Novel Rearrangements in the Reactions Directed Toward Preparation of Spiro-N,N-ketals: Reactions of Naphthalene-1,8-diamine with Ninhydrin and Isatin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
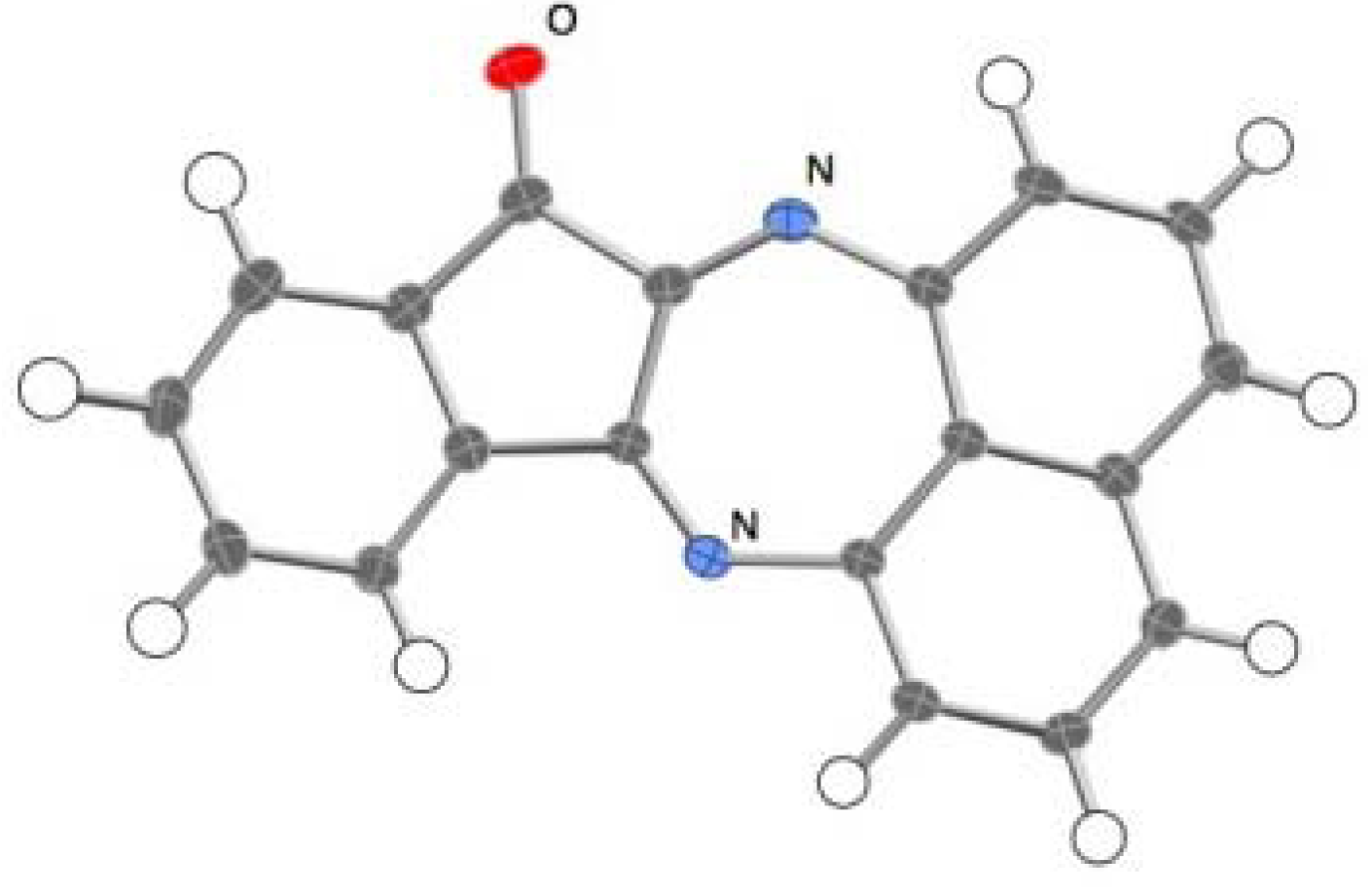
2. Results and Discussion
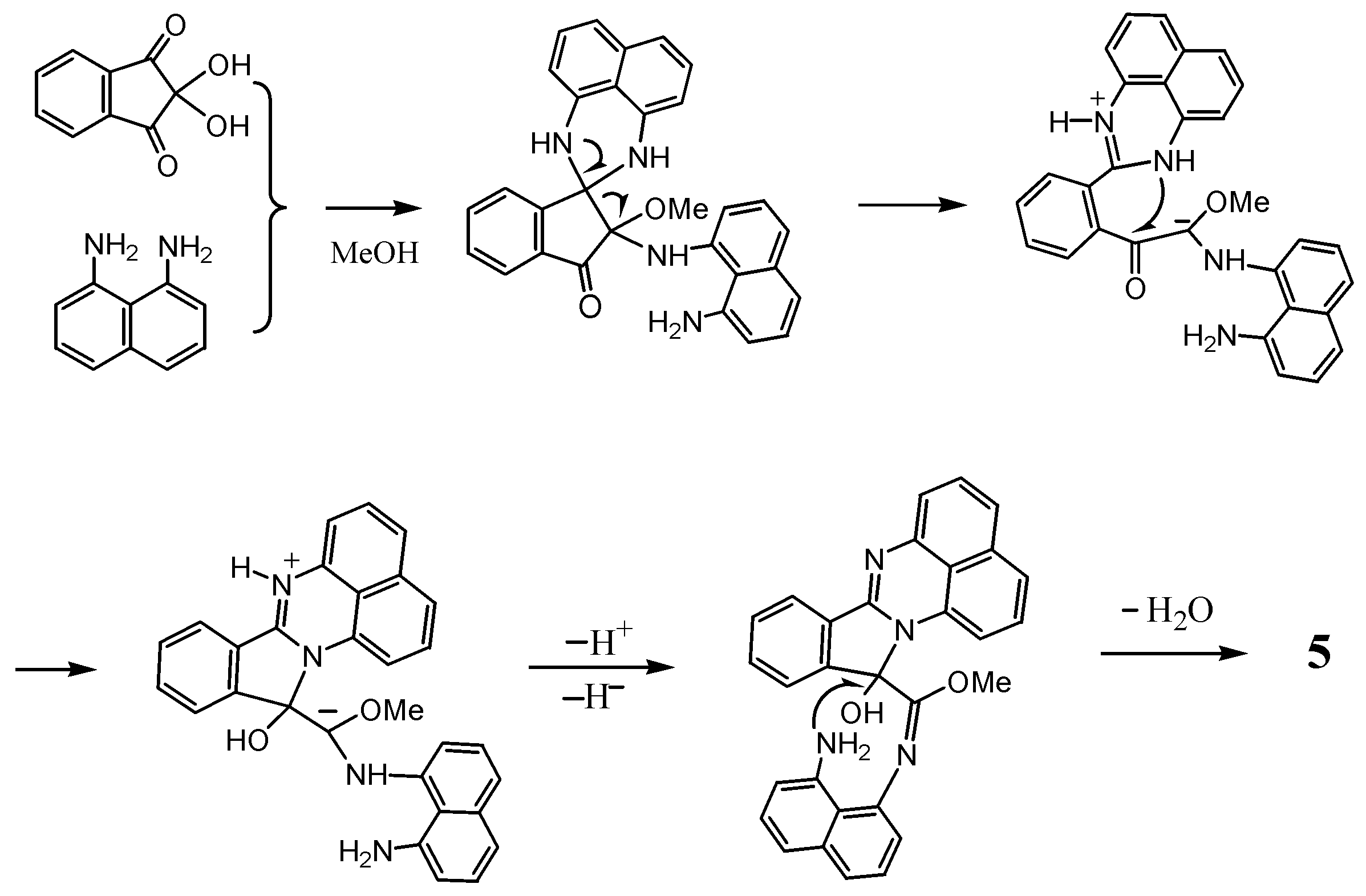
