Antimycobacterial Assessment of Salicylanilide Benzoates including Multidrug-Resistant Tuberculosis Strains


Abstract
:1. Introduction
2. Results and Discussion
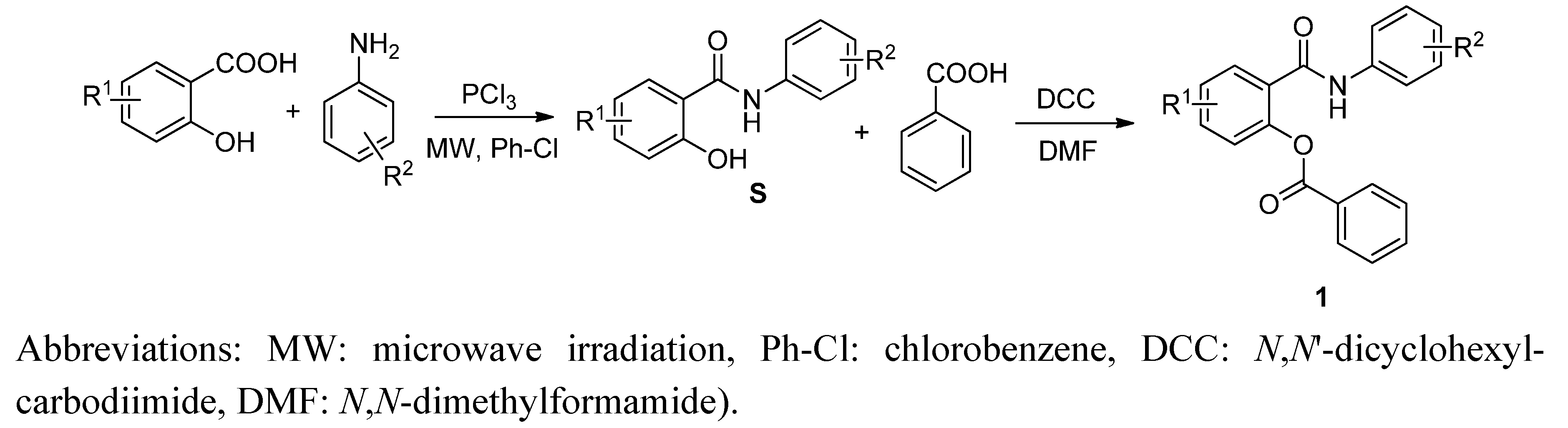
2.1. Chemistry
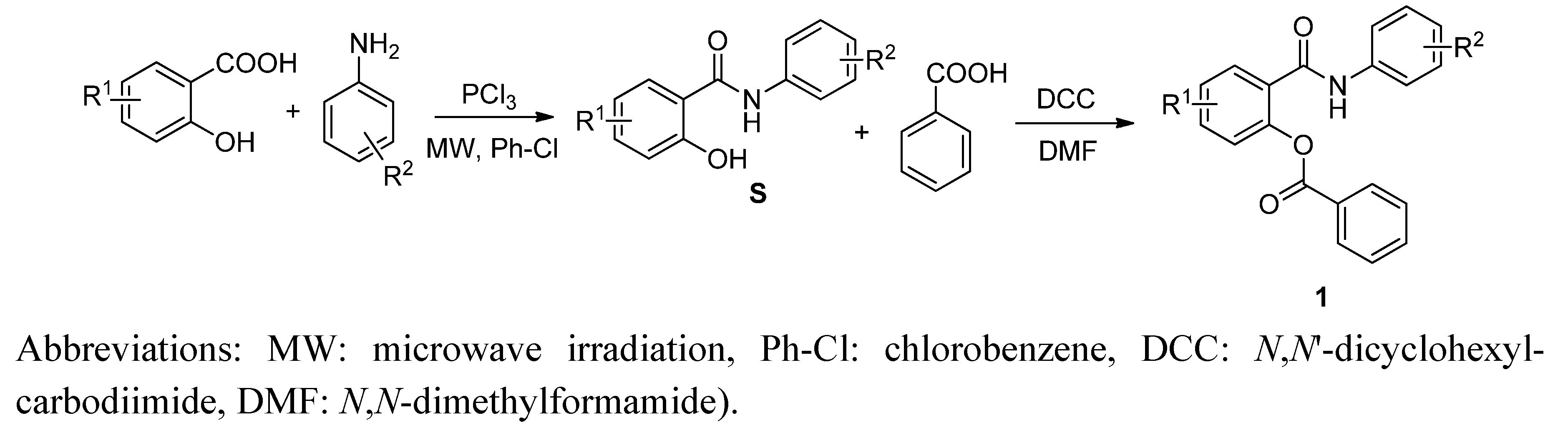
2.2. In Vitro Antimycobacterial Evaluation

{kind=link}
 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC [μmol/L] | |||||||||||||
| R1 | R2 | M. tuberculosis 331/88 | M. avium 330/88 | M. kansasii 235/80 | M. kansasii 6509/96 | ||||||||
| 14 d | 21 d | 14 d | 21 d | 7 d | 14 d | 21 d | 7 d | 14 d | 21 d | ||||
| 1a | 4-Cl | 3-Cl | 2 | 2 | 4 | 8 | 2 | 4 | 8 | 4 | 4 | 8 | |
| 1b | 5-Cl | 3-Cl | 4 | 4 | 8 | 8 | 2 | 4 | 4 | 4 | 4 | 8 | |
| 1c | 4-Cl | 4-Cl | 4 | 4 | 8 | 16 | 4 | 8 | 8 | 4 | 8 | 8 | |
| 1d | 5-Cl | 4-Cl | 2 | 4 | 4 | 4 | 2 | 2 | 4 | 2 | 2 | 4 | |
| 1e | 4-Cl | 3-Br | 2 | 2 | 8 | 16 | 2 | 4 | 8 | 2 | 4 | 8 | |
| 1f | 5-Cl | 3-Br | 4 | 4 | 4 | 8 | 2 | 4 | 4 | 2 | 4 | 4 | |
| 1g | 4-Cl | 4-Br | 2 | 2 | 4 | 8 | 2 | 4 | 4 | 2 | 4 | 4 | |
| 1h | 5-Cl | 4-Br | 2 | 2 | 4 | 4 | 2 | 4 | 4 | 2 | 4 | 4 | |
| 1i | 4-Cl | 3-F | 4 | 4 | 8 | 16 | 2 | 4 | 8 | 2 | 4 | 4 | |
| 1j | 5-Cl | 3-F | 4 | 8 | 8 | 16 | 2 | 8 | 8 | 2 | 4 | 8 | |
| 1k | 4-Cl | 4-F | 4 | 4 | 8 | 16 | 2 | 8 | 8 | 2 | 4 | 4 | |
| 1l | 5-Cl | 4-F | 8 | 8 | 4 | 8 | 2 | 4 | 8 | 4 | 8 | 8 | |
| 1m | 4-Cl | 3,4-diCl | 1 | 1 | 8 | 8 | 2 | 4 | 4 | 1 | 2 | 2 | |
| 1n | 5-Cl | 3,4-diCl | 2 | 2 | 8 | 8 | 1 | 2 | 4 | 1 | 2 | 4 | |
| 1o | 4-Cl | 4-CF3 | 0.5 | 1 | 4 | 4 | 1 | 1 | 1 | 2 | 2 | 2 | |
| 1p | 5-Cl | 4-CF3 | 2 | 2 | 8 | 8 | 1 | 2 | 4 | 1 | 2 | 4 | |
| 1q | 4-Cl | 3-CF3 | 2 | 2 | 8 | 16 | 2 | 4 | 4 | 2 | 4 | 4 | |
| 1r | 4-Br | 4-CF3 | 1 | 1 | 4 | 4 | 2 | 2 | 2 | 2 | 2 | 2 | |
| INH | - | - | 0.5-1 | 0.5-1 | >250 | >250 | >250 | >250 | >250 | 2 | 4 | 4-8 | |
| PAS | - | - | 62.5 | 62.5 | 32 | 125 | 125 | 1000 | >1000 | 32 | 125 | 500 | |
| BA | - | - | >1000 | >1000 | >1000 | >1000 | 1000 | >1000 | >1000 | 250 | 1000 | 1000 | |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| MIC [μmol/L] | ||||||||
