Forward Chemical Genetics in Yeast for Discovery of Chemical Probes Targeting Metabolism

Abstract
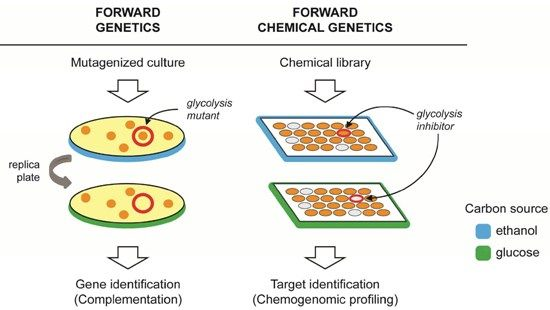
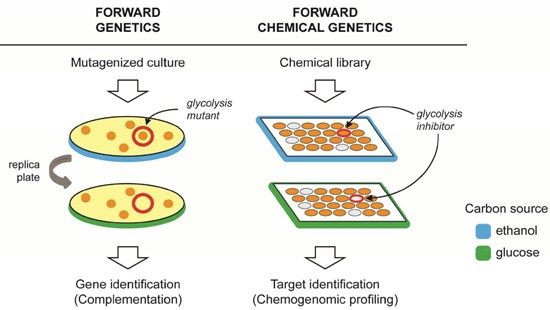
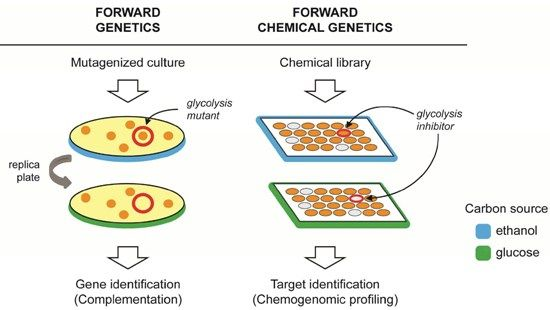
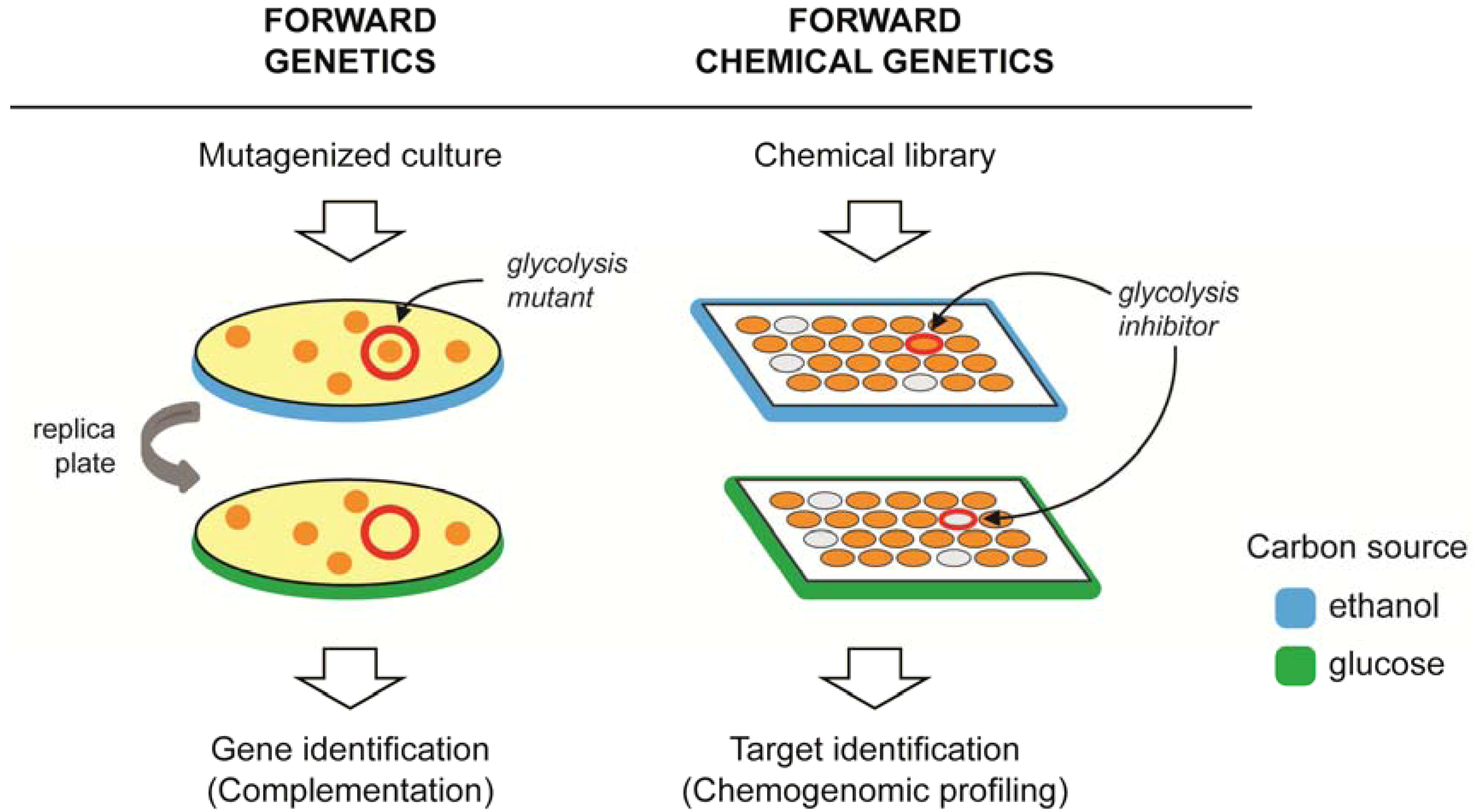
:
{kind=link}
{kind=link}
{kind=link}
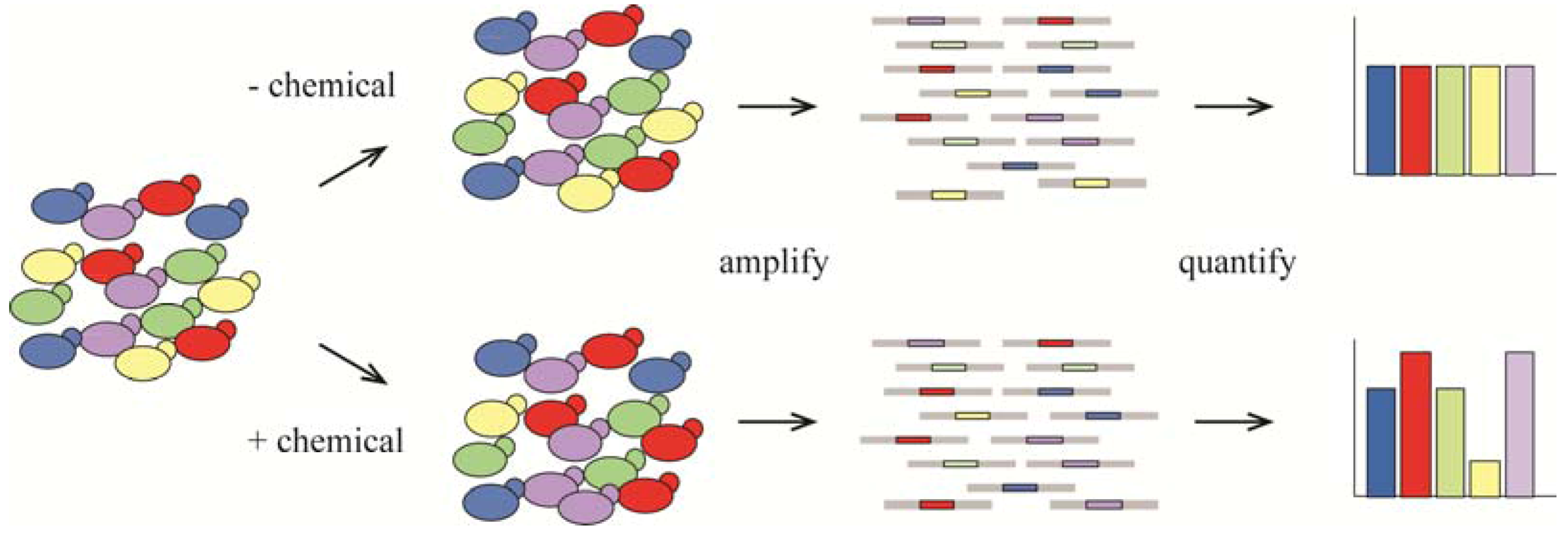{kind=link}
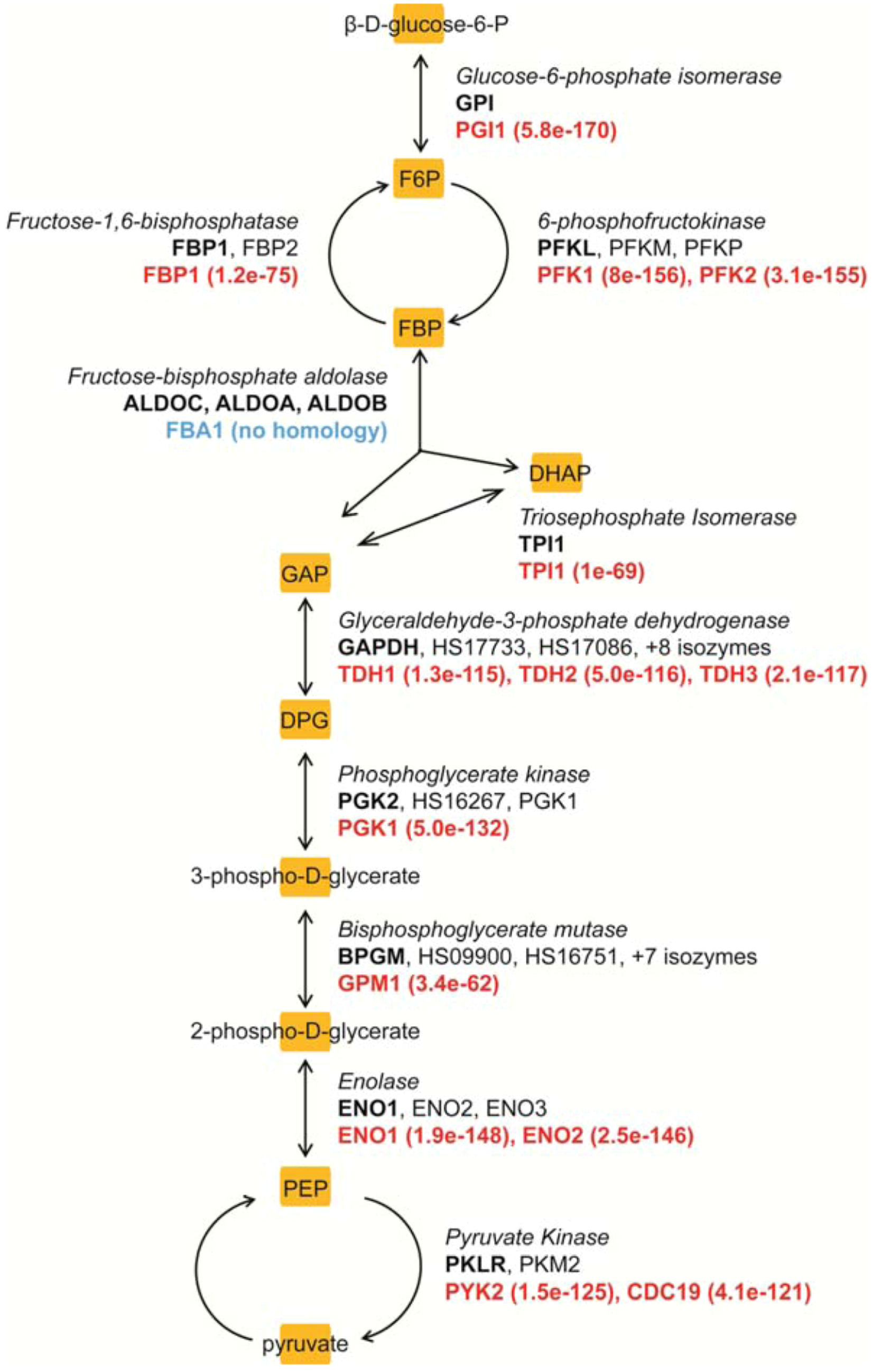{kind=link}
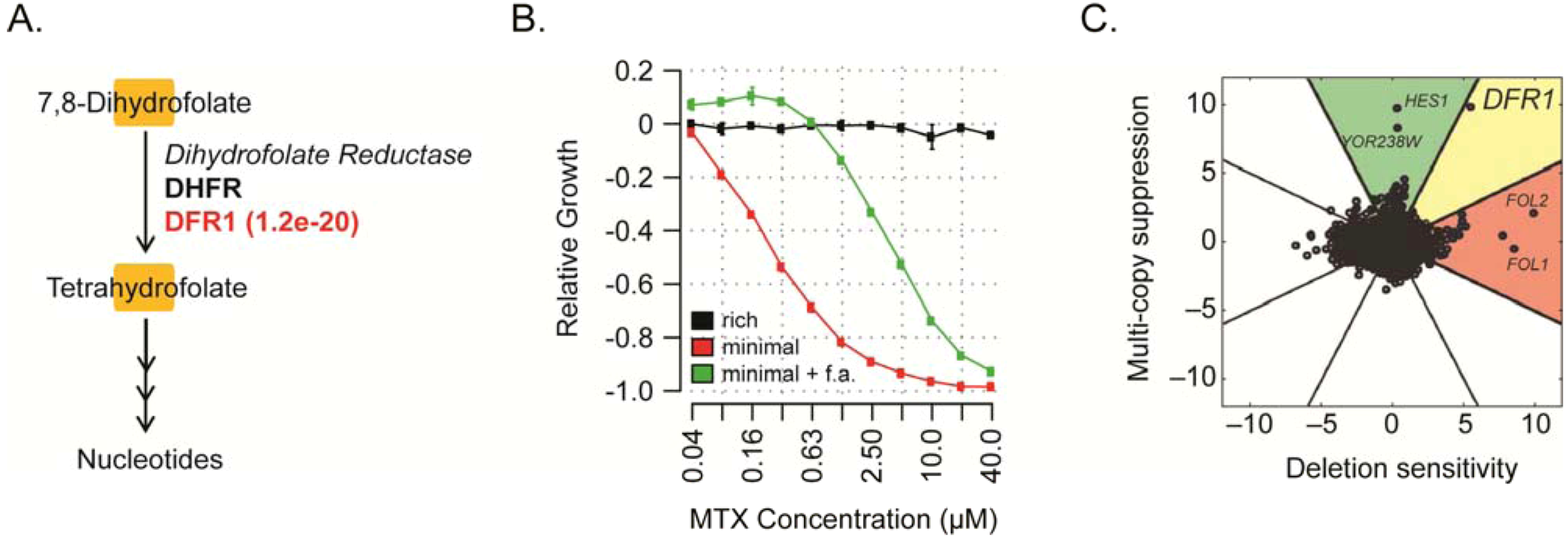{kind=link}
1. Phenotypic vs. Target-based Screening for Drug Discovery
2. Forward Genetics and Forward Chemical Genetics in Yeast
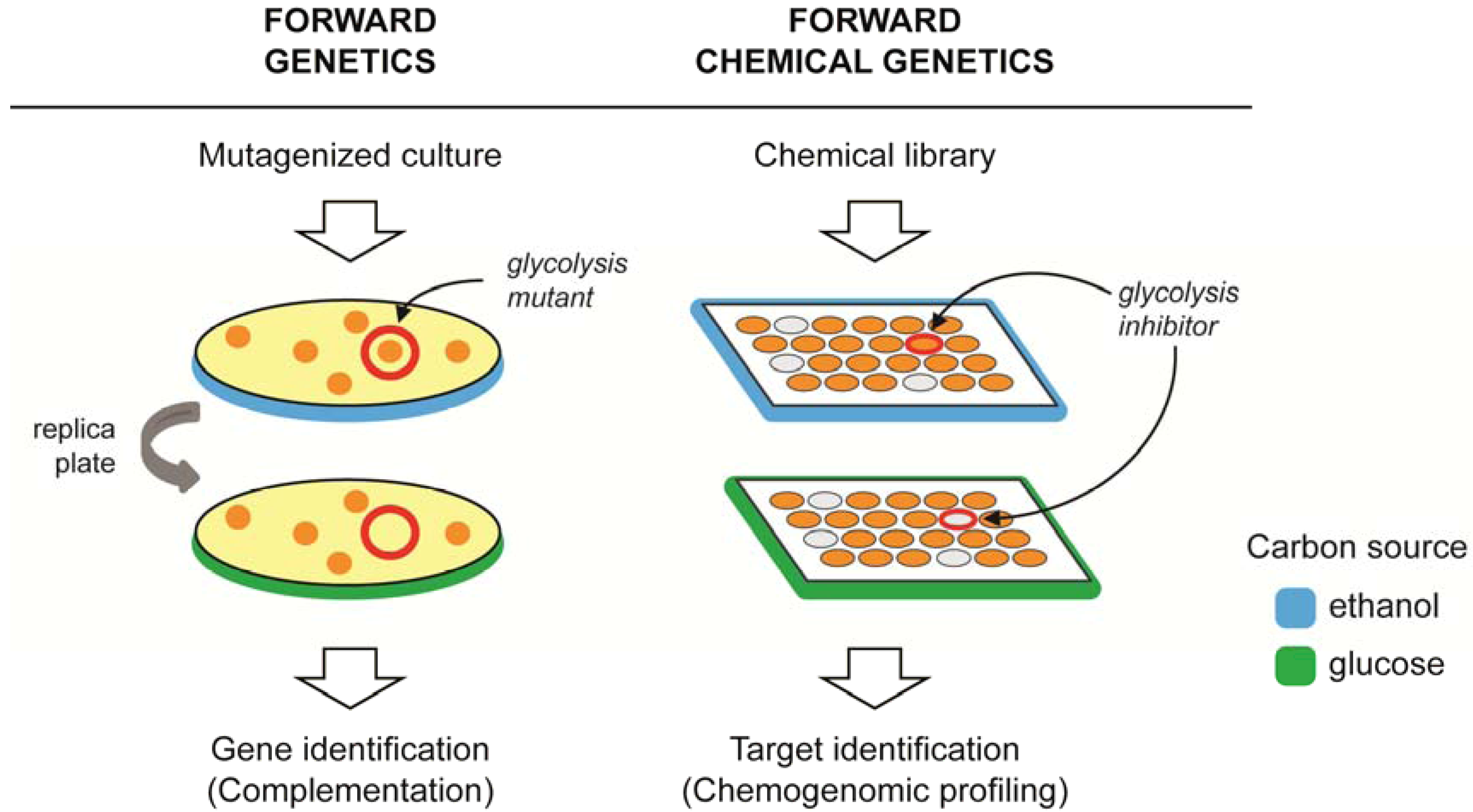
3. Target Identification Strategies in Yeast
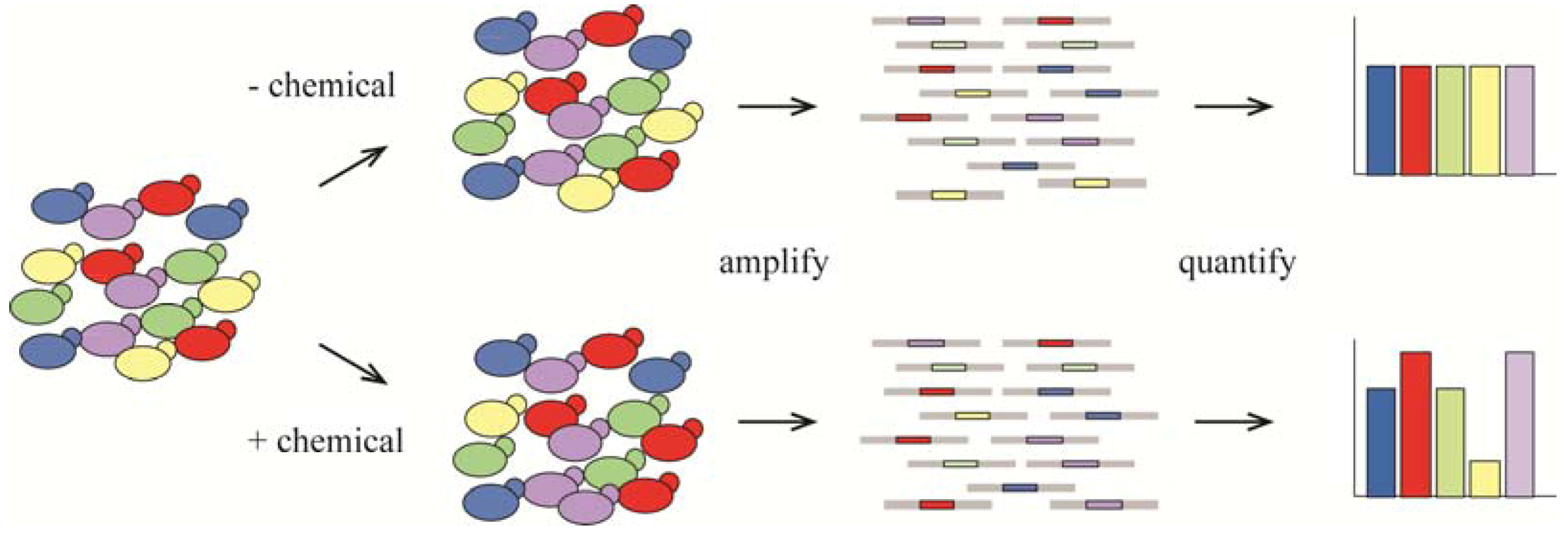
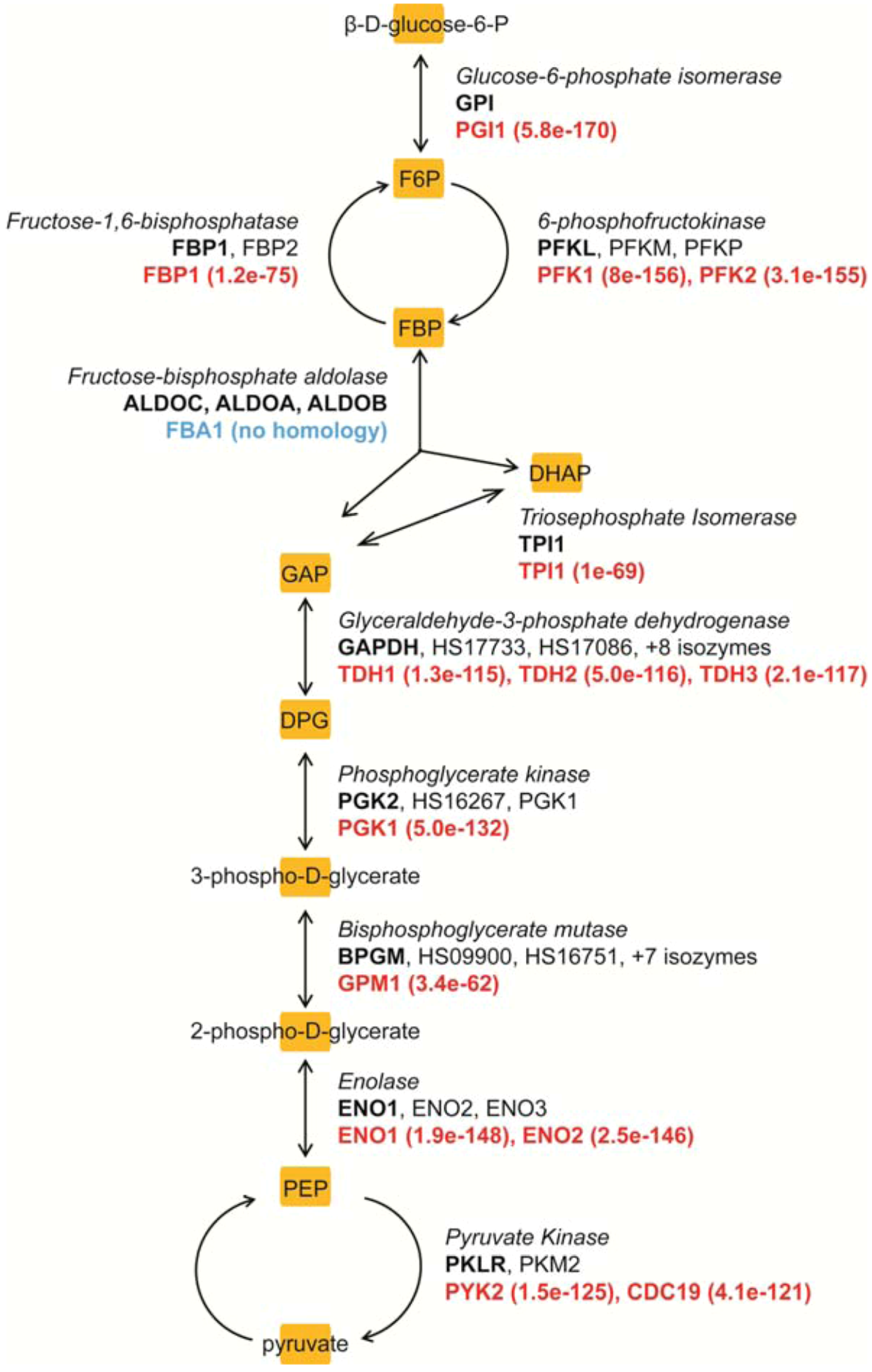
4. Metabolism as a Target for Cancer Therapy
5. Yeast as a Model for Discovery of Probes Targeting Tumor Metabolism
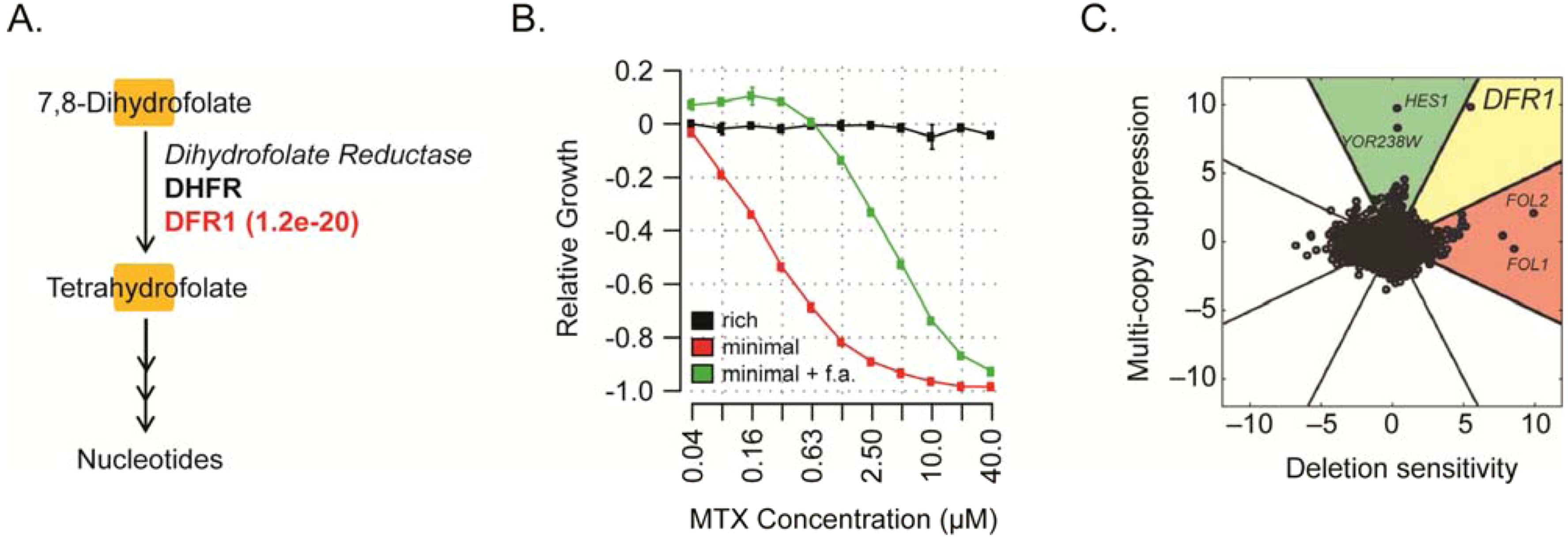
6. Conclusions and Perspectives
Acknowledgements
References
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef]
- Sams-Dodd, F. Target-based drug discovery: is something wrong? Drug Discov. Today 2005, 10, 139–147. [Google Scholar] [CrossRef]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Botstein, D.; Chervitz, S.A.; Cherry, J.M. Yeast as a model organism. Science 1997, 277, 1259–1260. [Google Scholar]
- Botstein, D.; Fink, G.R. Yeast: an experimental organism for 21st Century biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef]
- Norbury, C.; Nurse, P. Animal cell cycles and their control. Annu. Rev. Biochem. 1992, 61, 441–470. [Google Scholar] [CrossRef]
- Nurse, P. Cyclin dependent kinases and cell cycle control (nobel lecture). ChemBioChem 2002, 3, 596–603. [Google Scholar] [CrossRef]
- Prakash, S.; Sung, P.; Prakash, L. DNA repair genes and proteins of Saccharomyces cerevisiae. Annu. Rev. Genet. 1993, 27, 33–70. [Google Scholar] [CrossRef]
- Steinmetz, L.M.; Scharfe, C.; Deutschbauer, A.M.; Mokranjac, D.; Herman, Z.S.; Jones, T.; Chu, A.M.; Giaever, G.; Prokisch, H.; Oefner, P.J.; et al. Systematic screen for human disease genes in yeast. Nat. Genet. 2002, 31, 400–404. [Google Scholar]
- Game, J.C.; Mortimer, R.K. A genetic study of x-ray sensitive mutants in yeast. Mutat. Res. 1974, 24, 281–292. [Google Scholar]
- Basile, G.; Aker, M.; Mortimer, R.K. Nucleotide sequence and transcriptional regulation of the yeast recombinational repair gene RAD51. Mol. Cell. Biol. 1992, 12, 3235–3246. [Google Scholar]