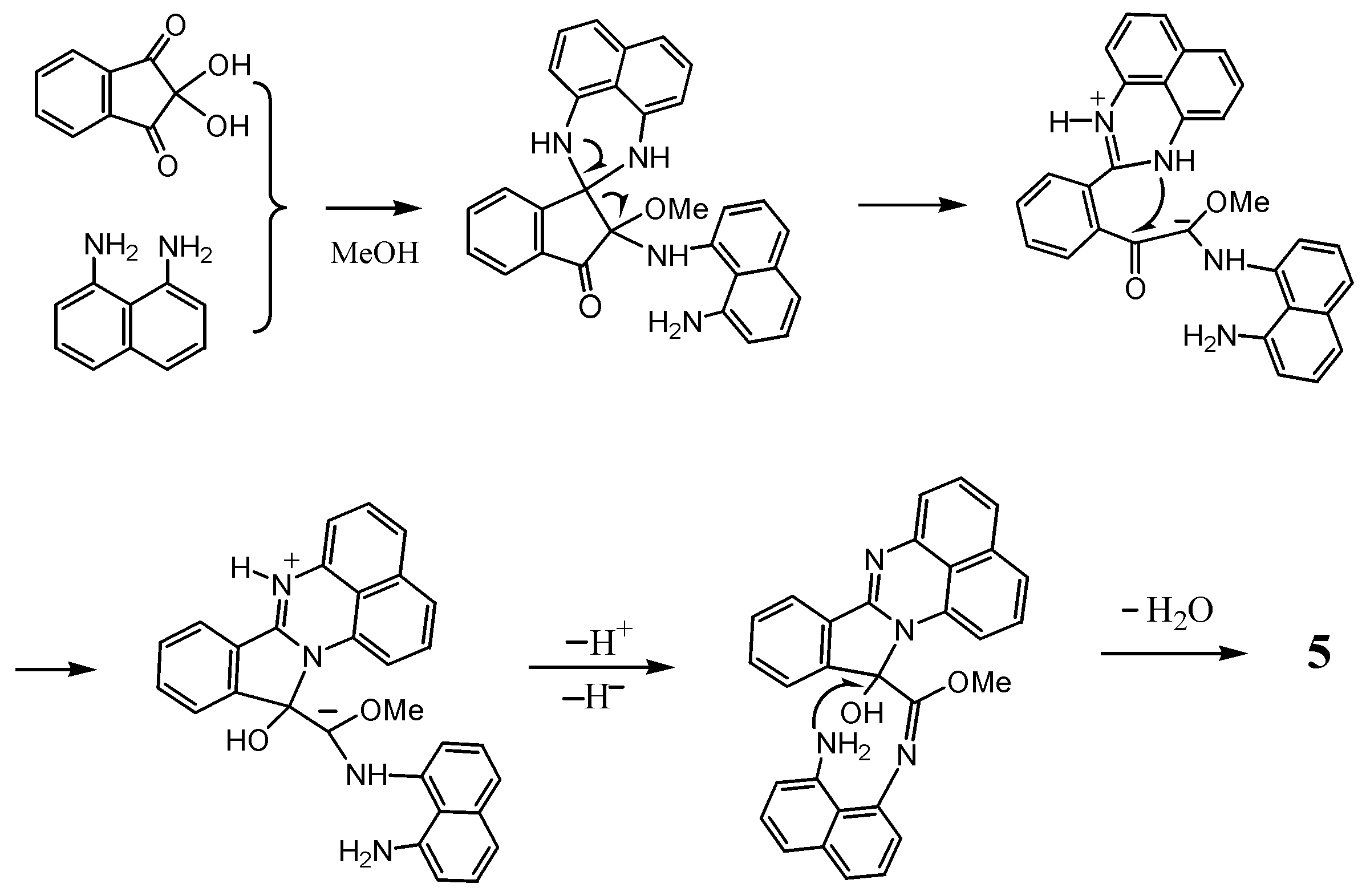
2.1. Reaction with Ninhydrin

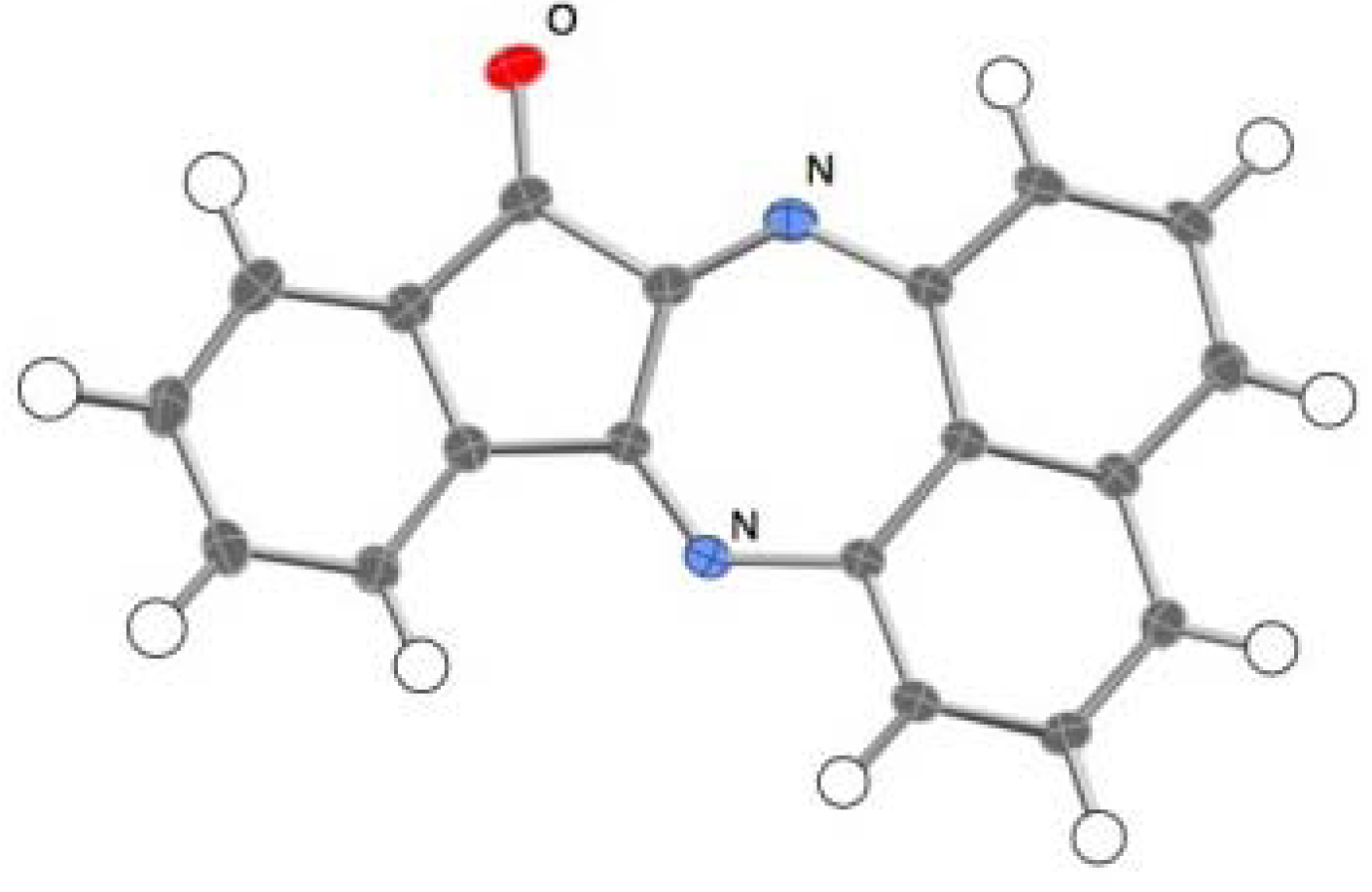