| R1 | R2 | M. tuberculosis 331/88 | M. avium 330/88 | M. kansasii 235/80 | ||||
| 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | |||
| S-a | 5-Cl | 3-Cl | 4 | 8 | 8 | 16 | 4 | 8 |
| S-b | 4-Cl | 3-Cl | 4 | 4 | 16 | 16 | 4 | 8 |
| S-c | 5-Cl | 4-Cl | 4 | 4 | 8 | 8 | 8 | 8 |
| S-d | 4-Cl | 4-Cl | 4 | 4 | 8 | 8 | 4 | 8 |
| S-e | 5-Cl | 3-Br | NT | NT | NT | NT | NT | NT |
| S-f | 4-Cl | 3-Br | NT | NT | NT | NT | NT | NT |
| S-g | 5-Cl | 4-Br | 8 | 16 | 8 | 8 | 4 | 4 |
| S-h | 4-Cl | 4-Br | 4 | 4 | 16 | 16 | 4 | 4 |
| S-i | 5-Cl | 3-F | 8 | 8 | 31 | 31 | 8 | 8 |
| S-j | 4-Cl | 3-F | 8 | 16 | 32 | 32 | 32 | 32 |
| S-k | 5-Cl | 4-F | 16 | 16 | 16 | 16 | 4 | 4 |
| S-l | 4-Cl | 4-F | 16 | 16 | 32 | 32 | 16 | 32 |
| S-m | 5-Cl | 3,4-diCl | 4 | 8 | 16 | 16 | 4 | 4 |
| S-n | 4-Cl | 3,4-diCl | 4 | 4 | 16 | 16 | 8 | 8 |
| S-o | 5-Cl | 4-CF3 | 2 | 2 | 8 | 8 | 1 | 1 |
| S-p | 4-Cl | 4-CF3 | 4 | 4 | 8 | 8 | 4 | 4 |
| S-q | 5-Cl | 3-CF3 | NT | NT | NT | NT | NT | NT |
| S-r | 4-Br | 4-CF3 | 1 | 1 | 1 | 1 | 2 | 4 |
2.3. In Vitro Activity against Drug-Resistant Tuberculosis Strains
| MIC [μmol/L] | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M. tuberculosis 234/2005 | M. tuberculosis 53/2009 | M. tuberculosis Praha 1 | M. tuberculosis Praha 131 | M. tuberculosis 7357/1998 | M. tuberculosis 9449/2006 | ||||||||
| 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | 14 d | 21 d | ||
| 1m | 0.25 | 0.5 | 0.5 | 1 | 0.5 | 0.5 | 0.5 | 1 | 0.5 | 0.5 | 0.5 | 1 | |
| 1n | 0.25 | 0.5 | 1 | 2 | 1 | 1 | 0.5 | 1 | 0.5 | 1 | 2 | 2 | |
| 1o | 0.25 | 0.25 | 0.5 | 1 | 0.5 | 0.5 | 0.25 | 0.5 | 0.25 | 0.5 | 0.5 | 0.5 | |
| 1r | 0.5 | 0.5 | 1 | 2 | 1 | 1 | 0.5 | 1 | 0.25 | 0.5 | 1 | 1 | |
| INH | 14.6 | 14.6 | 14.6 | 14.6 | 14.6 | 14.6 | 14.6 | 14.6 | 14.6 | 14.6 | 58.3 | 58.3 | |
2.4. Cytotoxicity Evaluation
| IC50 [µmol/L] Hep G2 | SI for M. tuberculosis 331/88 | SI for MDR-TB strains | SI for XDR-TB strain | ||||