- Marton, M.J.; DeRisi, J.L.; Bennett, H.A.; Iyer, V.R.; Meyer, M.R.; Roberts, C.J.; Stoughton, R.; Burchard, J.; Slade, D.; Dai, H.; et al. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat. Med. 1998, 4, 1293–1301. [Google Scholar] [CrossRef]
- Stockwell, B.R. Chemical genetics: ligand-based discovery of gene function. Nat. Rev. Genet. 2000, 1, 116–125. [Google Scholar] [CrossRef]
- Lokey, R.S. Forward chemical genetics: progress and obstacles on the path to a new pharmacopoeia. Curr. Opin. Chem. Biol. 2003, 7, 91–96. [Google Scholar] [CrossRef]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Veronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; Andre, B.; et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef]
- Ho, C.H.; Magtanong, L.; Barker, S.L.; Gresham, D.; Nishimura, S.; Natarajan, P.; Koh, J.L.; Porter, J.; Gray, C.A.; Andersen, R.J.; et al. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat. Biotechnol. 2009, 27, 369–377. [Google Scholar] [CrossRef]
- Lam, K.B.; Marmur, J. Isolation and characterization of Saccharomyces cerevisiae glycolytic pathway mutants. J. Bacteriol. 1977, 130, 746–749. [Google Scholar]
- Grozinger, C.M.; Chao, E.D.; Blackwell, H.E.; Moazed, D.; Schreiber, S.L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001, 276, 38837–38843. [Google Scholar]
- Bach, S.; Talarek, N.; Andrieu, T.; Vierfond, J.M.; Mettey, Y.; Galons, H.; Dormont, D.; Meijer, L.; Cullin, C.; Blondel, M. Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nat. Biotechnol. 2003, 21, 1075–1081. [Google Scholar]
- Chuprina, A.; Lukin, O.; Demoiseaux, R.; Buzko, A.; Shivanyuk, A. Drug- and lead-likeness, target class, and molecular diversity analysis of 7.9 million commercially available organic compounds provided by 29 suppliers. J. Chem. Inf. Model. 2010, 50, 470–479. [Google Scholar]
- Tan, D.S. Diversity-oriented synthesis: Exploring the intersections between chemistry and biology. Nat. Chem. Biol. 2005, 1, 74–84. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- CJ, O.C.; Beckmann, H.S.; Spring, D.R. Diversity-oriented synthesis: producing chemical tools for dissecting biology. Chem. Soc. Rev. 2012, 41, 4444–4456. [Google Scholar] [CrossRef]
- Young, K.; Lin, S.; Sun, L.; Lee, E.; Modi, M.; Hellings, S.; Husbands, M.; Ozenberger, B.; Franco, R. Identification of a calcium channel modulator using a high throughput yeast two-hybrid screen. Nat. Biotechnol. 1998, 16, 946–950. [Google Scholar] [CrossRef]