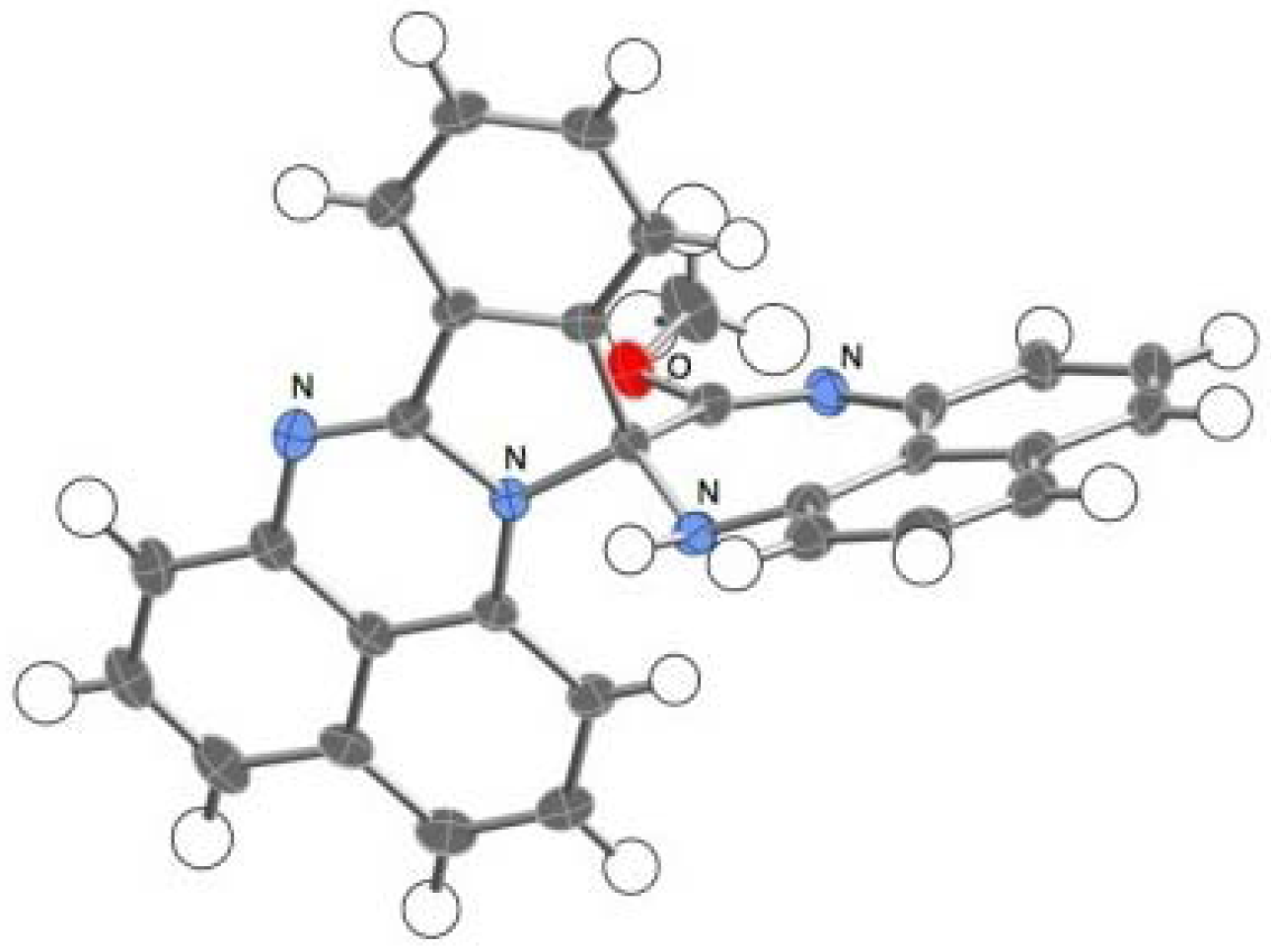
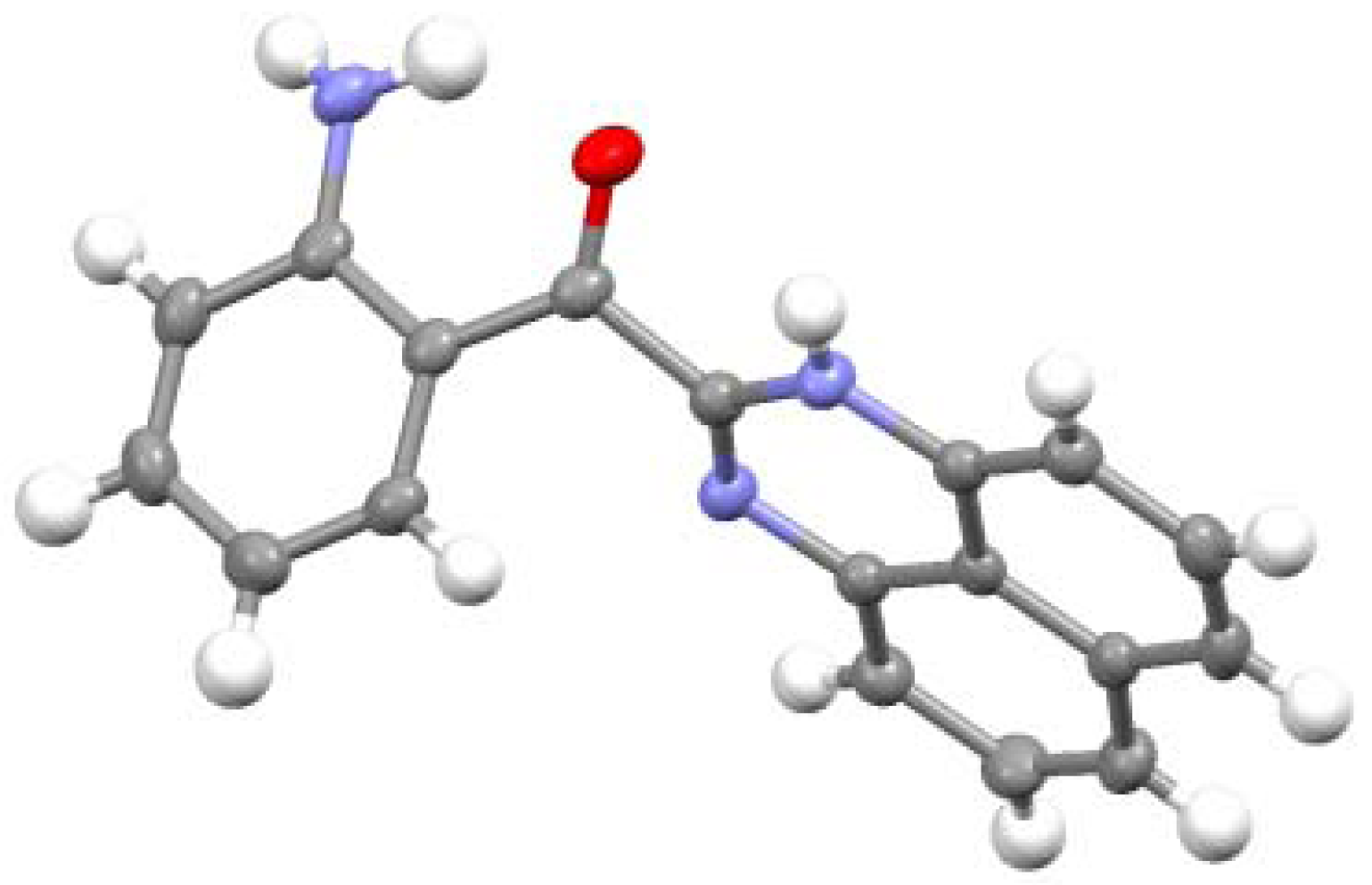
