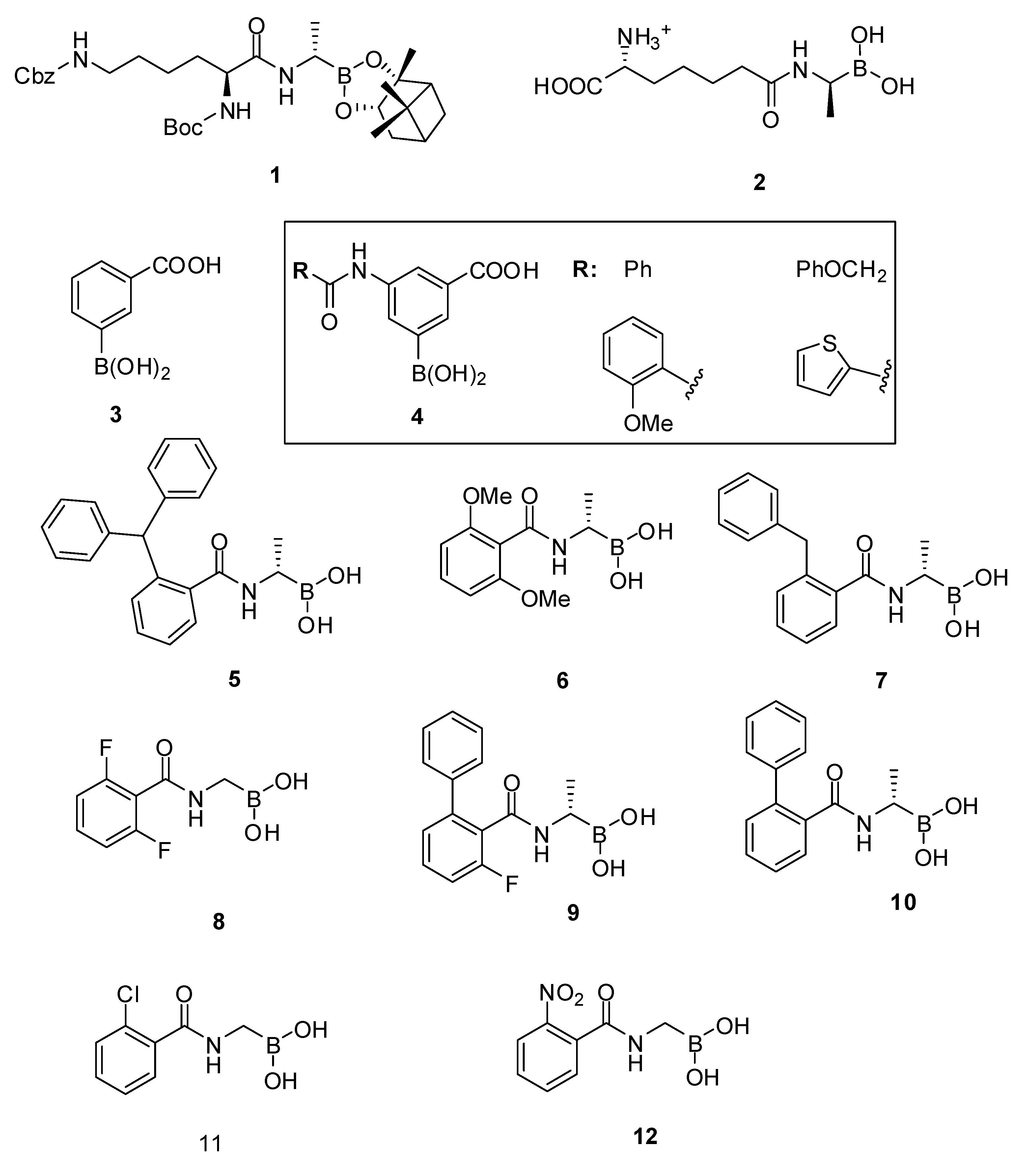
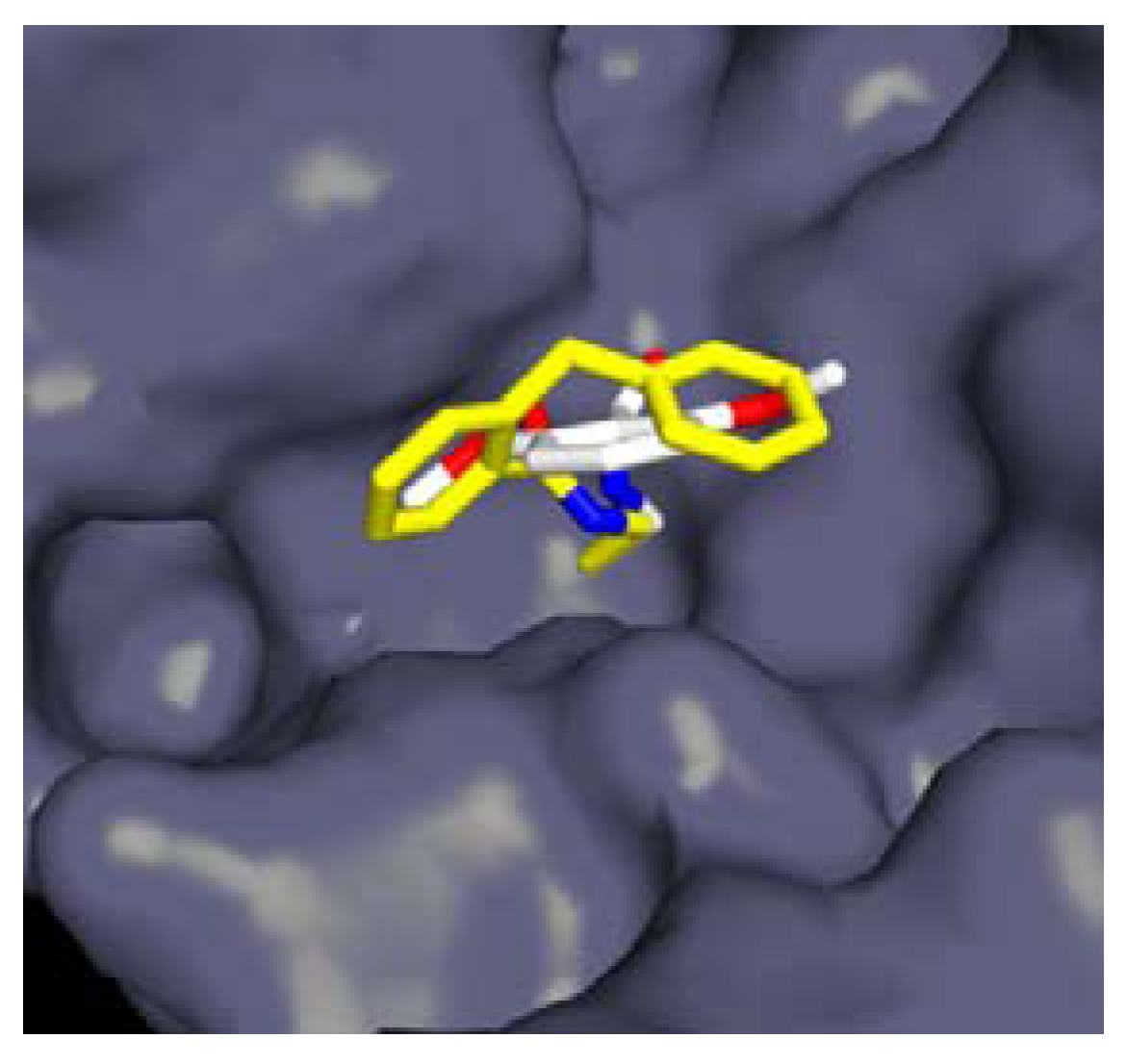

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Compound | |
|---|---|---|
| 9 | 10 | |
| Bacillus subtilis ATCC 6633 | 16 | 32 |
| Listeria monocytogenes ATCC 14780 | 32 | 64 |
| Enterococcus hirae ATCC 8790 | 32 | 32 |
| Staphylococcus aureus ATCC25923 | 32 | 64 |
| Staphylococcus aureus ATCC 43300 (MRSA) | 32 | 128 |
3.1.2. Carbonyl Compounds
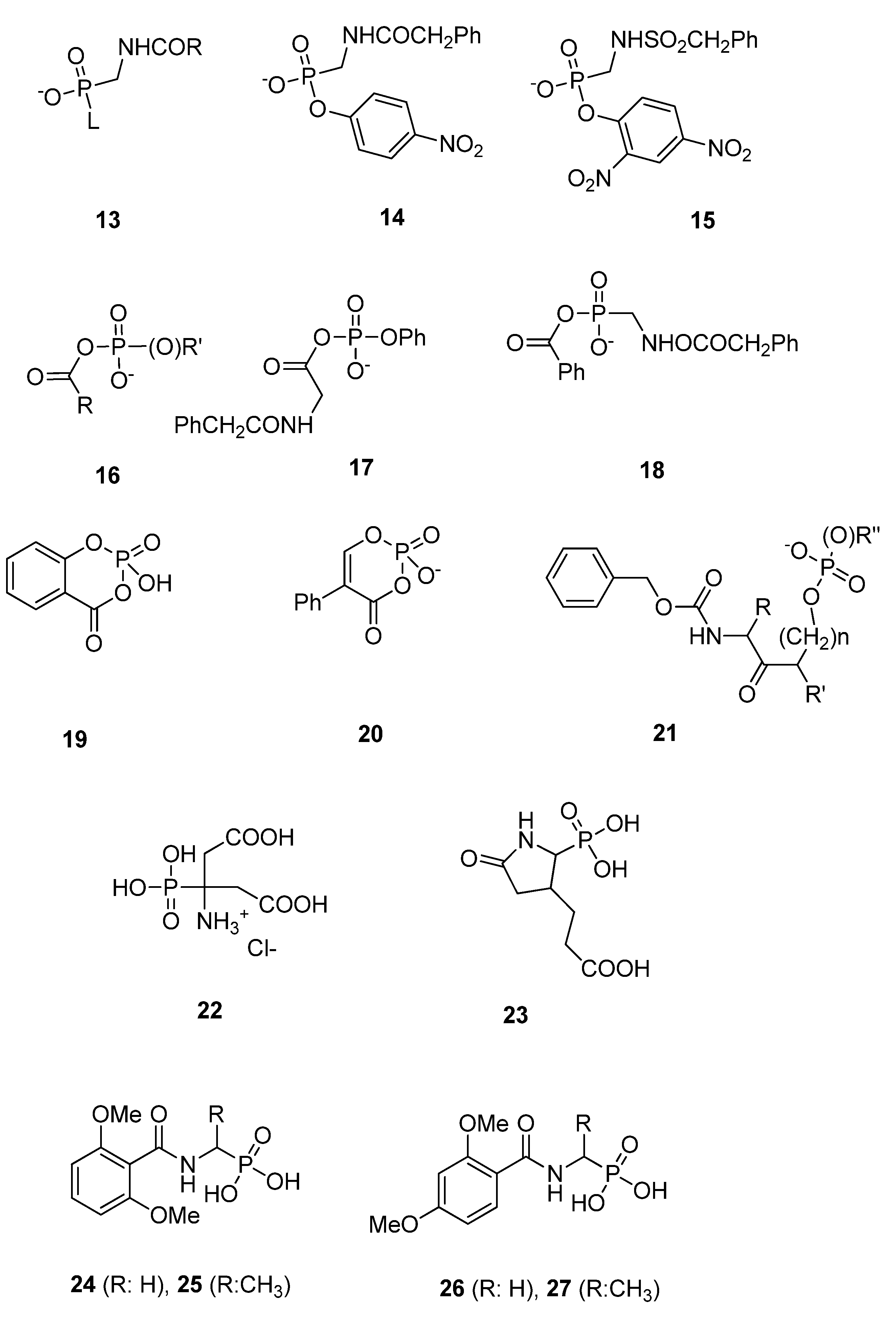
3.1.3. Phosph(on)ates
3.2. Substrate Analogs
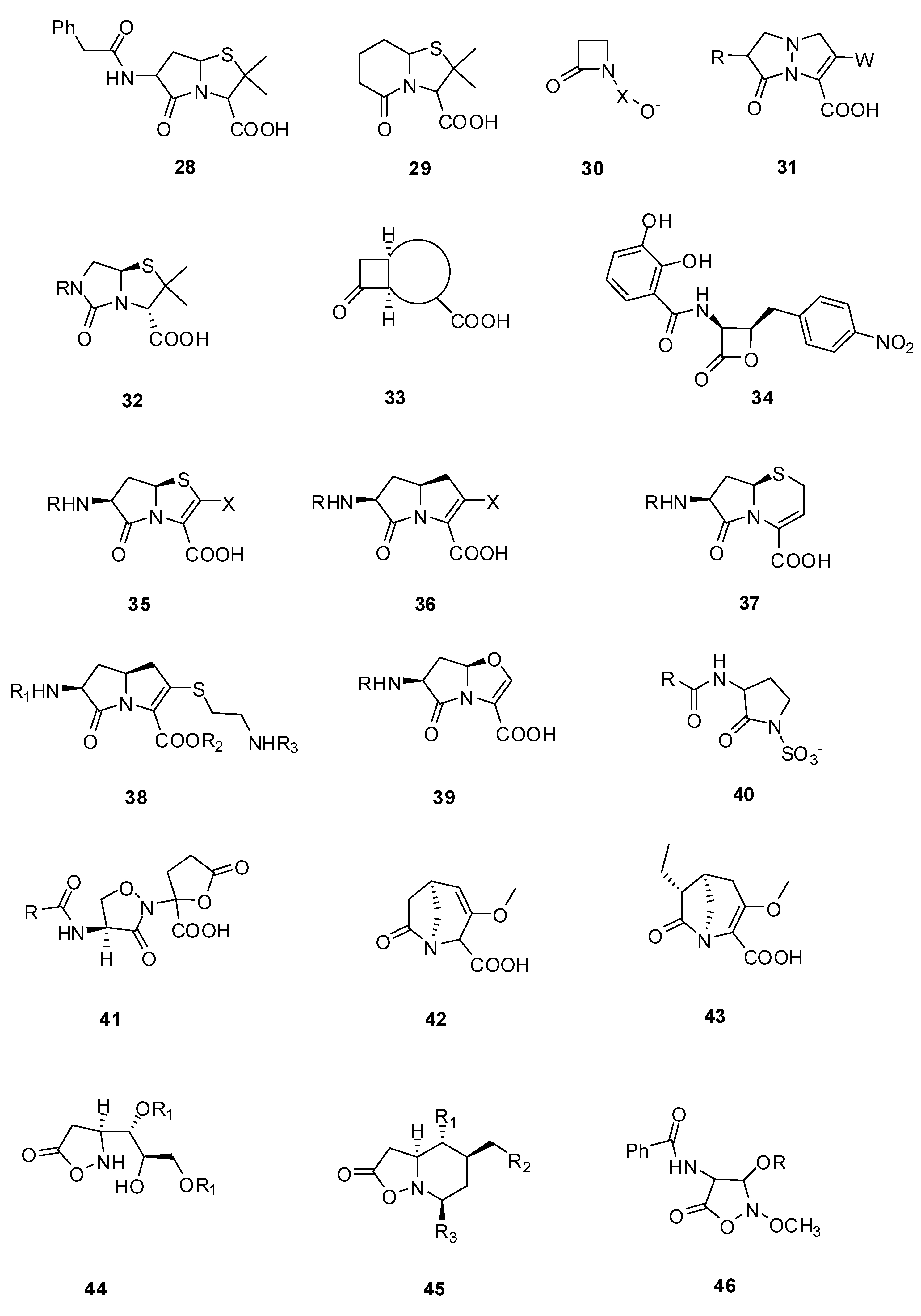
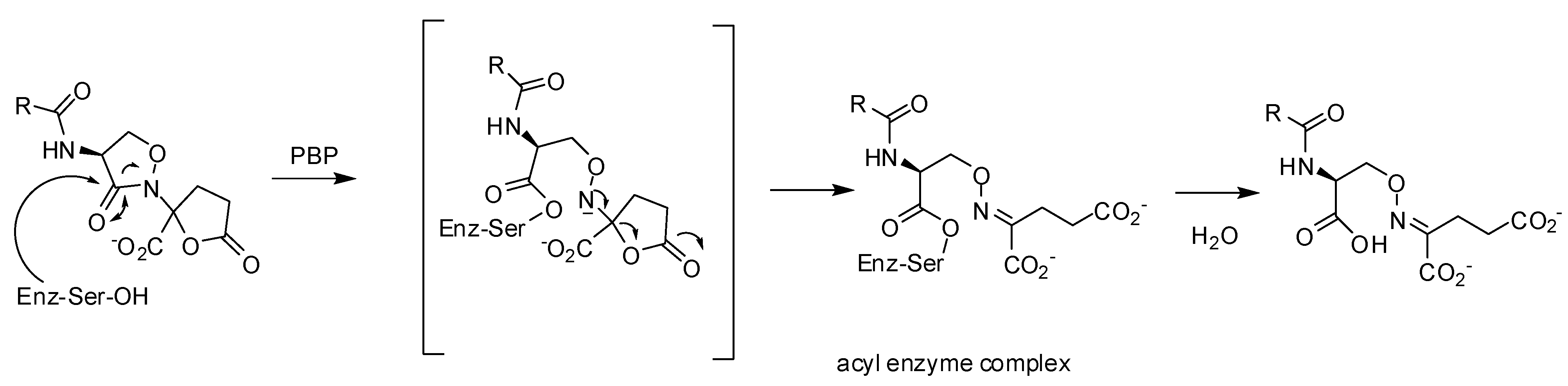
3.2.1. Pyrazolidinones

| Organism | Compound | ||
|---|---|---|---|
| 47 | 48 | 49 | |
| Staphylococcus aureus (X1.1) | 32 | 32 | >128 |
| Streptococcus pyogenes (C203) | 0.5 | 0.13 | 1 |
| Haemophilis influenzae (76) | 8 | 0.5 | 0.03 |
| Escherichia coli (EC14) | 2 | 0.06 | 0.03 |
| Klebsiella pneumoniae (X26) | 2 | 0.13 | 0.13 |
| Enterobacter cloacae (EB5) | 16 | 0.25 | 0.13 |
3.2.2. Lactivicin Analogs

| Organism | Compound | ||||
|---|---|---|---|---|---|
| 50 | 51 | 52 | 53 | 54 | |
| Staphylococcus aureus (FDA 209P) | 3.1 | 0.2 | 0.4 | 1.6 | 0.4 |
| Escherichia coli (O111) | 100 | 3.1 | 6.3 | 1.6 | 3.1 |
| Klebsiella pneumoniae (DT) | 100 | 3.1 | 25 | 3.1 | 6.3 |
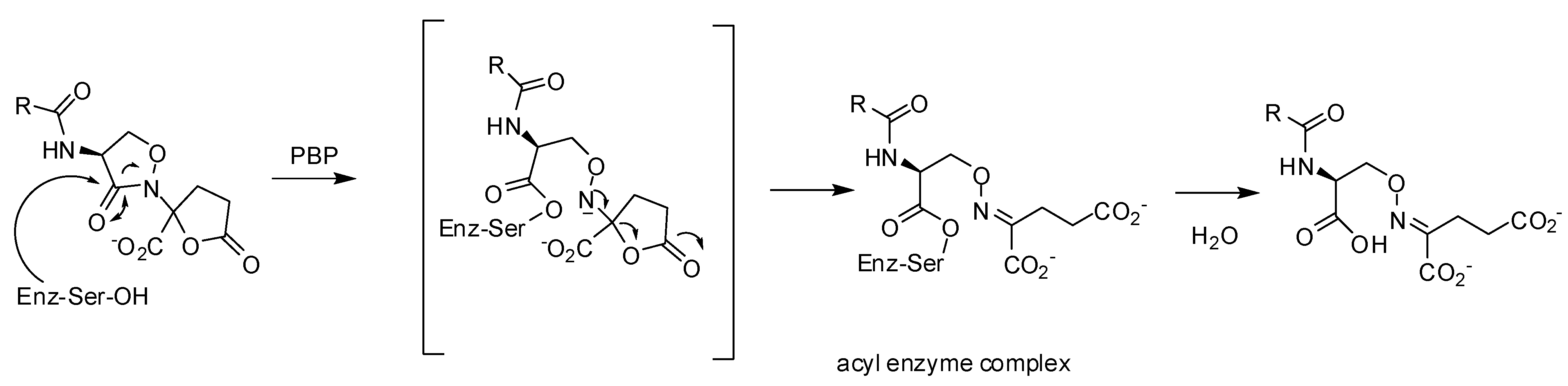
3.3. Non-covalent Inhibitors
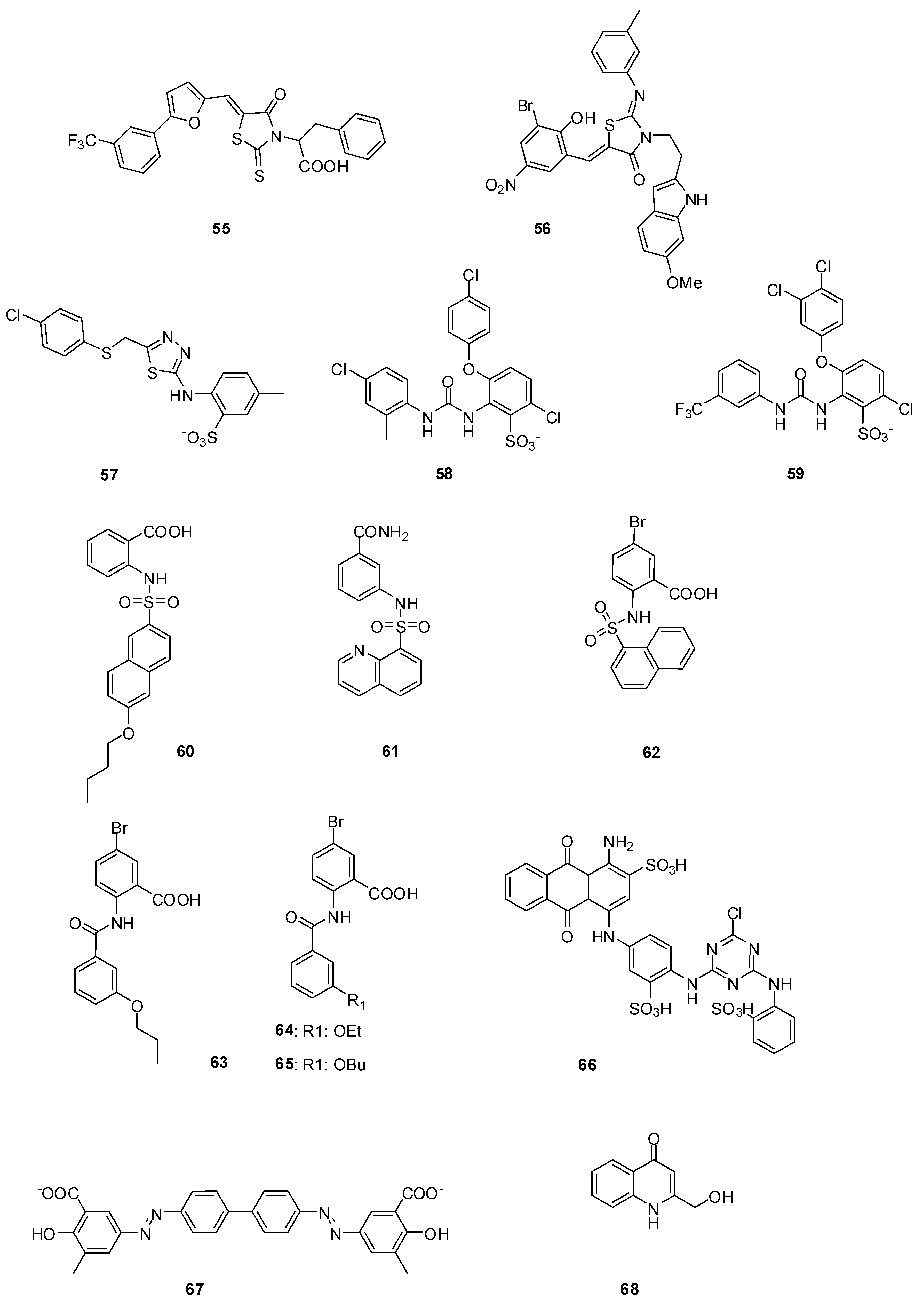
3.3.1. Arylalkylidene Rhodanines and Arylalkylidene Iminotriazolidenes
3.3.2. Aminothiadiazole and Ortho-phenoxyldiphenylurea Derivatives
| Molecule | PBP2aIC50 [µM] | PBP5fmIC50 [µM] | 5204 PBP2xIC50 [µM] |
|---|---|---|---|
| 57 1 | nd | nd | 219 |
| 58 1 | nd | nd | 71 |
| 59 1 | nd | nd | 72 |
| 60 1 | 97 | no inhibition at 1 mM | 391 |
| 61 1 | residual activity *: 58% | 930 | no inhibition at 1 mM |
| 62 1 | 80 | no inhibition at 1 mM | nd |
| 63 1 | 230 | residual activity *: 72% | 155 |
| 64 1 | 490 | residual activity *: 83% | nd |
| 65 1 | 352 | residual activity *: 85% | nd |
| 66 2 | 24 | nd | nd |
| 67 2 | 13 | nd | nd |
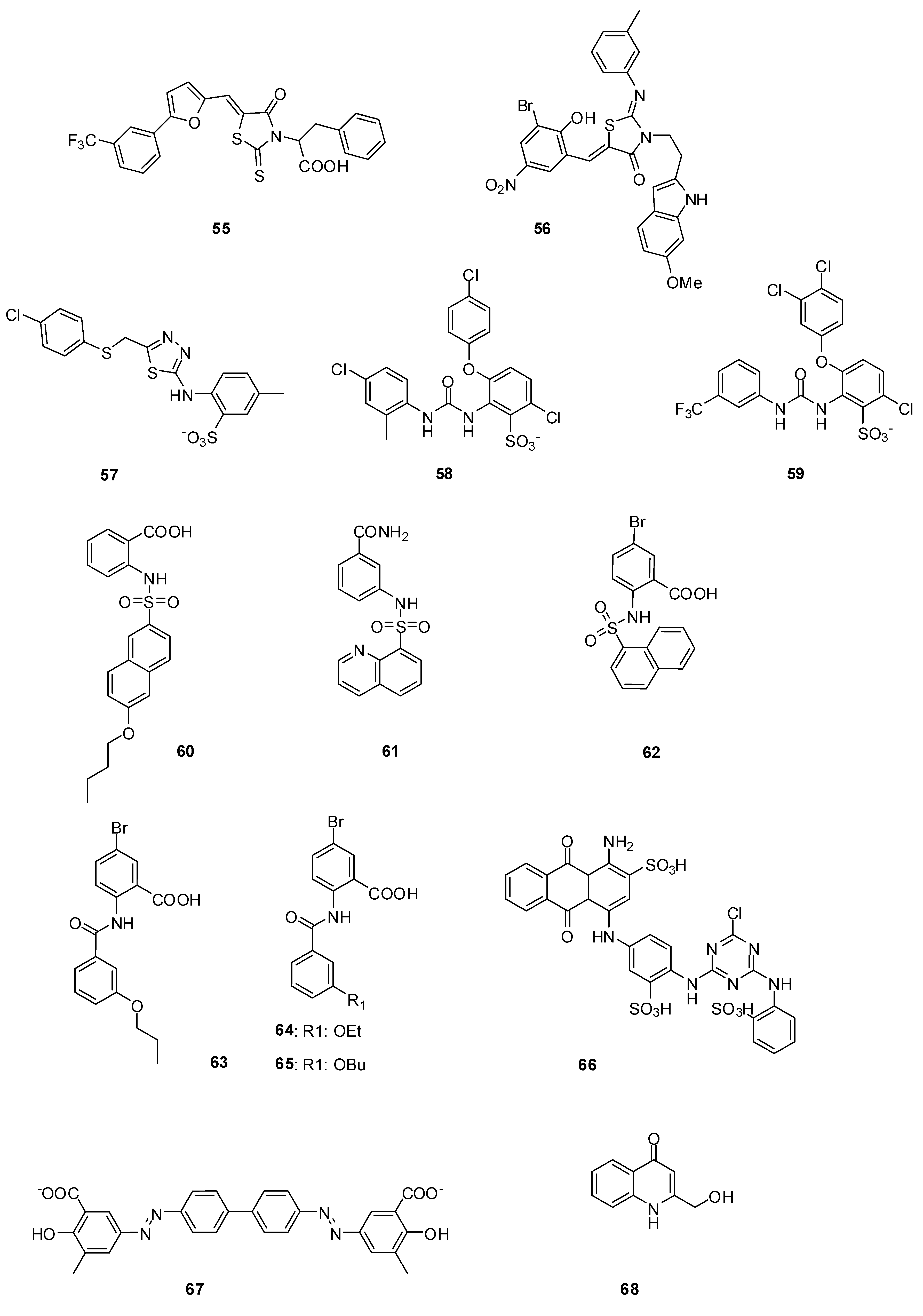
3.3.3. Naphthalene Sulfonamides
3.3.4. Anthranilic Acids
3.3.5. Cibacron Blue and Erie Yellow
3.3.6. Cyclic Peptide
3.3.7. 4-Quinolones
4. Conclusions
Acknowledgments
References
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, Present, And future. Antimicrob. Agents Ch. 2011, 55, 4943–4960. [Google Scholar] [CrossRef]
- Demain, A.L.; Sanchez, S. Microbial drug discovery: 80 Years of progress. J. Antibiot. 2009, 62, 5–16. [Google Scholar] [CrossRef]
- Rammelkamp, C.H.; Maxon, T. Resistance of Staphylococcus aureus to the Action of Penicillin. Proc. Soc. Exp. Biol. Med. 1942, 51, 386–389. [Google Scholar]
- Jovetic, S.; Zhu, Y.; Marcone, G.L.; Marinelli, F.; Tramper, J. beta-Lactam and glycopeptide antibiotics: First and last line of defense? Trends Biotechnol. 2010, 28, 596–604. [Google Scholar] [CrossRef]
- Chambers, H.F. Methicillin resistance in staphylococci: Molecular and biochemical basis and clinical implications. Clin. Microbiol. Rev. 1997, 10, 781–791. [Google Scholar]
- Drawz, S.M.; Bonomo, R.A. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- Barlow, M. What antimicrobial resistance has taught us about horizontal gene transfer. Methods Mol. Biol. 2009, 532, 397–411. [Google Scholar] [CrossRef]
