Synthesis, Cyclopolymerization and Cyclo-Copolymerization of 9-(2-Diallylaminoethyl)adenine and Its Hydrochloride Salt

Abstract
:1. Introduction
2. Results and Discussion




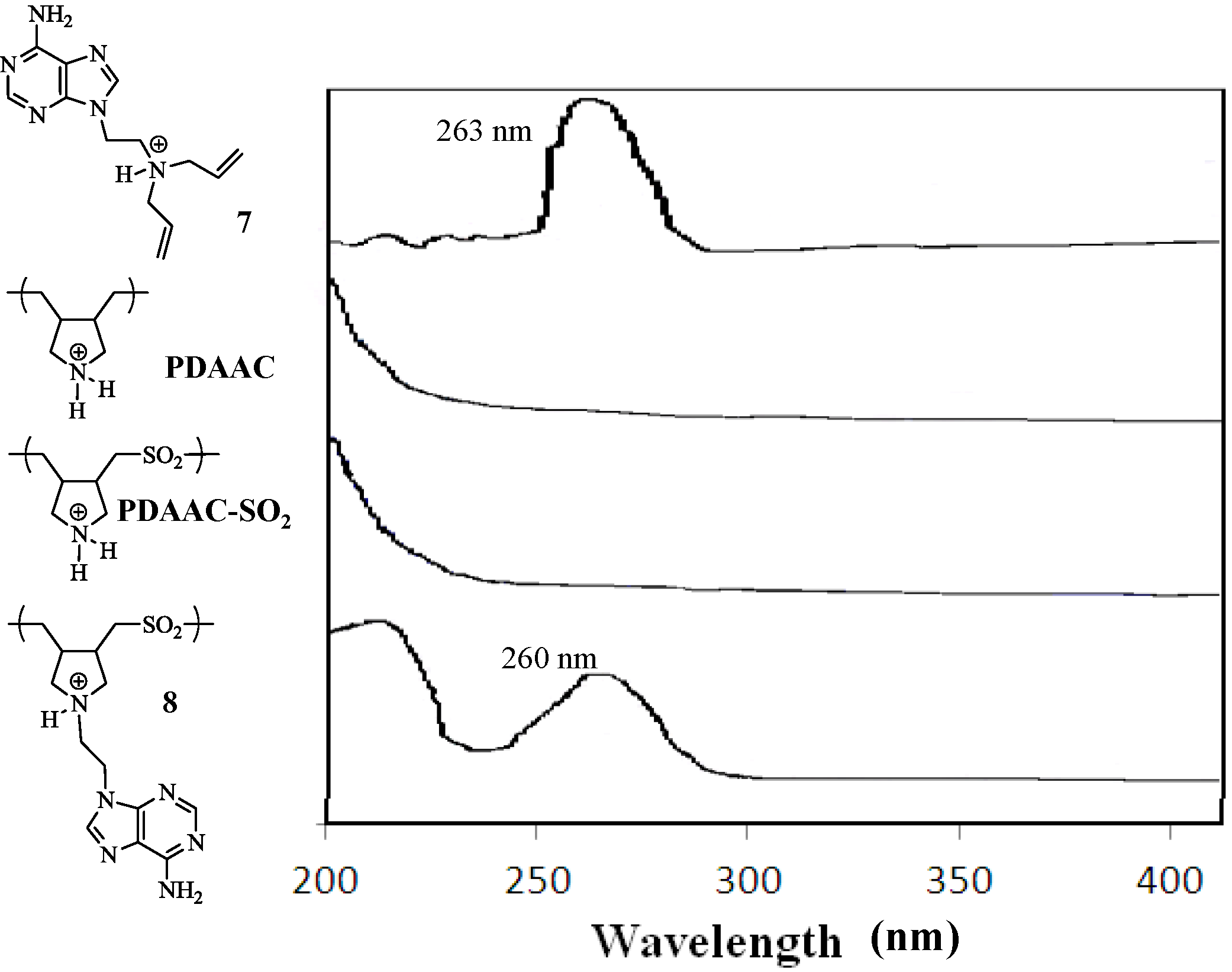{kind=link}
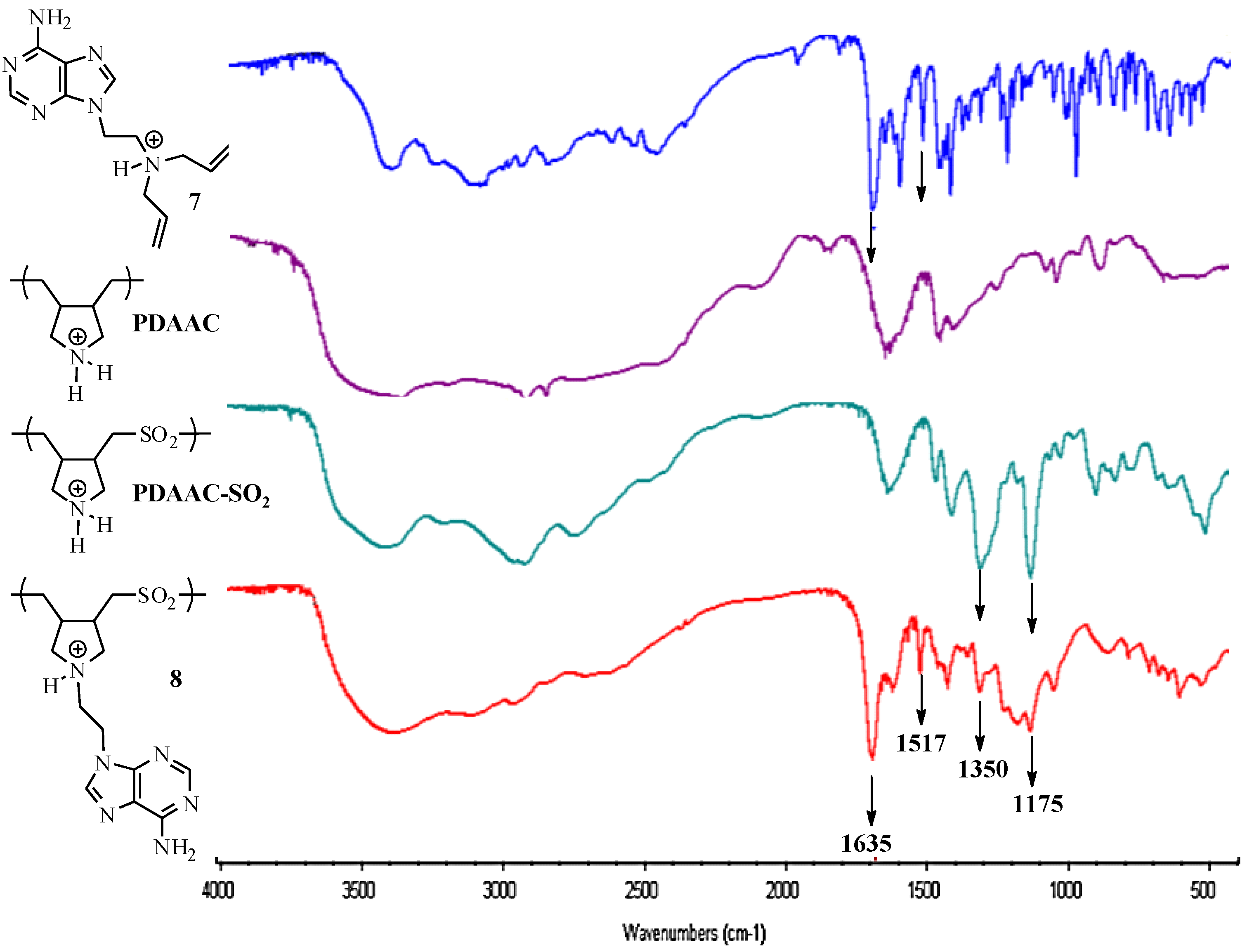{kind=link}
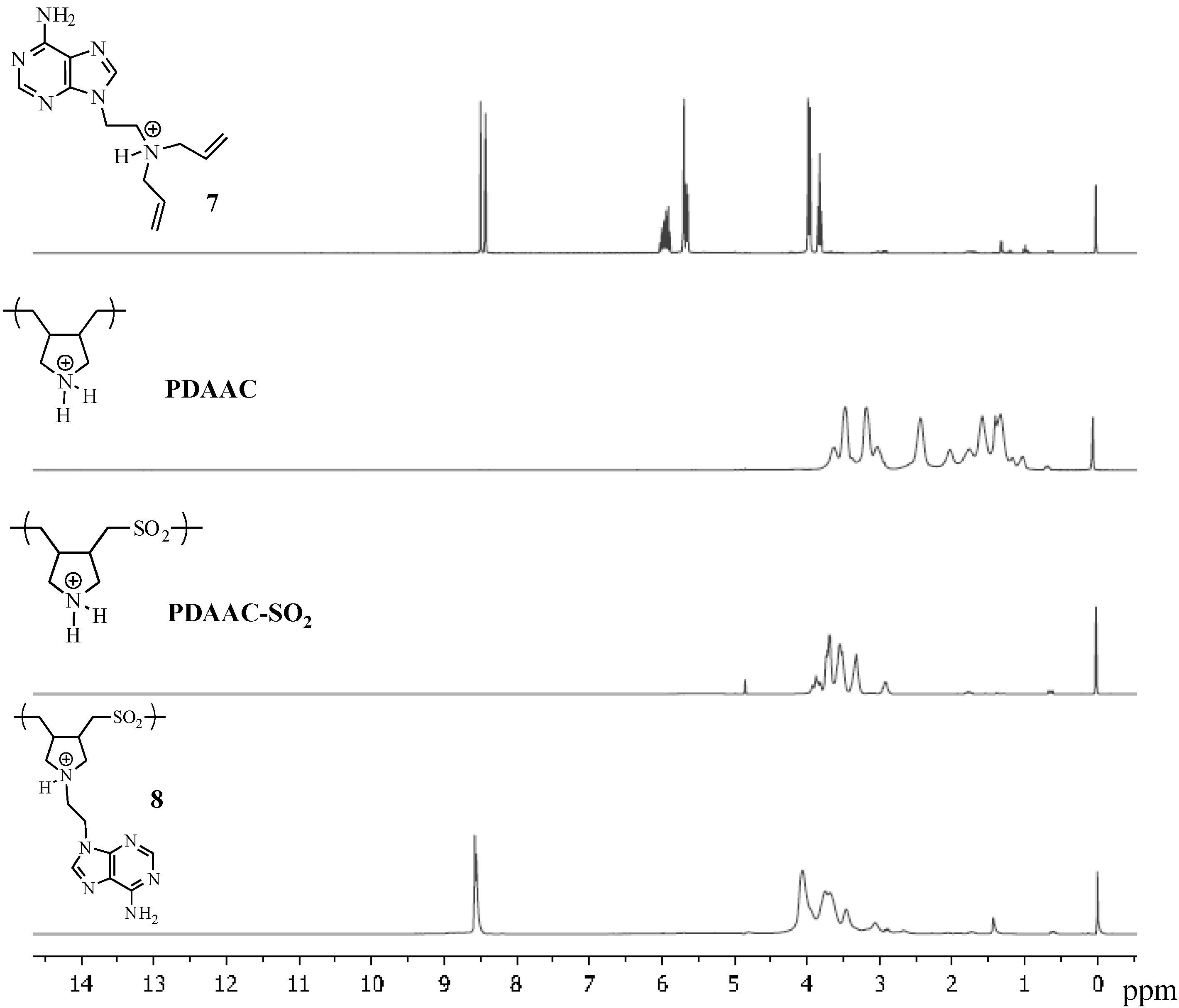{kind=link}
{kind=link}
{kind=link}
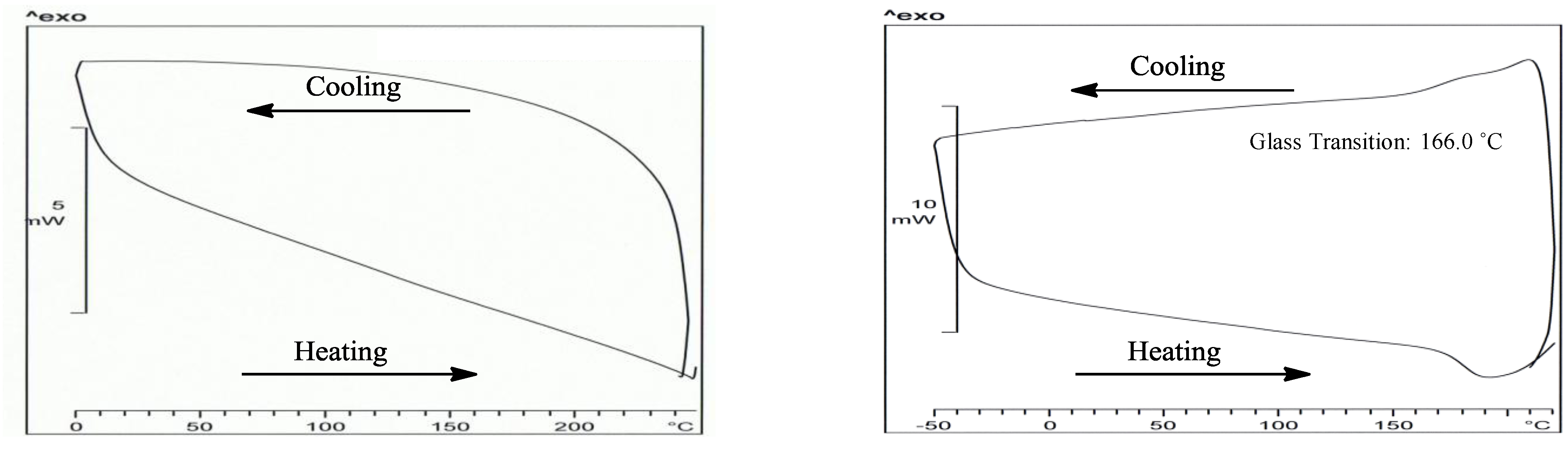{kind=link}
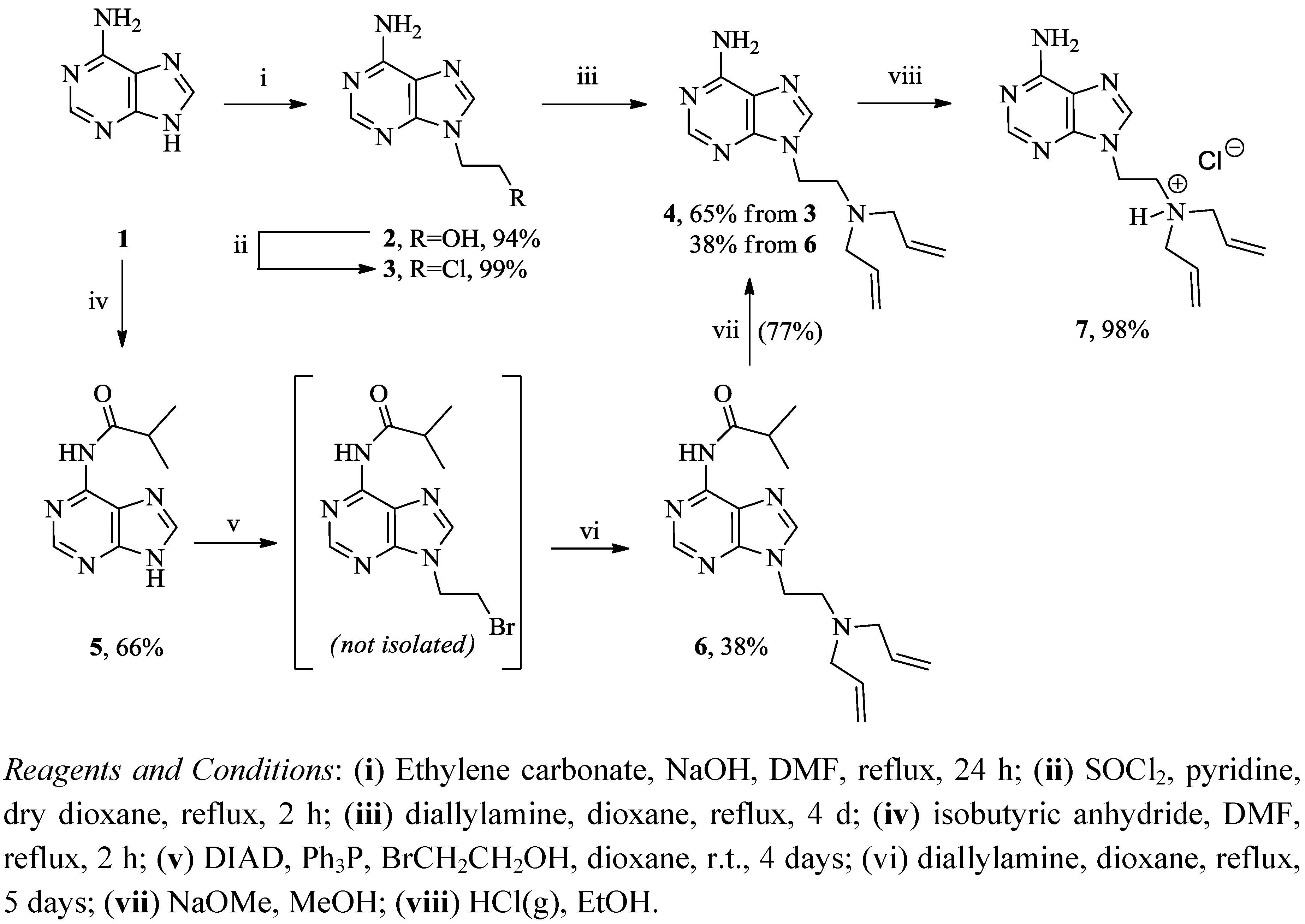{kind=link}
{kind=link}
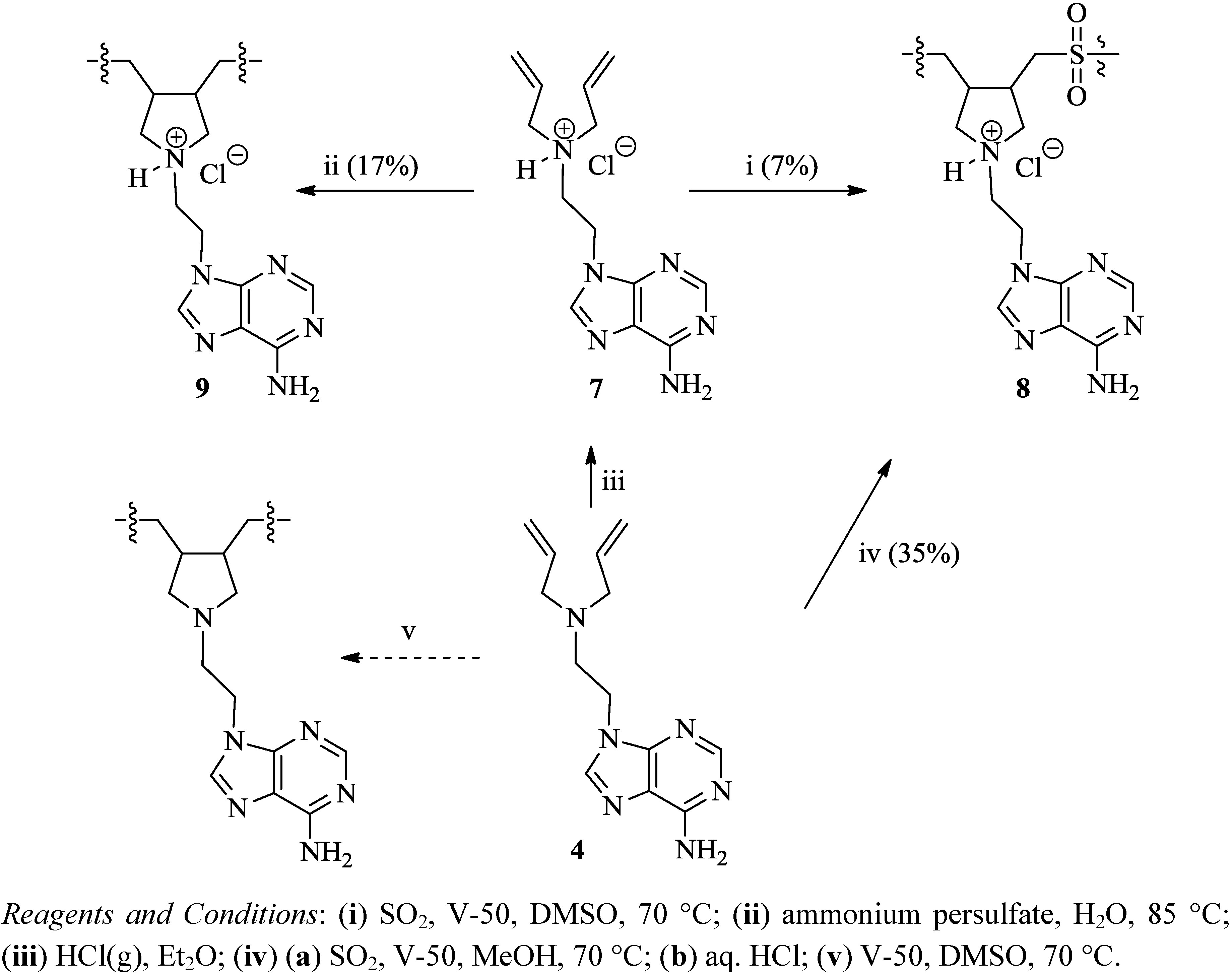{kind=link}
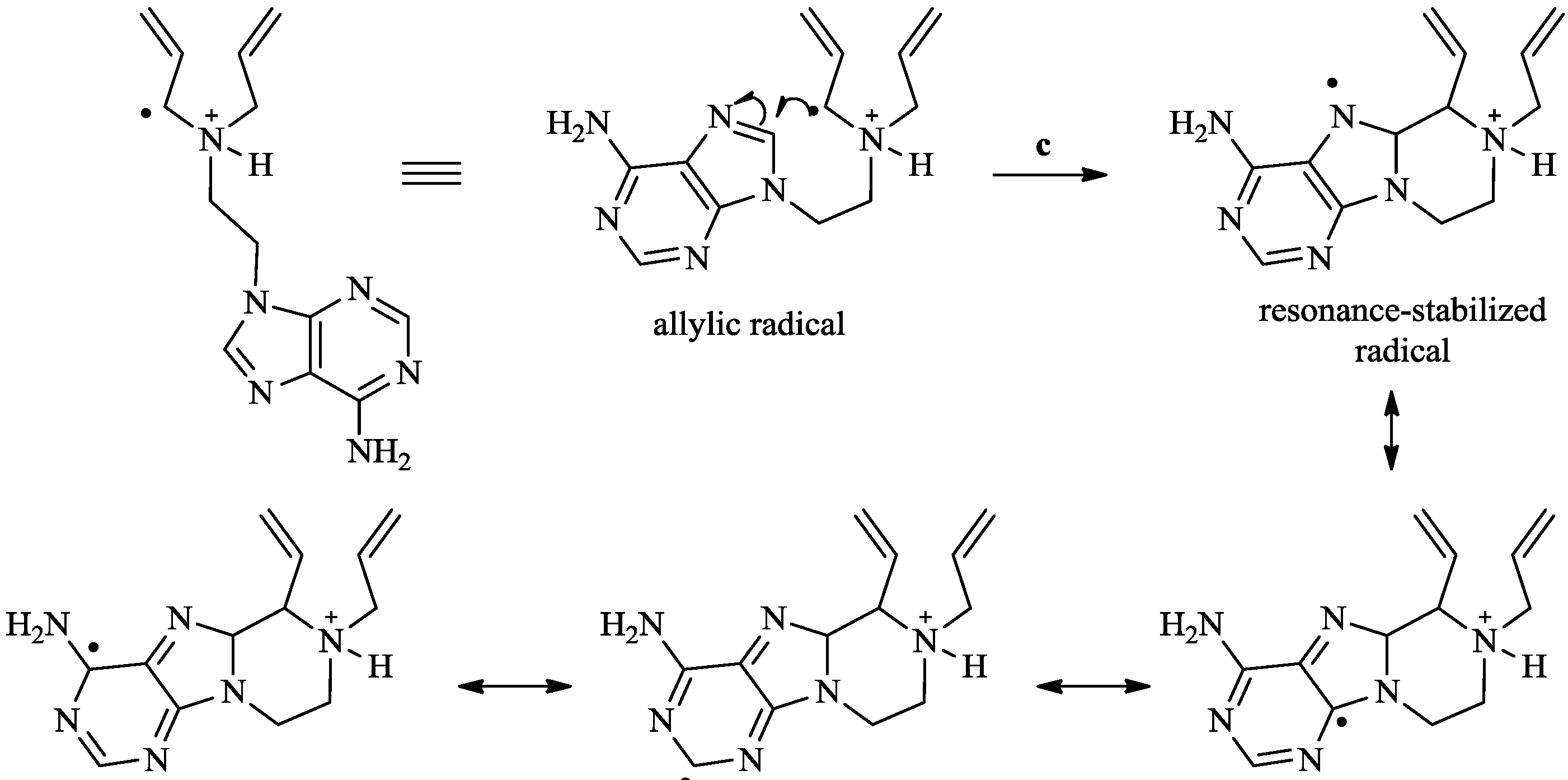{kind=link}
| Entry | Initiator b | Monomer | Solvent | Monomer | T | Yield | Mn d | Mw | Mz | PDI | dn/dc | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | (°C) | (%) c | (kDa) | (kDa) | (kDa) | ||||||
| (mL/g) | ||||||||||||
| 1 | V-50 | DAAC | H2O | - | 70 | 67 | 14.1 | 28.8 | 54.5 | 1.98 | 0.173 | |
| 2 | TBHP | 7 | H2O | - | 80 | 3.7 | - | - | - | - | - | |
| 3 | V-50 | 7 | H2O | - | 70 | 5 | - | - | - | - | - | |
| 4 | APS | 7 | H2O | - | 85 | 17 | 2 | 2.5 | 3 | 1.25 | 0.183 | |
| 5 | V-50 | DAAC | MeOH | SO2 | 70 | 92 | 1350 | 1700 | 2190 | 1.32 | 0.181 | |
| 6 | V-50 | 7 | MeOH | SO2 | 70 | trace | - | - | - | - | - | |
| 7 | V-50 | 7 | DMSO | SO2 | 70 | 7 | - | - | - | - | - | |
| 8 | V-50 | 4 | MeOH | SO2 | 70 | 35 | 6 | 6.2 | 6.3 | 1.03 | 0.183 | |
| 9 | VPE-0201 | 4 | MeOH | SO2 | 70 | 10.6 | - | - | - | - | - | |
| 10 | V-50 | 4 | DMSO | SO2 | 70 | trace | - | - | - | - | - | |