- Gassner, N.C.; Tamble, C.M.; Bock, J.E.; Cotton, N.; White, K.N.; Tenney, K.; St Onge, R.P.; Proctor, M.J.; Giaever, G.; Nislow, C.; et al. Accelerating the discovery of biologically active small molecules using a high-throughput yeast halo assay. J. Nat. Prod. 2007, 70, 383–390. [Google Scholar] [CrossRef]
- Couplan, E.; Aiyar, R.S.; Kucharczyk, R.; Kabala, A.; Ezkurdia, N.; Gagneur, J.; St Onge, R.P.; Salin, B.; Soubigou, F.; Le Cann, M.; et al. A yeast-based assay identifies drugs active against human mitochondrial disorders. Proc. Natl. Acad. Sci. USA 2011, 108, 11989–11994. [Google Scholar]
- Wallace, I.M.; Urbanus, M.L.; Luciani, G.M.; Burns, A.R.; Han, M.K.; Wang, H.; Arora, K.; Heisler, L.E.; Proctor, M.; St Onge, R.P.; et al. Compound prioritization methods increase rates of chemical probe discovery in model organisms. Chem. Biol. 2011, 18, 1273–1283. [Google Scholar]
- St Onge, R.P.; Mani, R.; Oh, J.; Proctor, M.; Fung, E.; Davis, R.W.; Nislow, C.; Roth, F.P.; Giaever, G. Systematic pathway analysis using high-resolution fitness profiling of combinatorial gene deletions. Nat. Genet. 2007, 39, 199–206. [Google Scholar]
- Schlecht, U.; Miranda, M.; Suresh, S.; Davis, R.W.; St Onge, R.P. Multiplex assay for condition-dependent changes in protein-protein interactions. Proc. Natl. Acad. Sci. USA 2012, 109, 9213–9218. [Google Scholar]
- Bauer, B.E.; Wolfger, H.; Kuchler, K. Inventory and function of yeast ABC proteins: about sex, stress, pleiotropic drug and heavy metal resistance. Biochim. Biophys. Acta 1999, 1461, 217–236. [Google Scholar]
- Rogers, B.; Decottignies, A.; Kolaczkowski, M.; Carvajal, E.; Balzi, E.; Goffeau, A. The pleitropic drug ABC transporters from Saccharomyces cerevisiae. J. Mol. Microbiol. Biotechnol. 2001, 3, 207–214. [Google Scholar]
- Suzuki, Y.; St Onge, R.P.; Mani, R.; King, O.D.; Heilbut, A.; Labunskyy, V.M.; Chen, W.; Pham, L.; Zhang, L.V.; Tong, A.H.; et al. Knocking out multigene redundancies via cycles of sexual assortment and fluorescence selection. Nat. Methods 2011, 8, 159–164. [Google Scholar]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 546, 563–567. [Google Scholar]
- Shoemaker, D.D.; Lashkari, D.A.; Morris, D.; Mittmann, M.; Davis, R.W. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat. Genet. 1996, 14, 450–456. [Google Scholar] [CrossRef]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef]
- Pierce, S.E.; Fung, E.L.; Jaramillo, D.F.; Chu, A.M.; Davis, R.W.; Nislow, C.; Giaever, G. A unique and universal molecular barcode array. Nat. Methods 2006, 3, 601–603. [Google Scholar]