- Pernot, L.; Chesnel, L.; Le Gouellec, A.; Croize, J.; Vernet, T.; Dideberg, O.; Dessen, A. A PBP2x from a clinical isolate of Streptococcus pneumoniae exhibits an alternative mechanism for reduction of susceptibility to beta-lactam antibiotics. J. Biol. Chem. 2004, 279, 16463–16470. [Google Scholar]
- Brannigan, J.A.; Tirodimos, I.A.; Zhang, Q.Y.; Dowson, C.G.; Spratt, B.G. Insertion of an extra amino acid is the main cause of the low affinity of penicillin-binding protein 2 in penicillin-resistant strains of Neisseria gonorrhoeae. Mol. Microbiol. 1990, 4, 913–919. [Google Scholar] [CrossRef]
- Dabernat, H.; Delmas, C.; Seguy, M.; Pelissier, R.; Faucon, G.; Bennamani, S.; Pasquier, C. Diversity of beta-lactam resistance-conferring amino acid substitutions in penicillin-binding protein 3 of Haemophilus influenzae. Antimicrob. Agents Ch. 2002, 46, 2208–2218. [Google Scholar]
- Hakenbeck, R. beta-lactam-resistant Streptococcus pneumoniae: Epidemiology and evolutionary mechanism. Chemotherapy 1999, 45, 83–94. [Google Scholar] [CrossRef]
- Lambert, P.A. Bacterial resistance to antibiotics: Modified target sites. Adv. Drug Deliv. Rev. 2005, 57, 1471–1485. [Google Scholar] [CrossRef]
- Doumith, M.; Ellington, M.J.; Livermore, D.M.; Woodford, N. Molecular mechanisms disrupting porin expression in ertapenem-resistant Klebsiella and Enterobacter spp. clinical isolates from the UK. J. Antimicrob. Chemother. 2009, 63, 659–667. [Google Scholar] [CrossRef]
- Livermore, D.M. Of Pseudomonas, Porins, Pumps and carbapenems. J. Antimicrob. Chemother. 2001, 47, 247–250. [Google Scholar] [CrossRef]
- Livermore, D.M. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 2009, 64, i29–i36, (Suppl. 1). [Google Scholar] [CrossRef]
- Rice, L.B. The clinical consequences of antimicrobial resistance. Curr. Opin. Microbiol. 2009, 12, 476–481. [Google Scholar] [CrossRef]
- Aarestrup, F. Sustainable farming: Get pigs off antibiotics. Nature 2012, 486, 465–466. [Google Scholar] [CrossRef]
- Infectious Diseases Society of America. The 10 x '20 Initiative: Pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin. Infect. Dis. 2010, 50, 1081–1083. [CrossRef]
- Devasahayam, G.; Scheld, W.M.; Hoffman, P.S. Newer antibacterial drugs for a new century. Expert Opin. Inv. Drugs 2010, 19, 215–234. [Google Scholar] [CrossRef]
- Courvalin, P. Predictable and unpredictable evolution of antibiotic resistance. J. Intern. Med. 2008, 264, 4–16. [Google Scholar]
- Bebrone, C.; Lassaux, P.; Vercheval, L.; Sohier, J.S.; Jehaes, A.; Sauvage, E.; Galleni, M. Current challenges in antimicrobial chemotherapy: Focus on beta-lactamase inhibition. Drugs 2010, 70, 651–679. [Google Scholar] [CrossRef]
- Pratt, R.F. Beta-lactamase inhibitors: Non-beta-lactams. In Beta-lactamases; Frére, J.-M., Ed.; Nova Science Publisher, Inc: Hauppauge, NY, USA, 2012; pp. 259–292. [Google Scholar]
- Coleman, K. Diazabicyclooctanes (DBOs): A potent new class of non-beta-lactam beta-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. [Google Scholar] [CrossRef]
- Endimiani, A.; Choudhary, Y.; Bonomo, R.A. In vitro activity of NXL104 in combination with beta-lactams against Klebsiella pneumoniae isolates producing KPC carbapenemases. Antimicrob. Agents Ch. 2009, 53, 3599–3601. [Google Scholar] [CrossRef]