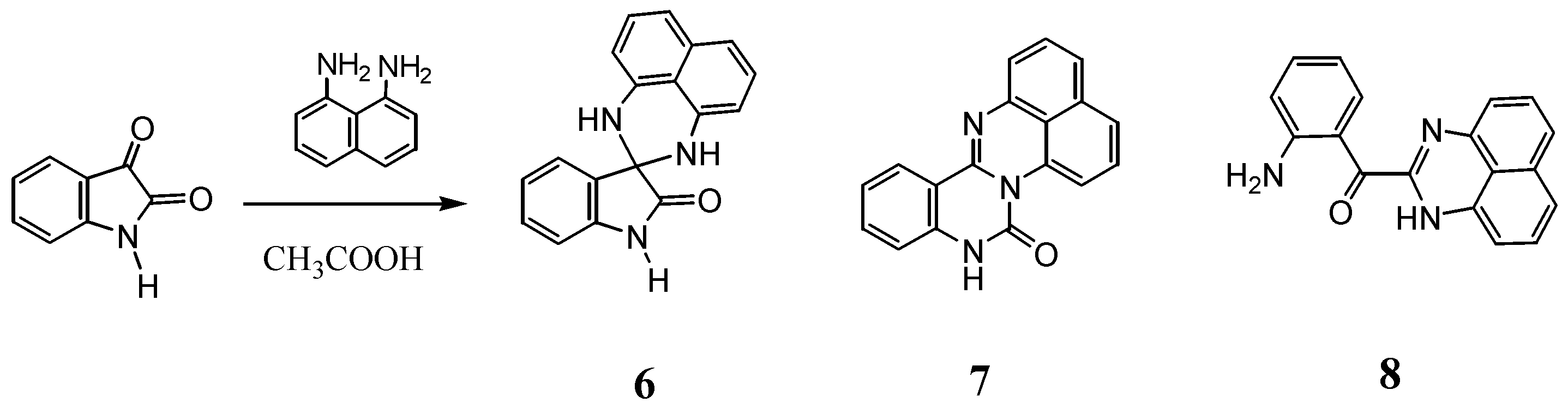
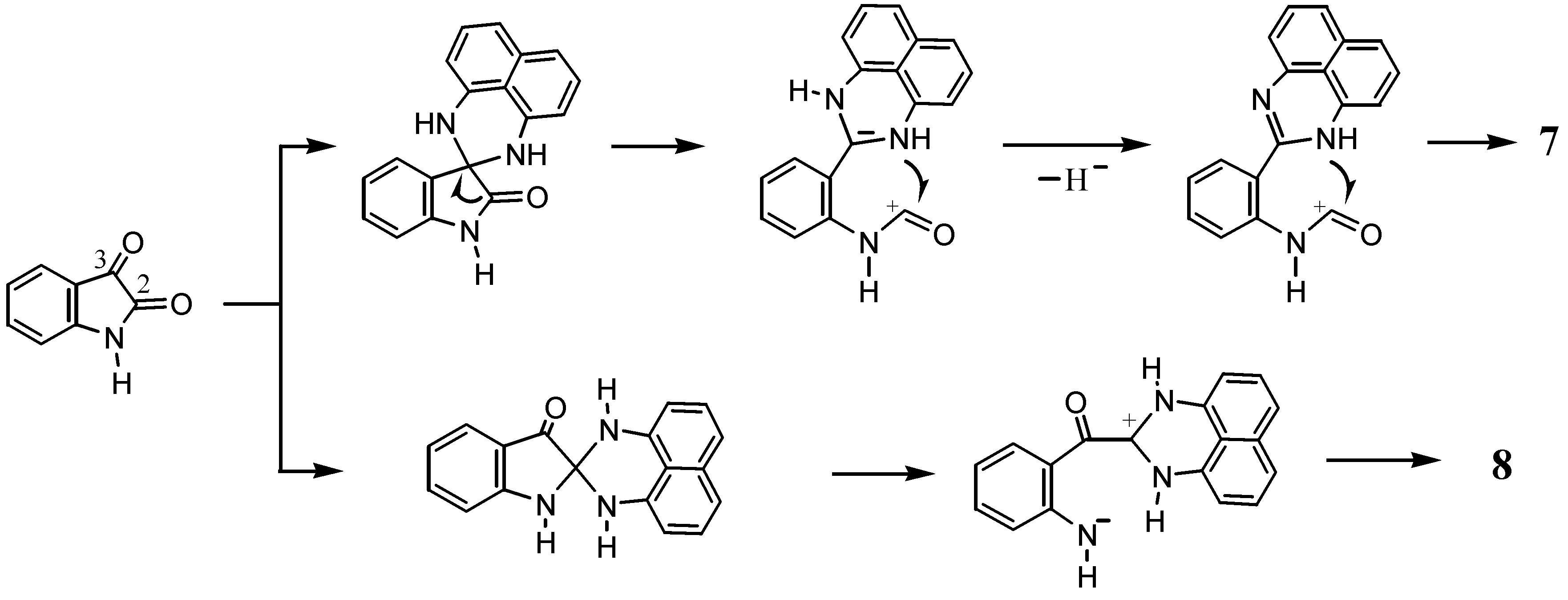
2.2. Reactions with Isatin