- Smith, A.M.; Heisler, L.E.; Mellor, J.; Kaper, F.; Thompson, M.J.; Chee, M.; Roth, F.P.; Giaever, G.; Nislow, C. Quantitative phenotyping via deep barcode sequencing. Genome Res. 2009, 19, 1836–1842. [Google Scholar] [CrossRef]
- Gresham, D.; Boer, V.M.; Caudy, A.; Ziv, N.; Brandt, N.J.; Storey, J.D.; Botstein, D. System-level analysis of genes and functions affecting survival during nutrient starvation in Saccharomyces cerevisiae. Genetics 2011, 187, 299–317. [Google Scholar] [CrossRef]
- Deutschbauer, A.; Price, M.N.; Wetmore, K.M.; Shao, W.; Baumohl, J.K.; Xu, Z.; Nguyen, M.; Tamse, R.; Davis, R.W.; Arkin, A.P. Evidence-based annotation of gene function in Shewanella oneidensis MR-1 using genome-wide fitness profiling across 121 conditions. PLoS Genet. 2011, 7, e1002385. [Google Scholar]
- Hensel, M.; Shea, J.E.; Gleeson, C.; Jones, M.D.; Dalton, E.; Holden, D.W. Simultaneous identification of bacterial virulence genes by negative selection. Science 1995, 269, 400–403. [Google Scholar]
- Xu, D.; Jiang, B.; Ketela, T.; Lemieux, S.; Veillette, K.; Martel, N.; Davison, J.; Sillaots, S.; Trosok, S.; Bachewich, C.; Bussey, H.; Youngman, P.; Roemer, T. Genome-wide fitness test and mechanism-of-action studies of inhibitory compounds in Candida albicans. PLoS Pathog. 2007, 3, e92. [Google Scholar] [CrossRef]
- Oh, J.; Fung, E.; Schlecht, U.; Davis, R.W.; Giaever, G.; St Onge, R.P.; Deutschbauer, A.; Nislow, C. Gene annotation and drug target discovery in Candida albicans with a tagged transposon mutant collection. PLoS Pathog. 2010, 6, e1001140. [Google Scholar]
- Andrusiak, K.; Piotrowski, J.S.; Boone, C. Chemical-genomic profiling: Systematic analysis of the cellular targets of bioactive molecules. Bioorg. Med. Chem. 2012, 20, 1952–1960. [Google Scholar] [CrossRef]
- Hoon, S.; St Onge, R.P.; Giaever, G.; Nislow, C. Yeast chemical genomics and drug discovery: an update. Trends Pharmacol. Sci. 2008, 29, 499–504. [Google Scholar]
- Smith, A.M.; Ammar, R.; Nislow, C.; Giaever, G. A survey of yeast genomic assays for drug and target discovery. Pharmacol. Ther. 2010, 127, 156–164. [Google Scholar] [CrossRef]
- Giaever, G.; Shoemaker, D.D.; Jones, T.W.; Liang, H.; Winzeler, E.A.; Astromoff, A.; Davis, R.W. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat. Genet. 1999, 21, 278–283. [Google Scholar] [CrossRef]
- Rine, J.; Hansen, W.; Hardeman, E.; Davis, R.W. Targeted selection of recombinant clones through gene dosage effects. Proc. Natl. Acad. Sci. USA 1983, 80, 6750–6754. [Google Scholar] [CrossRef]