- Stachyra, T.; Levasseur, P.; Pechereau, M.C.; Girard, A.M.; Claudon, M.; Miossec, C.; Black, M.T. In vitro activity of the {beta}-lactamase inhibitor NXL104 against KPC-2 carbapenemase and Enterobacteriaceae expressing KPC carbapenemases. J. Antimicrob. Chemother. 2009, 64, 326–329. [Google Scholar] [CrossRef]
- Llarrull, L.I.; Testero, S.A.; Fisher, J.F.; Mobashery, S. The future of the beta-lactams. Curr. Opin. Microbiol. 2010, 13, 551–557. [Google Scholar] [CrossRef]
- Theuretzbacher, U. Resistance drives antibacterial drug development. Curr. Opin. Pharmacol. 2011, 11, 433–438. [Google Scholar] [CrossRef]
- Queenan, A.M.; Shang, W.; Flamm, R.; Bush, K. Hydrolysis and inhibition profiles of beta-lactamases from molecular classes A to D with doripenem, imipenem, and meropenem. Antimicrob. Agents Ch. 2010, 54, 565–569. [Google Scholar] [CrossRef]
- Vidaillac, C.; Rybak, M.J. Ceftobiprole: First cephalosporin with activity against methicillin-resistant Staphylococcus aureus. Pharmacotherapy 2009, 29, 511–525. [Google Scholar] [CrossRef]
- Laudano, J.B. Ceftaroline fosamil: A new broad-spectrum cephalosporin. J. Antimicrob. Chemoth. 2011, 66, ii11–ii18, (Suppl. 3). [Google Scholar] [CrossRef]
- Vidaillac, C.; Leonard, S.N.; Sader, H.S.; Jones, R.N.; Rybak, M.J. In vitro activity of ceftaroline alone and in combination against clinical isolates of resistant gram-negative pathogens, including beta-lactamase-producing Enterobacteriaceae and Pseudomonas aeruginosa. Antimicrob. Agents Ch. 2009, 53, 2360–2366. [Google Scholar] [CrossRef]
- Sliwa, A.; Dive, G.; Habib Jiwan, J.-L.; Marchand-Brynaert, J. Cyclodimerization by ring-closing metathesis: Synthesis, Computational, And biological evaluation of novel bis-azetidinyl-macrocycles. Tetrahedron 2010, 66, 9519–9527. [Google Scholar] [CrossRef]
- Sliwa, A.; Dive, G.; Zervosen, A.; Verlaine, O.; Sauvage, E.; Marchand-Brynaert, J. Unprecedented inhibition of resistant penicillin binding proteins by bis-2-oxoazetidinyl macrocycles. Med. Chem. Commun. 2012, 3, 344–351. [Google Scholar] [CrossRef]
- Urbach, A.; Dive, G.; Marchand-Brynaert, J. Novel Large-Ring 1,3-Bridged 2-Azetidinones as Potential Inhibitors of Penicillin-Binding Proteins. Eur. J. Org. Chem. 2009, 2009, 1757–1770. [Google Scholar] [CrossRef]
- Urbach, A.; Dive, G.; Tinant, B.; Duval, V.; Marchand-Brynaert, J. Large ring 1,3-bridged 2-azetidinones: Experimental and theoretical studies. Eur. J. Med. Chem. 2009, 44, 2071–2080. [Google Scholar] [CrossRef]
- Lim, D.; Strynadka, N.C. Structural basis for the beta lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 2002, 9, 870–876. [Google Scholar]
- Dessen, A.; Mouz, N.; Gordon, E.; Hopkins, J.; Dideberg, O. Crystal structure of PBP2x from a highly penicillin-resistant Streptococcus pneumoniae clinical isolate: A mosaic framework containing 83 mutations. J. Biol. Chem. 2001, 276, 45106–45112. [Google Scholar]
- Sauvage, E.; Kerff, F.; Fonze, E.; Herman, R.; Schoot, B.; Marquette, J.P.; Taburet, Y.; Prevost, D.; Dumas, J.; Leonard, G.; et al. The 2.4-A crystal structure of the penicillin-resistant penicillin-binding protein PBP5fm from Enterococcus faecium in complex with benzylpenicillin. Cell. Mol. Life Sci. 2002, 59, 1223–1232. [Google Scholar] [CrossRef]