|---|---|---|---|---|---|---|---|
| 14 d | 21 d | 14 d | 21 d | 14 d | 14 d | ||
| 2m | 2.54 | 2.54 | 2.54 | 5.08–10.16 | 2.54–5.08 | 5.08 | 2.54 |
| 2o | 2.40 | 4.80 | 2.40 | 4.80–9.60 | 2.40–9.60 | 9.60 | 4.80 |
| 2r | 2.34 | 2.34 | 2.34 | 2.34–9.36 | 1.17–4.68 | 4.68 | 2.34 |
| S-m | 0.84 | 0.21 | 0.10 | - | - | - | - |
| S-o | 0.36 | 0.18 | 0.18 | - | - | - | - |
| S-r | 2.71 | 2.71 | 2.71 | - | - | - | - |
3. Experimental
3.1. Chemistry
3.2. In Vitro Antimycobacterial Susceptibility Testing
4. Conclusions
Acknowledgments
Conflict of Interest
- Sample Availability: Samples of the compounds S-a–S-r and 1a-r are available from the authors.
References
- Krátký, M.; Vinšová, J. Advances in the Development of Antituberculotics Acting on Multidrug-Resistant Strains. Chem. Listy 2010, 104, 998–1005. [Google Scholar]
- Sturdy, A.; Goodman, A.; Jose, R.J.; Loyse, A.; O’Donoghue, M.; Kon, O.M.; Dedicoat, M.J.; Harrison, T.S.; John, L.; Lipman, M.; et al. Multidrug-resistant tuberculosis (MDR-TB) treatment in the UK: A study of injectable use and toxicity in practice. J. Antimicrob. Chemother. 2011, 66, 1815–1820. [Google Scholar]
- La Rosa, V.; Poce, G.; Canseco, J.O.; Buroni, S.; Pasca, M.R.; Biava, M.; Raju, R.M.; Porretta, G.C.; Alfonso, S.; Battilocchio, C.; et al. MmpL3 Is the Cellular Target of the Antitubercular Pyrrole Derivative BM212. Antimicrob. Agents Chemother. 2012, 56, 324–331. [Google Scholar]
- Grzegorzewicz, A.E.; Pham, H.; Gundi, V.A.K.B.; Scherman, M.S.; North, E.J.; Hess, T.; Jones, V.; Gruppo, V.; Born, S.E.M.; Kordulakova, J.; et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012, 8, 334–341. [Google Scholar] [CrossRef]
- Batt, S.M.; Jabeen, T.; Bhowruth, V.; Quill, L.; Lund, P.A.; Eggeling, L.; Alderwick, L.J.; Futterer, K.; Besra, G.S. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 11354–11359. [Google Scholar]