- Giaever, G.; Flaherty, P.; Kumm, J.; Proctor, M.; Nislow, C.; Jaramillo, D.F.; Chu, A.M.; Jordan, M.I.; Arkin, A.P.; Davis, R.W. Chemogenomic profiling: Identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. USA 2004, 101, 793–798. [Google Scholar]
- Hoon, S.; Smith, A.M.; Wallace, I.M.; Suresh, S.; Miranda, M.; Fung, E.; Proctor, M.; Shokat, K.M.; Zhang, C.; Davis, R.W.; et al. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat. Chem. Biol. 2008, 4, 498–506. [Google Scholar] [CrossRef]
- Lum, P.Y.; Armour, C.D.; Stepaniants, S.B.; Cavet, G.; Wolf, M.K.; Butler, J.S.; Hinshaw, J.C.; Garnier, P.; Prestwich, G.D.; Leonardson, A.; et al. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell 2004, 116, 121–137. [Google Scholar] [CrossRef]
- Luesch, H.; Wu, T.Y.; Ren, P.; Gray, N.S.; Schultz, P.G.; Supek, F. A genome-wide overexpression screen in yeast for small-molecule target identification. Chem. Biol. 2005, 12, 55–63. [Google Scholar] [CrossRef]
- Baetz, K.; McHardy, L.; Gable, K.; Tarling, T.; Reberioux, D.; Bryan, J.; Andersen, R.J.; Dunn, T.; Hieter, P.; Roberge, M. Yeast genome-wide drug-induced haploinsufficiency screen to determine drug mode of action. Proc. Natl. Acad. Sci. USA 2004, 101, 4525–4530. [Google Scholar]
- Butcher, R.A.; Bhullar, B.S.; Perlstein, E.O.; Marsischky, G.; LaBaer, J.; Schreiber, S.L. Microarray-based method for monitoring yeast overexpression strains reveals small-molecule targets in TOR pathway. Nat. Chem. Biol. 2006, 2, 103–109. [Google Scholar] [CrossRef]
- Yan, Z.; Berbenetz, N.M.; Giaever, G.; Nislow, C. Precise gene-dose alleles for chemical genetics. Genetics 2009, 182, 623–626. [Google Scholar]
- Hillenmeyer, M.E.; Fung, E.; Wildenhain, J.; Pierce, S.E.; Hoon, S.; Lee, W.; Proctor, M.; St Onge, R.P.; Tyers, M.; Koller, D.; et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 2008, 320, 362–365. [Google Scholar] [CrossRef]
- Kapitzky, L.; Beltrao, P.; Berens, T.J.; Gassner, N.; Zhou, C.; Wuster, A.; Wu, J.; Babu, M.M.; Elledge, S.J.; Toczyski, D.; et al. Cross-species chemogenomic profiling reveals evolutionarily conserved drug mode of action. Mol. Syst. Biol. 2010, 6, 451. [Google Scholar]
- Parsons, A.B.; Lopez, A.; Givoni, I.E.; Williams, D.E.; Gray, C.A.; Porter, J.; Chua, G.; Sopko, R.; Brost, R.L.; Ho, C.H.; et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 2006, 126, 611–625. [Google Scholar] [CrossRef]