- Frere, J.M.; Marchot, P. Inactivators in competition: How to deal with them ... and not! Biochem. Pharmacol. 2005, 70, 1417–1423. [Google Scholar] [CrossRef]
- Frère, J.-M.; Nguyen-Disteche, M.; Coyette, J.; Joris, B. Mode of action: Interaction with penicillin binding proteins. In The Chemistry of beta-lactams; Page, M., Ed.; Chapman and Hall: Glasgow, Scotland, 1992; pp. 148–195. [Google Scholar]
- Fuad, N.; Frere, J.M.; Ghuysen, J.M.; Duez, C.; Iwatsubo, M. Mode of interaction between beta-lactam antibiotics and the exocellular DD-carboxypeptidase—transpeptidase from Streptomyces R39. Biochem. J. 1976, 155, 623–629. [Google Scholar]
- Jamin, M.; Damblon, C.; Millier, S.; Hakenbeck, R.; Frere, J.M. Penicillin-binding protein 2x of Streptococcus pneumoniae: Enzymic activities and interactions with beta-lactams. Biochem. J. 1993, 292, 735–741. [Google Scholar]
- Jamin, M.; Hakenbeck, R.; Frere, J.M. Penicillin binding protein 2x as a major contributor to intrinsic beta-lactam resistance of Streptococcus pneumoniae. FEBS Lett. 1993, 331, 101–104. [Google Scholar] [CrossRef]
- Leyh-Bouille, M.; Nguyen-Disteche, M.; Pirlot, S.; Veithen, A.; Bourguignon, C.; Ghuysen, J.M. Streptomyces K15 DD-peptidase-catalysed reactions with suicide beta-lactam carbonyl donors. Biochem. J. 1986, 235, 177–182. [Google Scholar]
- Lakaye, B.; Damblon, C.; Jamin, M.; Galleni, M.; Lepage, S.; Joris, B.; Marchand-Brynaert, J.; Frydrych, C.; Frere, J.M. Synthesis, Purification and kinetic properties of fluorescein-labelled penicillins. Biochem. J. 1994, 300, 141–145. [Google Scholar]
- Zhao, G.; Meier, T.I.; Kahl, S.D.; Gee, K.R.; Blaszczak, L.C. BOCILLIN FL, A sensitive and commercially available reagent for detection of penicillin-binding proteins. Antimicrob. Agents Ch. 1999, 43, 1124–1128. [Google Scholar]
- Dargis, M.; Malouin, F. Use of biotinylated beta-lactams and chemiluminescence for study and purification of penicillin-binding proteins in bacteria. Antimicrob. Agents Ch. 1994, 38, 973–980. [Google Scholar]
- Toney, J.H.; Hammond, G.G.; Leiting, B.; Pryor, K.D.; Wu, J.K.; Cuca, G.C.; Pompliano, D.L. Soluble penicillin-binding protein 2a: Beta-lactam binding and inhibition by non-beta-lactams using a 96-well format. Anal. Biochem. 1998, 255, 113–119. [Google Scholar]
- Stefanova, M.; Bobba, S.; Gutheil, W.G. A microtiter plate-based beta-lactam binding assay for inhibitors of high-molecular-mass penicillin-binding proteins. Anal. Biochem. 2010, 396, 164–166. [Google Scholar] [CrossRef]
- Bobba, S.; Ponnaluri, V.K.; Mukherji, M.; Gutheil, W.G. Microtiter plate-based assay for inhibitors of penicillin-binding protein 2a from methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Ch. 2011, 55, 2783–2787. [Google Scholar] [CrossRef]
- Inglis, S.R.; Strieker, M.; Rydzik, A.M.; Dessen, A.; Schofield, C.J. A boronic-acid-based probe for fluorescence polarization assays with penicillin binding proteins and beta-lactamases. Anal. Biochem. 2012, 420, 41–47. [Google Scholar]