| 11 | V-50 | 4 | DMSO | - | 70 | trace | - | - | - | - | - |

| PDAAC-SO2 | 8 | |||||
|---|---|---|---|---|---|---|
| Solvent | Є a | bp (°C) | Cold b | Hot c | Cold b | Hot c |
| Formamide | 111 | 210 | + | + | + | + |
| Water | 78.4 | 100 | + | + | + | + |
| Formic Acid | 58.5 | 100–101 | + | + | + | + |
| DMSO | 47.2 | 189 | + | ± | − | ± |
| DMF | 38.3 | 153 | − | − | − | − |
| Ethylene glycol | 37.3 | 196–198 | ± | + | ± | + |
| Methanol | 32.3 | 65 | − | − | − | − |
| Ethanol | 24.3 | 78 | − | − | − | − |
| Acetone | 20.7 | 56 | − | − | − | − |
| Diglyme | 7 | 162 | − | − | − | − |
| Dioxane | 2.2 | 101 | − | − | − | − |
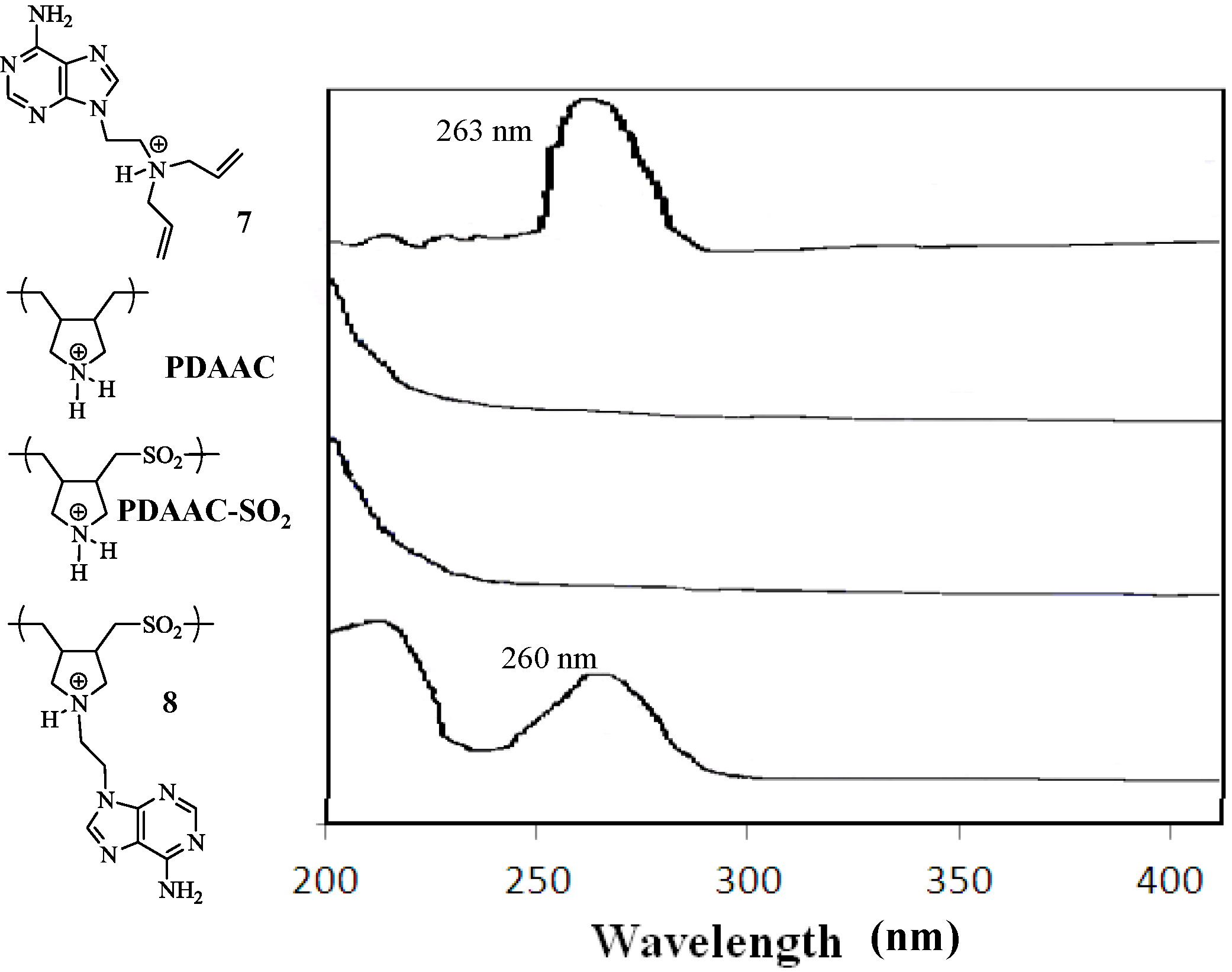





3. Experimental
3.1. General
3.2. Measurements
3.3. Procedure for Cyclopolymerization of DAAC with V-50
3.4. Procedure for Cyclo-Copolymerization of DAAC with V-50
3.5. Procedure for Cyclopolymerization of 7 with V-50
3.6. Procedure for Cyclopolymerization of 7 with Ammonium Persulfate
3.7. Procedure for the Cyclo-Copolymerization of 7 with V-50
3.8. Procedure for Cyclo-Copolymerization of 4 with V-50
4. Conclusions
Acknowledgements
References
- Han, M.J.; Chang, J.Y. Polynucleotide analogues. Adv. Polym. Sci. 2000, 153, 1–36. [Google Scholar] [CrossRef]
- Agrawal, S. Importance of nucleotide sequence and chemical modifications of antisense oligonucleotides. Biochim. Biophys. Acta 1999, 1489, 53–68. [Google Scholar] [CrossRef]