- Parsons, A.B.; Brost, R.L.; Ding, H.; Li, Z.; Zhang, C.; Sheikh, B.; Brown, G.W.; Kane, P.M.; Hughes, T.R.; Boone, C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotechnol. 2004, 22, 62–69. [Google Scholar] [CrossRef]
- Costanzo, M.; Baryshnikova, A.; Bellay, J.; Kim, Y.; Spear, E.D.; Sevier, C.S.; Ding, H.; Koh, J.L.; Toufighi, K.; Mostafavi, S.; et al. The genetic landscape of a cell. Science 2010, 327, 425–431. [Google Scholar] [CrossRef]
- Koh, J.L.; Ding, H.; Costanzo, M.; Baryshnikova, A.; Toufighi, K.; Bader, G.D.; Myers, C.L.; Andrews, B.J.; Boone, C. DRYGIN: A database of quantitative genetic interaction networks in yeast. Nucleic Acids Res. 2010, 38, D502–D507. [Google Scholar] [CrossRef]
- Licitra, E.J.; Liu, J.O. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 12817–12821. [Google Scholar] [CrossRef]
- Rix, U.; Superti-Furga, G. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 2009, 5, 616–624. [Google Scholar] [CrossRef]
- Tribouillard-Tanvier, D.; Dos Reis, S.; Gug, F.; Voisset, C.; Beringue, V.; Sabate, R.; Kikovska, E.; Talarek, N.; Bach, S.; Huang, C.; et al. Protein folding activity of ribosomal RNA is a selective target of two unrelated antiprion drugs. PLoS One 2008, 3, e2174. [Google Scholar]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.; Wu, R.P.; Gomez, F.; Loo, J.A.; et al. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar]
- Chan, J.N.; Vuckovic, D.; Sleno, L.; Olsen, J.B.; Pogoutse, O.; Havugimana, P.; Hewel, J.A.; Bajaj, N.; Wang, Y.; Musteata, M.F.; et al. Target identification by chromatographic co-elution: monitoring of drug-protein interactions without immobilization or chemical derivatization. Mol. Cell. Proteomics 2012, 11, M111–016642. [Google Scholar] [CrossRef]
- Tennant, D.A.; Duran, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Targeting cancer metabolism: a therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar]
- Gohil, V.M.; Sheth, S.A.; Nilsson, R.; Wojtovich, A.P.; Lee, J.H.; Perocchi, F.; Chen, W.; Clish, C.B.; Ayata, C.; Brookes, P.S.; et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat. Biotechnol. 2010, 28, 249–255. [Google Scholar]
- Scatena, R.; Bottoni, P.; Pontoglio, A.; Mastrototaro, L.; Giardina, B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2008, 17, 1533–1545. [Google Scholar] [CrossRef]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell. Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2009, 19, 17–24. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar]