- Contreras-Martel, C.; Amoroso, A.; Woon, E.C.; Zervosen, A.; Inglis, S.; Martins, A.; Verlaine, O.; Rydzik, A.M.; Job, V.; Luxen, A.; et al. Structure-guided design of cell wall biosynthesis inhibitors that overcome beta-lactam resistance in Staphylococcus aureus (MRSA). ACS Chem. Biol. 2011, 6, 943–951. [Google Scholar] [CrossRef]
- Zervosen, A.; Bouillez, A.; Herman, A.; Amoroso, A.; Joris, B.; Sauvage, E.; Charlier, P.; Luxen, A. Synthesis and evaluation of boronic acids as inhibitors of Penicillin Binding Proteins of classes A, B and C. Bioorg. Med. Chem. 2012, 20, 3915–3924. [Google Scholar] [CrossRef]
- Zervosen, A.; Lu, W.P.; Chen, Z.; White, R.E.; Demuth, T.P., Jr.; Frere, J.M. Interactions between penicillin-binding proteins (PBPs) and two novel classes of PBP inhibitors, Arylalkylidene rhodanines and arylalkylidene iminothiazolidin-4-ones. Antimicrob. Agents Ch. 2004, 48, 961–969. [Google Scholar] [CrossRef]
- Adam, M.; Damblon, C.; Plaitin, B.; Christiaens, L.; Frere, J.M. Chromogenic Depsipeptide Substrates for Beta-Lactamases and Penicillin-Sensitive DD-Peptidases. Biochem. J. 1990, 270, 525–529. [Google Scholar]
- Damblon, C.; Zhao, G.H.; Jamin, M.; Ledent, P.; Dubus, A.; Vanhove, M.; Raquet, X.; Christiaens, L.; Frere, J.M. Breakdown of the stereospecificity of DD-peptidases and beta-lactamases with thiolester substrates. Biochem. J. 1995, 309, 431–436. [Google Scholar]
- Sauvage, E.; Zervosen, A.; Dive, G.; Herman, R.; Amoroso, A.; Joris, B.; Fonze, E.; Pratt, R.F.; Luxen, A.; Charlier, P.; et al. Structural basis of the inhibition of class A beta-lactamases and penicillin-binding proteins by 6-beta-iodopenicillanate. J. Am. Chem. Soc. 2009, 131, 15262–15269. [Google Scholar]
- Zervosen, A.; Herman, R.; Kerff, F.; Herman, A.; Bouillez, A.; Prati, F.; Pratt, R.F.; Frere, J.M.; Joris, B.; Luxen, A.; et al. Unexpected tricovalent binding mode of boronic acids within the active site of a penicillin-binding protein. J. Am. Chem. Soc. 2011, 133, 10839–10848. [Google Scholar]
- Gao, W.; Xing, B.; Tsien, R.Y.; Rao, J. Novel fluorogenic substrates for imaging beta-lactamase gene expression. J. Am. Chem. Soc. 2003, 125, 11146–11147. [Google Scholar] [CrossRef]
- Xing, B.; Khanamiryan, A.; Rao, J. Cell-permeable near-infrared fluorogenic substrates for imaging beta-lactamase activity. J. Am. Chem. Soc. 2005, 127, 4158–4159. [Google Scholar] [CrossRef]
- Zlokarnik, G.; Negulescu, P.A.; Knapp, T.E.; Mere, L.; Burres, N.; Feng, L.; Whitney, M.; Roemer, K.; Tsien, R.Y. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science 1998, 279, 84–88. [Google Scholar] [CrossRef]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef]
- Shoichet, B.K. Screening in a spirit haunted world. Drug Discov. Today 2006, 11, 607–615. [Google Scholar] [CrossRef]
- Kraut, J. Serine proteases: Structure and mechanism of catalysis. Annu. Rev. Biochem. 1977, 46, 331–358. [Google Scholar] [CrossRef]
- Mattei, P.J.; Neves, D.; Dessen, A. Bridging cell wall biosynthesis and bacterial morphogenesis. Curr. Opin. Struct. Biol. 2010, 20, 749–755. [Google Scholar] [CrossRef]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef]
- Pechenov, A.; Stefanova, M.E.; Nicholas, R.A.; Peddi, S.; Gutheil, W.G. Potential transition state analogue inhibitors for the penicillin-binding proteins. Biochemistry 2003, 42, 579–588. [Google Scholar] [CrossRef]