- Uhlmann, E.; Peyman, A. Antisense oligonucleotides: A new therapeutic principle. Chem. Rev. 1990, 90, 543–584. [Google Scholar] [CrossRef]
- Milligan, J.F.; Matteucci, M.D.; Martin, J.C. Current concepts in antisense drug design. J. Med. Chem. 1993, 36, 1923–1937. [Google Scholar] [CrossRef]
- Miller, K.J.; Das, S.D. Antisense oligonucleotides: Strategies for delivery. Pharm. Sci. Technol. Today 1998, 1, 377–386. [Google Scholar] [CrossRef]
- Miller, P.S.; Hamma, T. Syntheses of alternating oligo-2'-O-methylribonucleoside methylphosphonates and their interactions with HIV TAR RNA. Biochemistry 1999, 38, 15333–15342. [Google Scholar] [CrossRef]
- Ottenbrite, R.M.; Ryan, W.S., Jr. Cyclopolymerization of N,N-dialkyldiallylammonium halides: A review and use analysis. Ind. Eng. Chem. Prod. Res. Develop. 1980, 19, 528–532. [Google Scholar] [CrossRef]
- Solomon, D.H.; Hawthorne, D.G. Cyclopolymerization of diallylamines. J. Macromol. Sci. Rev. Macromol. Chem. 1976, C15, 143–164. [Google Scholar]
- Wang, G.J.; Engberts, J.B.F.N. Non-cross-linked and cross-linked poly(alkylmethyldiallylammonium halides): Synthesis and aggregation behavior. J. Phys. Org. Chem. 1998, 11, 305–320. [Google Scholar]
- Ai, H.; Fang, M.; Jones, S.A.; Lvov, Y.M. Electrostatic layer-by-layer nanoassembly on biological microtemplates: Platelets. Biomacromolecules 2002, 3, 560–564. [Google Scholar] [CrossRef]