- Szeliga, M.; Obara-Michlewska, M. Glutamine in neoplastic cells: focus on the expression and roles of glutaminases. Neurochem. Int. 2009, 55, 71–75. [Google Scholar] [CrossRef]
- Masetti, R.; Pession, A. First-line treatment of acute lymphoblastic leukemia with pegasparaginase. Biologics 2009, 3, 359–368. [Google Scholar]
- Ni, Y.; Schwaneberg, U.; Sun, Z.H. Arginine deiminase, a potential anti-tumor drug. Cancer Lett. 2008, 261, 1–11. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef]
- Li, J.N.; Gorospe, M.; Chrest, F.J.; Kumaravel, T.S.; Evans, M.K.; Han, W.F.; Pizer, E.S. Pharmacological inhibition of fatty acid synthase activity produces both cytostatic and cytotoxic effects modulated by p53. Cancer Res. 2001, 61, 1493–1499. [Google Scholar]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2011, 1807, 568–576. [Google Scholar] [CrossRef]
- Peregrin-Alvarez, J.M.; Sanford, C.; Parkinson, J. The conservation and evolutionary modularity of metabolism. Genome Biol. 2009, 10, R63. [Google Scholar] [CrossRef]
- Perocchi, F.; Jensen, L.J.; Gagneur, J.; Ahting, U.; von Mering, C.; Bork, P.; Prokisch, H.; Steinmetz, L.M. Assessing systems properties of yeast mitochondria through an interaction map of the organelle. PLoS Genet. 2006, 2, e170. [Google Scholar] [CrossRef]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar]
- Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, Not sugar, Is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [Google Scholar]
- Diaz-Ruiz, R.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Tumor cell energy metabolism and its common features with yeast metabolism. Biochim. Biophys. Acta 2009, 1796, 252–265. [Google Scholar]
- Turcotte, B.; Liang, X.B.; Robert, F.; Soontorngun, N. Transcriptional regulation of nonfermentable carbon utilization in budding yeast. FEMS Yeast Res. 2010, 10, 2–13. [Google Scholar] [CrossRef]
- Breslow, D.K.; Cameron, D.M.; Collins, S.R.; Schuldiner, M.; Stewart-Ornstein, J.; Newman, H.W.; Braun, S.; Madhani, H.D.; Krogan, N.J.; Weissman, J.S. A comprehensive strategyenabling high-resolution functional analysis of the yeast genome. Nat. Methods 2008, 5, 711–718. [Google Scholar]
- Walling, J. From methotrexate to pemetrexed and beyond. A review of the pharmacodynamic and clinical properties of antifolates. Invest. New Drugs 2006, 24, 37–77. [Google Scholar] [CrossRef]