- Nicola, G.; Peddi, S.; Stefanova, M.; Nicholas, R.A.; Gutheil, W.G.; Davies, C. Crystal structure of Escherichia coli penicillin-binding protein 5 bound to a tripeptide boronic acid inhibitor: A role for Ser-110 in deacylation. Biochemistry 2005, 44, 8207–8217. [Google Scholar]
- Dzhekieva, L.; Rocaboy, M.; Kerff, F.; Charlier, P.; Sauvage, E.; Pratt, R.F. Crystal structure of a complex between the Actinomadura R39 DD-peptidase and a peptidoglycan-mimetic boronate inhibitor: Interpretation of a transition state analogue in terms of catalytic mechanism. Biochemistry 2010, 49, 6411–6419. [Google Scholar] [CrossRef]
- Inglis, S.R.; Zervosen, A.; Woon, E.C.; Gerards, T.; Teller, N.; Fischer, D.S.; Luxen, A.; Schofield, C.J. Synthesis and evaluation of 3-(dihydroxyboryl)benzoic acids as D,D-carboxypeptidase R39 inhibitors. J. Med. Chem. 2009, 52, 6097–6106. [Google Scholar] [CrossRef]
- Woon, E.C.Y.; Zervosen, A.; Sauvage, E.; Simmons, K.J.; Zivec, M.; Inglis, S.R.; Fishwick, C.W.G.; Gobec, S.; Charlier, P.; Luxen, A.; et al. Structure Guided Development of Potent Reversibly Binding Penicillin Binding Protein Inhibitors. ACS Med. Chem. Lett. 2011, 2, 219–223. [Google Scholar] [CrossRef]
- Trippier, P.C.; McGuigan, C. Boronic acids in medicinal chemistry: Anticancer, Antibacterial and antiviral applications. Med. Chem. Commun. 2010, 1, 183–198. [Google Scholar]
- Li, N.; Rahil, J.; Wright, M.E.; Pratt, R.F. Structure-activity studies of the inhibition of serine beta-lactamases by phosphonate monoesters. Bioorg. Med. Chem. 1997, 5, 1783–1788. [Google Scholar] [CrossRef]
- Pratt, R.F. Inhibition of a class C beta-lactamase by a specific phosphonate monoester. Science 1989, 246, 917–919. [Google Scholar]
- Rahil, J.; Pratt, R.F. Phosphonate monoester inhibitors of class A beta-lactamases. Biochem. J. 1991, 275, 793–795. [Google Scholar]
- Chen, C.C.; Rahil, J.; Pratt, R.F.; Herzberg, O. Structure of a phosphonate-inhibited beta-lactamase: An analog of the tetrahedral transition state/intermediate of beta-lactam hydrolysis. J. Mol. Biol. 1993, 234, 165–178. [Google Scholar] [CrossRef]
- Lobkovsky, E.; Billings, E.M.; Moews, P.C.; Rahil, J.; Pratt, R.F.; Knox, J.R. Crystallographic structure of a phosphonate derivative of the Enterobacter cloacae P99 cephalosporinase: Mechanistic interpretation of a beta-lactamase transition-state analog. Biochemistry 1994, 33, 6762–6772. [Google Scholar]
- Maveyraud, L.; Pratt, R.F.; Samama, J.P. Crystal structure of an acylation transition-state analog of the TEM-1 beta-lactamase: Mechanistic implications for class A beta-lactamases. Biochemistry 1998, 37, 2622–2628. [Google Scholar]
- Morrison, M.J.; Li, N.; Pratt, R.F. Inverse acyl phosph(on)ates: Substrates or inhibitors of beta-lactam-recognizing enzymes? Bioorg. Chem. 2001, 29, 271–281. [Google Scholar] [CrossRef]
- Pratt, R.F.; Hammar, N.J. Salicyloyl Cyclic Phosphate, a “Penicillin-Like” Inhibitor of β-Lactamases. J. Am. Chem. Soc. 1998, 120, 3004–3006. [Google Scholar] [CrossRef]
- Silvaggi, N.R.; Kaur, K.; Adediran, S.A.; Pratt, R.F.; Kelly, J.A. Toward Better Antibiotics: Crystallographic Studies of a Novel Class of DD-Peptidase/β-Lactamase Inhibitors. Biochemistry 2004, 43, 7046–7053. [Google Scholar]