- Cook, J.L. Nontuberculous mycobacteria: Opportunistic environmental pathogens for predisposed hosts. Br. Med. Bull. 2010, 96, 45–59. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J. Salicylanilide Ester Prodrugs as Potential Antimicrobial Agents—A Review. Curr. Pharm. Des. 2011, 17, 3494–3505. [Google Scholar] [CrossRef]
- Vinsova, J.; Imramovsky, A.; Buchta, V.; Ceckova, M.; Dolezal, M.; Staud, F.; Jampilek, J.; Kaustova, J. Salicylanilide Acetates: Synthesis and Antibacterial Evaluation. Molecules 2007, 12, 1–12. [Google Scholar] [CrossRef]
- Férriz, J.M.; Vávrová, K.; Kunc, F.; Imramovský, A.; Stolaříková, J.; Vavříková, E.; Vinšová, J. Salicylanilide carbamates: Antitubercular agents active against multidrug-resistant Mycobacterium tuberculosis strains. Bioorg. Med. Chem. 2010, 18, 1054–1061. [Google Scholar]
- Krátký, M.; Vinšová, J.; Buchta, V.; Horvati, K.; Bösze, S.; Stolaříková, J. New amino acid esters ofsalicylanilides active against MDR-TB and other microbes. Eur. J. Med. Chem. 2010, 45, 6106–6113. [Google Scholar] [CrossRef]
- Imramovský, A.; Vinšová, J.; Férriz, J.M.; Doležal, R.; Jampílek, J.; Kaustová, J.; Kunc, F. New antituberculotics originated from salicylanilides with promising in vitro activity against atypical mycobacterial strains. Bioorg. Med. Chem. 2009, 17, 3572–3579. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Rodriguez, N.G.; Stolaříková, J. Antimycobacterial Activity of Salicylanilide Benzenesulfonates. Molecules 2012, 17, 492–503. [Google Scholar] [CrossRef]
- Mathew, R.; Kruthiventi, A.K.; Prasad, J.V.; Kumar, S.P.; Srinu, G.; Chatterji, D. Inhibition of Mycobacterial Growth by Plumbagin Derivatives. Chem. Biol. Drug Des. 2010, 76, 34–42. [Google Scholar] [CrossRef]
- Muddassar, M.; Jang, J.W.; Gon, H.S.; Cho, Y.S.; Kim, E.E.; Keum, K.C.; Oh, T.; Cho, S.N.; Pae, A.N. Identification of novel antitubercular compounds through hybrid virtual screening approach. Bioorg. Med. Chem. 2010, 18, 6914–6921. [Google Scholar] [CrossRef]