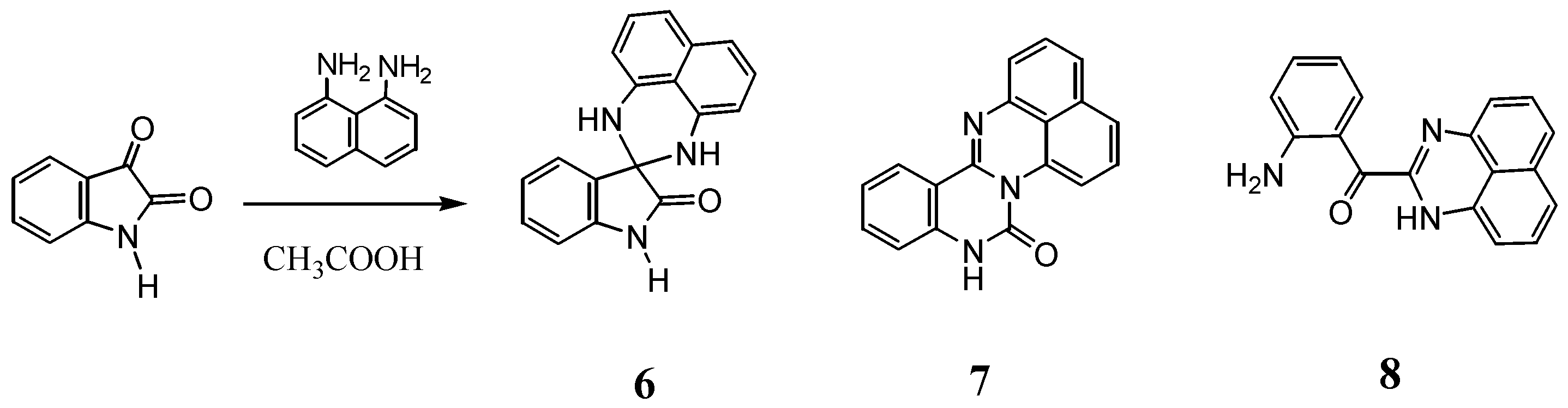
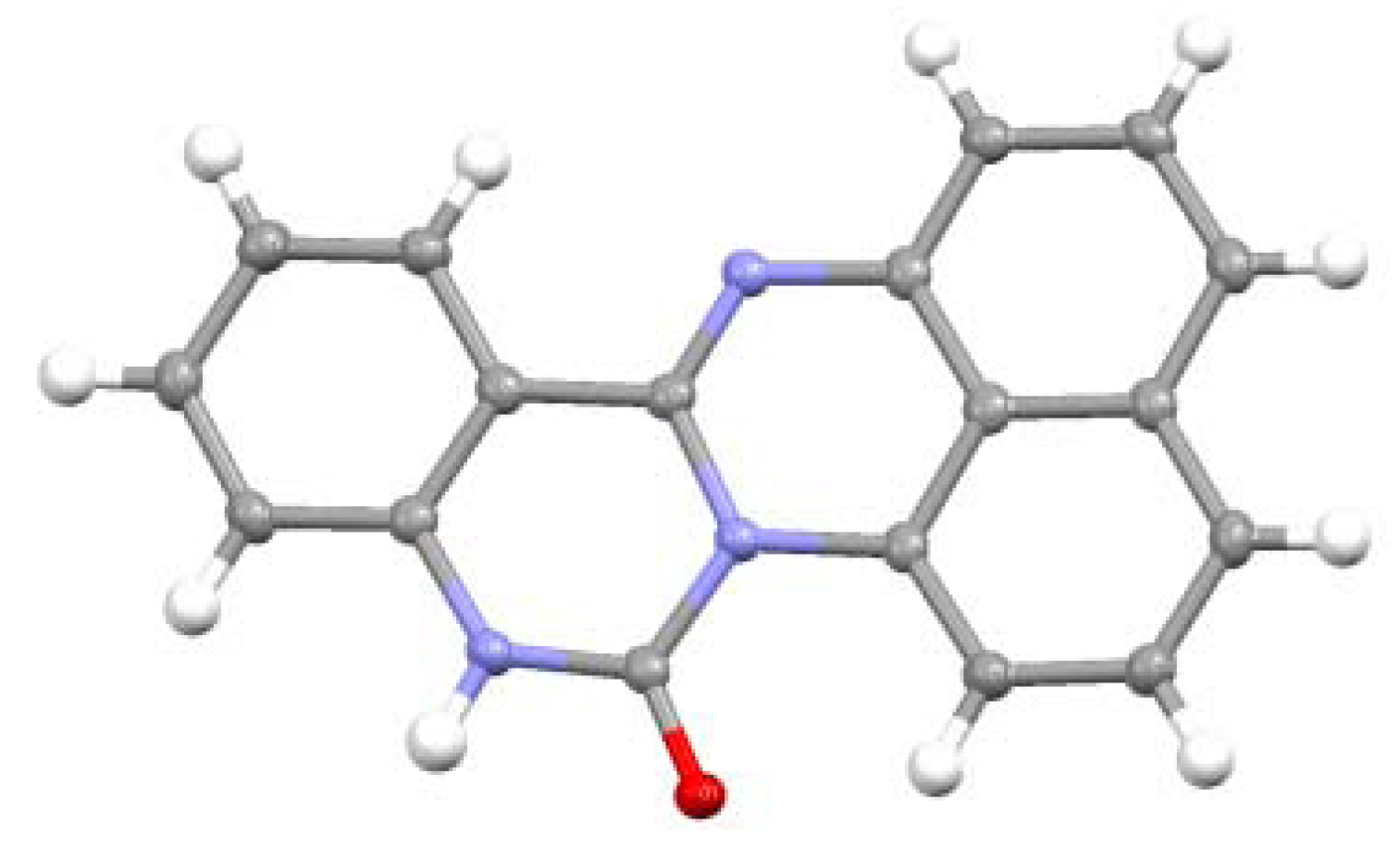

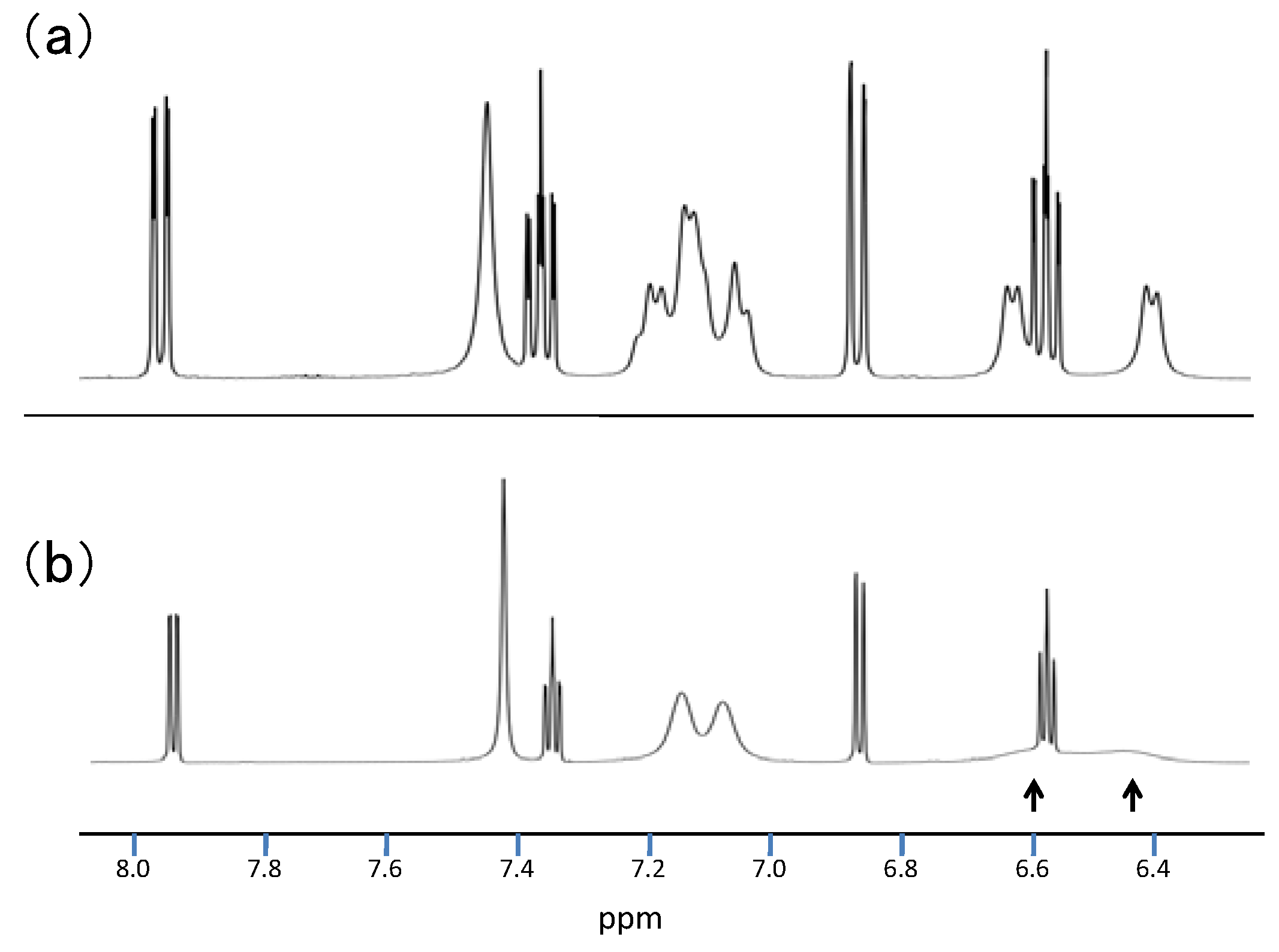