- Perumal, S.K.; Pratt, R.F. Synthesis and evaluation of ketophosph(on)ates as beta-lactamase inhibitors. J. Org. Chem. 2006, 71, 4778–4785. [Google Scholar] [CrossRef]
- Stefanova, M.E.; Davies, C.; Nicholas, R.A.; Gutheil, W.G. pH, Inhibitor, And substrate specificity studies on Escherichia coli penicillin-binding protein 5. Biochim. Biophys. Acta 2002, 1597, 292–300. [Google Scholar] [CrossRef]
- Beck, J.; Gharbi, S.; Herteg-Fernea, A.; Vercheval, L.; Bebrone, C.; Lassaux, P.; Zervosen, A.; Marchand-Brynaert, J. Aminophosphonic Acids and Aminobis(phosphonic acids) as Potential Inhibitors of Penicillin-Binding Proteins. Eur. J. Org. Chem. 2009, 85–97. [Google Scholar]
- Jungheim, L.N.; Ternansky, R.J. Non-beta-lactam mimics of beta-lactam antibiotics. In The Chemistry of Beta-Lactams; Page, M.I., Ed.; Chapman and Hall: London, UK, 1992; pp. 306–324. [Google Scholar]
- Imming, P.; Klar, B.; Dix, D. Hydrolytic stability versus ring size in lactams: Implications for the development of lactam antibiotics and other serine protease inhibitors. J. Med. Chem. 2000, 43, 4328–4331. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Lynch, G.P.; Pitlik, J. Gamma-lactam analogues of beta-lactam antibiotics. J. Antibiot. 1991, 44, 1–24. [Google Scholar] [CrossRef]
- Marchand-Brynaert, J.; Ghosez, L. Non-beta-lactam analogs of penicillins and cephalosporins. In Recent Progress in the Chemical Synthesis of Antibiotics; Ohno, M., Lukais, G., Eds.; Springer-Verlag: Berlin, Germany, 1990; pp. 729–794. [Google Scholar]
- Aszodi, J.; Rowlands, D.A.; Mauvais, P.; Collette, P.; Bonnefoy, A.; Lampilas, M. Design and synthesis of bridged gamma-lactams as analogues of beta-lactam antibiotics. Bioorg. Med. Chem. Lett. 2004, 14, 2489–2492. [Google Scholar]
- Panfil, I.; Urbańczyk-Lipkowska, Z.; Chmielewski, M. Isoxazolidin-5-one analogs of β-lactam antibiotics. Carbohyd. Res. 1998, 306, 505–515. [Google Scholar] [CrossRef]
- Cao, X.; Iqbal, A.; Patel, A.; Gretz, P.; Huang, G.; Crowder, M.; Day, R.A. 3-alkoxy-5-isoxazolidinones mimic beta-lactams. Biochem. Biophys. Res. Commun. 2003, 311, 267–271. [Google Scholar] [CrossRef]
- Bonnefoy, A.; Dupuis-Hamelin, C.; Steier, V.; Delachaume, C.; Seys, C.; Stachyra, T.; Fairley, M.; Guitton, M.; Lampilas, M. In vitro activity of AVE1330A, An innovative broad-spectrum non-beta-lactam beta-lactamase inhibitor. J. Antimicrob. Chemother. 2004, 54, 410–417. [Google Scholar] [CrossRef]
- Allen, N.E.; Hobbs, J.N., Jr.; Preston, D.A.; Turner, J.R.; Wu, C.Y. Antibacterial properties of the bicyclic pyrazolidinones. J. Antibiot. 1990, 43, 92–99. [Google Scholar] [CrossRef]
- Harada, S.; Tsubotani, S.; Hida, T.; Ono, H.; Okazaki, H. Structure of lactivicin, An antibiotic having a new nucleus and similar biological activities to β-lactam antibiotics. Tetrahedron Lett. 1986, 27, 6229–6232. [Google Scholar] [CrossRef]
- Nozaki, Y.; Katayama, N.; Harada, S.; Ono, H.; Okazaki, H. Lactivicin, A naturally occurring non-beta-lactam antibiotic having beta-lactam-like action: Biological activities and mode of action. J. Antibiot. 1989, 42, 84–93. [Google Scholar] [CrossRef]
- Nozaki, Y.; Katayama, N.; Ono, H.; Tsubotani, S.; Harada, S.; Okazaki, H.; Nakao, Y. Binding of a non-beta-lactam antibiotic to penicillin-binding proteins. Nature 1987, 325, 179–180. [Google Scholar] [CrossRef]