- Rullens, F.; Vuillaume, P.Y.; Moussa, A.; Habib-Jiwan, J.L.; Laschewsky, A. Ordered polyelectrolyte “Multilayers”. 7. Hybrid films self-Assembled from fluorescent and smectogenic poly(diallylammonium) salts and delaminated clay. Chem. Mater. 2006, 18, 3078–3087. [Google Scholar] [CrossRef]
- Pei, R.; Cui, X.; Yang, X.; Wang, E. Assembly of alternating polycation and DNA multilayer films by electrostatic layer-by-layer adsorption. Biomacromolecules 2001, 2, 463–468. [Google Scholar] [CrossRef]
- Schuller, W.H.; Price, J.A.; Moore, S.T.; Thomas, W.M. Soluble copolymers of diallyl monomers. J. Chem. Eng. Data 1959, 4, 273–276. [Google Scholar] [CrossRef]
- Armentrout, R.S.; McCormick, C.L. Amphoteric cyclocopolymers with sulfobetaine units: Phase behavior in aqueous media and solubilization of p-Cresol in microdomains. Macromolecules 2000, 33, 2944–2951. [Google Scholar] [CrossRef]
- Ali, S.A.; Saeed, M.T. Synthesis and corrosion inhibition study of some 1,6-hexanediamine-based N,N-diallyl quaternary ammonium salts and their polymers. Polymer 2001, 42, 2785–2794. [Google Scholar] [CrossRef]
- Weber, W.; Rinderknecht, M.; Daoud-El Baba, M.; de Glutz, F.N.; Aubel, D.; Fussenegger, M. CellMAC: A novel technology for encapsulation of mammalian cells in cellulose sulfate/pDADMAC capsules assembled on a transient alginate/Ca2+ scaffold. J. Biotechnol. 2004, 114, 315–326. [Google Scholar]
- Sawada, H.; Tanba, K.; Tomita, T.; Kawase, T.; Baba, M.; Ide, T. Antibacterial activity of fluoroalkylated allyl- and diallyl-ammonium chloride oligomers. J. Fluorine Chem. 1997, 84, 141–144. [Google Scholar] [CrossRef]