- Gu, P.; Constantino, L.; Zhang, Y. Enhancement of the antituberculosis activity of weak acids by inhibitors of energy metabolism but not by anaerobiosis suggests that weak acids act differently from the front-line tuberculosis drug pyrazinamide. J. Med. Microbiol. 2008, 57, 1129–1134. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Novotná, E.; Mandíková, J.; Wsól, V.; Trejtnar, F.; Ulmann, V.; Stolaříková, J.; Fernandes, S.; Bhat, S.; Liu, J.O. Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase. Tuberculosis 2012, 92, 434–439. [Google Scholar] [CrossRef]
- Krátký, M.; Vinšová, J.; Buchta, V. In Vitro Antibacterial and Antifungal Activity of Salicylanilide Benzoates. ScientificWorldJournal 2012, 2012, 290628. [Google Scholar]
- Waisser, K.; Bureš, O.; Holý, P.; Kuneš, J.; Oswald, R.; Jirásková, L.; Pour, M.; Klimešová, V.; Kubicová, L.; Kaustová, J. Relationship between the Structure and Antimycobacterial Activity of Substituted Salicylanilides. Arch. Pharm. Pharm. Med. Chem. 2003, 336, 53–71. [Google Scholar] [CrossRef]
- Hilliard, J.J.; Goldschmidt, R.M.; Licata, L.; Baum, E.Z.; Bush, K. Multiple Mechanisms of Action for Inhibitors of Histidine Protein Kinases from Bacterial Two-Component Systems. Antimicrob. Agents Chemother. 1999, 43, 1693–1699. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krátký, M.; Vinšová, J.; Stolaříková, J. Antimycobacterial Assessment of Salicylanilide Benzoates including Multidrug-Resistant Tuberculosis Strains. Molecules 2012, 17, 12812-12820. https://doi.org/10.3390/molecules171112812
Krátký M, Vinšová J, Stolaříková J. Antimycobacterial Assessment of Salicylanilide Benzoates including Multidrug-Resistant Tuberculosis Strains. Molecules. 2012; 17(11):12812-12820. https://doi.org/10.3390/molecules171112812
Chicago/Turabian StyleKrátký, Martin, Jarmila Vinšová, and Jiřina Stolaříková. 2012. "Antimycobacterial Assessment of Salicylanilide Benzoates including Multidrug-Resistant Tuberculosis Strains" Molecules 17, no. 11: 12812-12820. https://doi.org/10.3390/molecules171112812