- Harada, S.; Tsubotani, S.; Hida, T.; Koyana, K.; Kondo, M.; Ono, H. Chemistry of a new antibiotic: Lactivicin. Tetrahedron 1988, 44, 6589–6606. [Google Scholar] [CrossRef]
- Natsugari, H.; Kawano, Y.; Morimoto, A.; Yoshioka, K.; Ochiai, M. Synthesis of lactivicin and its derivatives. J. Chem. Soc. Chem. Commun. 1987, 62–63. [Google Scholar]
- Tamura, N.; Matsushita, Y.; Kawano, Y.; Yoshioka, K. Synthesis and antibacterial activity of lactivicin derivatives. Chem. Pharm. Bull. 1990, 38, 116–122. [Google Scholar] [CrossRef]
- Macheboeuf, P.; Fischer, D.S.; Brown, T., Jr.; Zervosen, A.; Luxen, A.; Joris, B.; Dessen, A.; Schofield, C.J. Structural and mechanistic basis of penicillin-binding protein inhibition by lactivicins. Nat. Chem. Biol. 2007, 3, 565–569. [Google Scholar] [CrossRef]
- Grant, E.B.; Guiadeen, D.; Baum, E.Z.; Foleno, B.D.; Jin, H.; Montenegro, D.A.; Nelson, E.A.; Bush, K.; Hlasta, D.J. The synthesis and SAR of rhodanines as novel class C beta-lactamase inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 2179–2182. [Google Scholar] [CrossRef]
- Miguet, L.; Zervosen, A.; Gerards, T.; Pasha, F.A.; Luxen, A.; Disteche-Nguyen, M.; Thomas, A. Discovery of new inhibitors of resistant Streptococcus pneumoniae penicillin binding protein (PBP) 2x by structure-based virtual screening. J. Med. Chem. 2009, 52, 5926–5936. [Google Scholar]
- Turk, S.; Verlaine, O.; Gerards, T.; Zivec, M.; Humljan, J.; Sosic, I.; Amoroso, A.; Zervosen, A.; Luxen, A.; Joris, B.; et al. New noncovalent inhibitors of penicillin-binding proteins from penicillin-resistant bacteria. PLoS One 2011, 6, e19418. [Google Scholar]
- Sosic, I.; Turk, S.; Sinreith, M.; Trost, N.; Verlaine, O.; Amoroso, A.; Zervosen, A.; Luxen, A.; Joris, B.; Gobec, S. Exploration of the chemical space of novel naphthalene-sulfonamide and anthranilic acid-based inhibitors of penicillin-binding proteins. Acta Chim. Slov. 2012, 59, 380–388. [Google Scholar]
- Phichith, D.; Bun, S.; Padiolleau-Lefevre, S.; Guellier, A.; Banh, S.; Galleni, M.; Frere, J.M.; Thomas, D.; Friboulet, A.; Avalle, B. Novel peptide inhibiting both TEM-1 beta-lactamase and penicillin-binding proteins. FEBS J. 2010, 277, 4965–4972. [Google Scholar] [CrossRef]
- Shilabin, A.G.; Dzhekieva, L.; Misra, P.; Jayaram, B.; Pratt, R.F. 4-Quinolones as Noncovalent Inhibitors of High Molecular Mass Penicillin-Binding Proteins. ACS Med. Chem. Lett. 2012, 3, 592–595. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zervosen, A.; Sauvage, E.; Frère, J.-M.; Charlier, P.; Luxen, A. Development of New Drugs for an Old Target — The Penicillin Binding Proteins. Molecules 2012, 17, 12478-12505. https://doi.org/10.3390/molecules171112478
Zervosen A, Sauvage E, Frère J-M, Charlier P, Luxen A. Development of New Drugs for an Old Target — The Penicillin Binding Proteins. Molecules. 2012; 17(11):12478-12505. https://doi.org/10.3390/molecules171112478
Chicago/Turabian StyleZervosen, Astrid, Eric Sauvage, Jean-Marie Frère, Paulette Charlier, and André Luxen. 2012. "Development of New Drugs for an Old Target — The Penicillin Binding Proteins" Molecules 17, no. 11: 12478-12505. https://doi.org/10.3390/molecules171112478