- Chen, J.C.; Yeh, J.T.; Chen, C.C. Crosslinking of cotton cellulose in the presence of alkyl diallyl ammonium salts. Part I. Physical properties and agent distribution. J. Appl. Polym. Sci. 2003, 90, 1662–1669. [Google Scholar] [CrossRef]
- Timofeeva, L.M.; Kleshcheva, N.A.; Vasilieva, Y.A.; Gromova, G.L.; Timofeeva, G.I.; Filatova, M.P. Effect of dielectric properties and structure of aqueous solutions of diallylammonium salts on their reactivity in radical polymerization . Polym. Sci. Ser. A 2005, 47, 273–282. [Google Scholar]
- Timofeeva, L.M.; Vasilieva, Y.A.; Klescheva, N.A.; Gromova, G.L.; Timofeeva, G.I.; Rebrov, A.I.; Topchiev, D.A. Effect of dielectric and structural properties of solutions on the polymerizability of diallylammonium-type monomers. Phys. Chem. 2006, 406, 53–56. [Google Scholar]
- Timofeeva, L.M.; Kleshcheva, N.A.; Moroz, A.F.; Didenko, L.V. Secondary and tertiary polydiallylammonium salts: Novel polymers with high antimicrobial activity. Biomacromolecules 2009, 1, 2976–2986. [Google Scholar]
- Tsai, J.Y.; Bouhadir, K.H.; Zhou, J.L.; Webb, T.R.; Sun, Y.; Shevlin, P.B. Synthesis of purine- and pyrimidine-substituted heptadienes. The stereochemistry of cyclization and cyclopolymerization products. J. Org. Chem. 2003, 68, 1235–1241. [Google Scholar] [CrossRef]
- Shatila, R.; Bouhadir, K. Two simple protocols for the preparation of diallylaminoethyl-substituted nucleic bases: A comparison. Tetrahedron Lett. 2006, 47, 1767–1770. [Google Scholar] [CrossRef]