- Osborn, M.J.; Freeman, M.; Huennekens, F.M. Inhibition of dihydrofolic reductase by aminopterin and amethopterin. Proc. Soc. Exp. Biol. Med. 1958, 97, 429–431. [Google Scholar]
- Huang, T.; Barclay, B.J.; Kalman, T.I.; von Borstel, R.C.; Hastings, P.J. The phenotype of a dihydrofolate reductase mutant of Saccharomyces cerevisiae. Gene 1992, 121, 167–171. [Google Scholar] [CrossRef]
- Keshava, C.; Keshava, N.; Whong, W.Z.; Nath, J.; Ong, T.M. Inhibition of methotrexate-induced chromosomal damage by folinic acid in V79 cells. Mutat. Res. 1998, 397, 221–228. [Google Scholar] [CrossRef]
- Treco, D.A.; Reynolds, A.; Lundblad, V. Growth and manipulation of yeast. Curr. Protoc. Protein Sci. 2001, Appendix 4, Appendix 4L. [Google Scholar]
- Lagosky, P.A.; Taylor, G.R.; Haynes, R.H. Molecular characterization of the Saccharomyces cerevisiae dihydrofolate reductase gene (DFR1). Nucleic Acids Res. 1987, 15, 10355–10371. [Google Scholar] [CrossRef]
- Lardenois, A.; Liu, Y.; Walther, T.; Chalmel, F.; Evrard, B.; Granovskaia, M.; Chu, A.; Davis, R.W.; Steinmetz, L.M.; Primig, M. Execution of the meiotic noncoding RNA expression program and the onset of gametogenesis in yeast require the conserved exosome subunit Rrp6. Proc. Natl. Acad. Sci. USA 2011, 108, 1058–1063. [Google Scholar]
- Pronk, J.T. Auxotrophic yeast strains in fundamental and applied research. Appl. Environ. Microbiol. 2002, 68, 2095–2100. [Google Scholar] [CrossRef]
- Wenzel, T.J.; van den Berg, M.A.; Visser, W.; van den Berg, J.A.; Steensma, H.Y. Characterization of Saccharomyces cerevisiae mutants lacking the E1 alpha subunit of the pyruvate dehydrogenase complex. Eur. J. Biochem. 1992, 209, 697–705. [Google Scholar] [CrossRef]
- Frye, S.V. The art of the chemical probe. Nat. Chem. Biol. 2010, 6, 159–161. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
St.Onge, R.; Schlecht, U.; Scharfe, C.; Evangelista, M. Forward Chemical Genetics in Yeast for Discovery of Chemical Probes Targeting Metabolism. Molecules 2012, 17, 13098-13115. https://doi.org/10.3390/molecules171113098
St.Onge R, Schlecht U, Scharfe C, Evangelista M. Forward Chemical Genetics in Yeast for Discovery of Chemical Probes Targeting Metabolism. Molecules. 2012; 17(11):13098-13115. https://doi.org/10.3390/molecules171113098
Chicago/Turabian StyleSt.Onge, Robert, Ulrich Schlecht, Curt Scharfe, and Marie Evangelista. 2012. "Forward Chemical Genetics in Yeast for Discovery of Chemical Probes Targeting Metabolism" Molecules 17, no. 11: 13098-13115. https://doi.org/10.3390/molecules171113098