- Ustyuzhanin, G.E.; Kolomeitseva, V.V.; Tikhomirova-Sidorova, N.S. Hydroxyethylation of uracil, adenine, and cytosine with ethylene carbonate. Chem. Heterocycl. Compd. 1978, 14, 562–566. [Google Scholar] [CrossRef]
- Vivekanadam, T.S.; Gopala, A.; Vasudevan, T.; Umapathy, S. Sonochemical cyclopolymerization of diallylamine. Eur. Polym. J. 2000, 36, 385–392. [Google Scholar] [CrossRef]
- Vivekanadam, T.S.; Gopala, A.; Vasudevan, T.; Umapathy, S. Sonochemical cyclopolymerization of diallylamine in the presence of peroxomonosulfate. J. Appl. Polym. Sci. 2005, 98, 1548–1553. [Google Scholar] [CrossRef]
- Harada, S.; Katayama, M. The cyclo-copolymerization of diallyl compound and sulfur dioxide. I. Diallylamine hydrochloride and sulfur dioxide. Macromol. Chem. Phys. 1966, 90, 177–186. [Google Scholar] [CrossRef]
- Ueda, N.; Kondo, K.; Kono, M.; Takemoto, K.; Imoto, M. Vinyl polymerization. 217. Vinyl compounds of nucleic acid basis. I. Synthesis of N-vinylthymine, and N-vinyladenine. Macromol. Chem. Phys. 1968, 120, 13–20. [Google Scholar] [CrossRef]
- Zhou, J.; Shevlin, P.B. A short synthesis of 1-vinyluracil and 1-vinylthymine. Synth. Commun. 1997, 27, 3591–3597. [Google Scholar] [CrossRef]
- Pitha, J.; Ts’o, P.O.P. N-Vinyl derivatives of substituted pyrimidines and purines. J. Org. Chem. 1968, 33, 1341–1344. [Google Scholar] [CrossRef]
- Akashi, M.; Inaki, Y.; Takemoto, K. Functional monomers and polymers, 31. On the stereoregularity of vinyl polymers containing nucleic acid bases. Macromol. Chem. Phys. 1977, 178, 353–364. [Google Scholar] [CrossRef]
- Butler, G. Cyclopolymerization and cyclocopolymerization. Acc. Chem. Res. 1982, 15, 370–378. [Google Scholar] [CrossRef]
- Tuzun, N.S.; Aviyente, V.; Houk, K.N. A theoretical study on the mechanism of the cyclopolymerization of diallyl monomers. J. Org. Chem. 2003, 68, 6369–6374. [Google Scholar] [CrossRef]
- Al-Muallem, H.; Wazeer, M.I.M.; Ali, S.A. Synthesis and solution properties of a new pH-responsive polymer containing amino acid residues. Polymer 2002, 43, 4285–4295. [Google Scholar] [CrossRef]
- Vasilieva, Y.A.; Kleshcheva, N.A.; Gromova, G.L.; Rebrov, A.I.; Filatova, M.P.; Krut’ko, E.B.; Timofeeva, L.M.; Topchiev, D.A. Synthesis of high-molecular-weight polyamine by radical polymerization of N,N-diallyl-N-methylamine. Russ. Chem. Bull. 2000, 49, 431–437. [Google Scholar] [CrossRef]
- Tuzun, N.S.; Aviyente, V. Modeling the cyclopolymerization of diallyl ether and methyl α-[(allyloxy)methyl]acrylate. Int. J. Quantum Chem. 2007, 107, 894–906. [Google Scholar] [CrossRef]
- Litt, M.; Eirich, F.R. Polymerization of allyl acetate. J. Polym. Sci. 1960, 45, 379–396. [Google Scholar] [CrossRef]
- Ali, S.A.; Wazeer, M.I.M.; Ahmed, S.Z. Piperazine-based homo- and copolymers containing trivalent and quaternary nitrogen functionalities. J. Appl. Polym. Sci. 1998, 69, 1329–1334. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization, 4th ed; Wiley-Interscience: Hoboken, NJ, USA, 2004; pp. 144–166. [Google Scholar]
- Gorbunova, M.; Vorob’eva, A.; Muslukhov, R. NMR for determining the structure of new polysulfones. Int. J. Polym. Anal. Charact. 2009, 14, 575–587. [Google Scholar] [CrossRef]
- Gorbunova, M.; Vorob’eva, A.; Tolstikov, A.; Monakov, Y. New N-allylated monomers in the synthesis of practical valuable high-molecular-weight compounds. Polym. Adv. Technol. 2009, 20, 209–215. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compounds 2–9 are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bouhadir, K.H.; Abramian, L.; Ezzeddine, A.; Usher, K.; Vladimirov, N. Synthesis, Cyclopolymerization and Cyclo-Copolymerization of 9-(2-Diallylaminoethyl)adenine and Its Hydrochloride Salt. Molecules 2012, 17, 13290-13306. https://doi.org/10.3390/molecules171113290
Bouhadir KH, Abramian L, Ezzeddine A, Usher K, Vladimirov N. Synthesis, Cyclopolymerization and Cyclo-Copolymerization of 9-(2-Diallylaminoethyl)adenine and Its Hydrochloride Salt. Molecules. 2012; 17(11):13290-13306. https://doi.org/10.3390/molecules171113290
Chicago/Turabian StyleBouhadir, Kamal H., Lara Abramian, Alaa Ezzeddine, Karyn Usher, and Nikolay Vladimirov. 2012. "Synthesis, Cyclopolymerization and Cyclo-Copolymerization of 9-(2-Diallylaminoethyl)adenine and Its Hydrochloride Salt" Molecules 17, no. 11: 13290-13306. https://doi.org/10.3390/molecules171113290