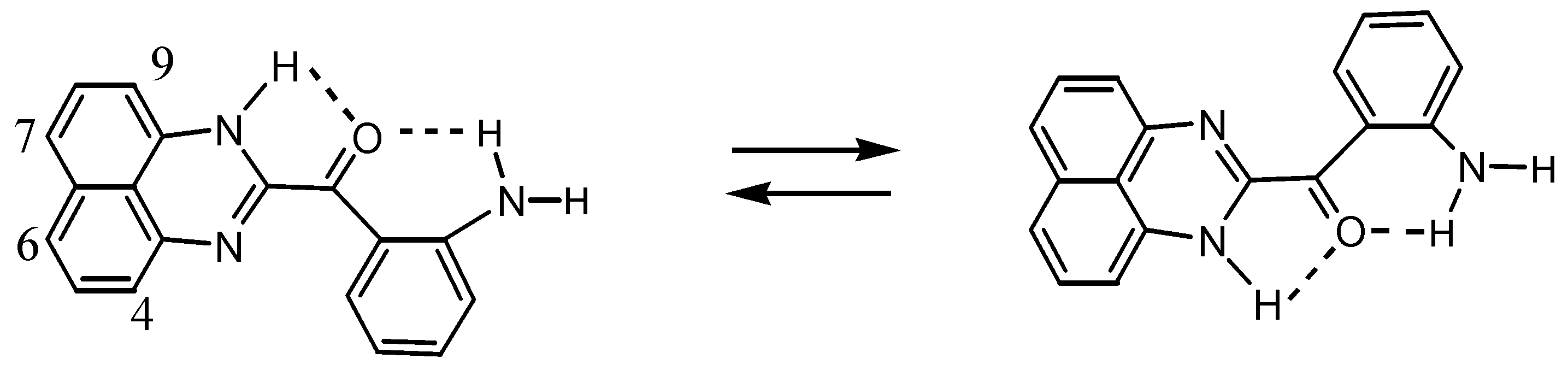
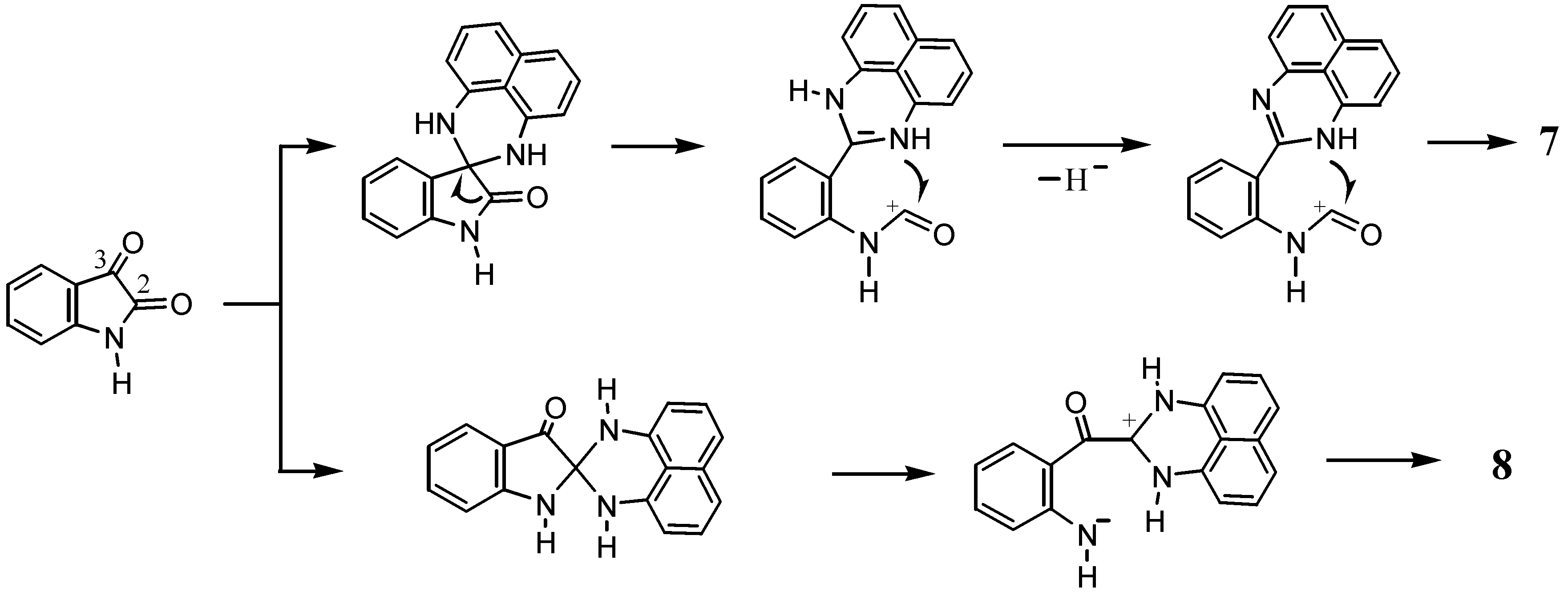
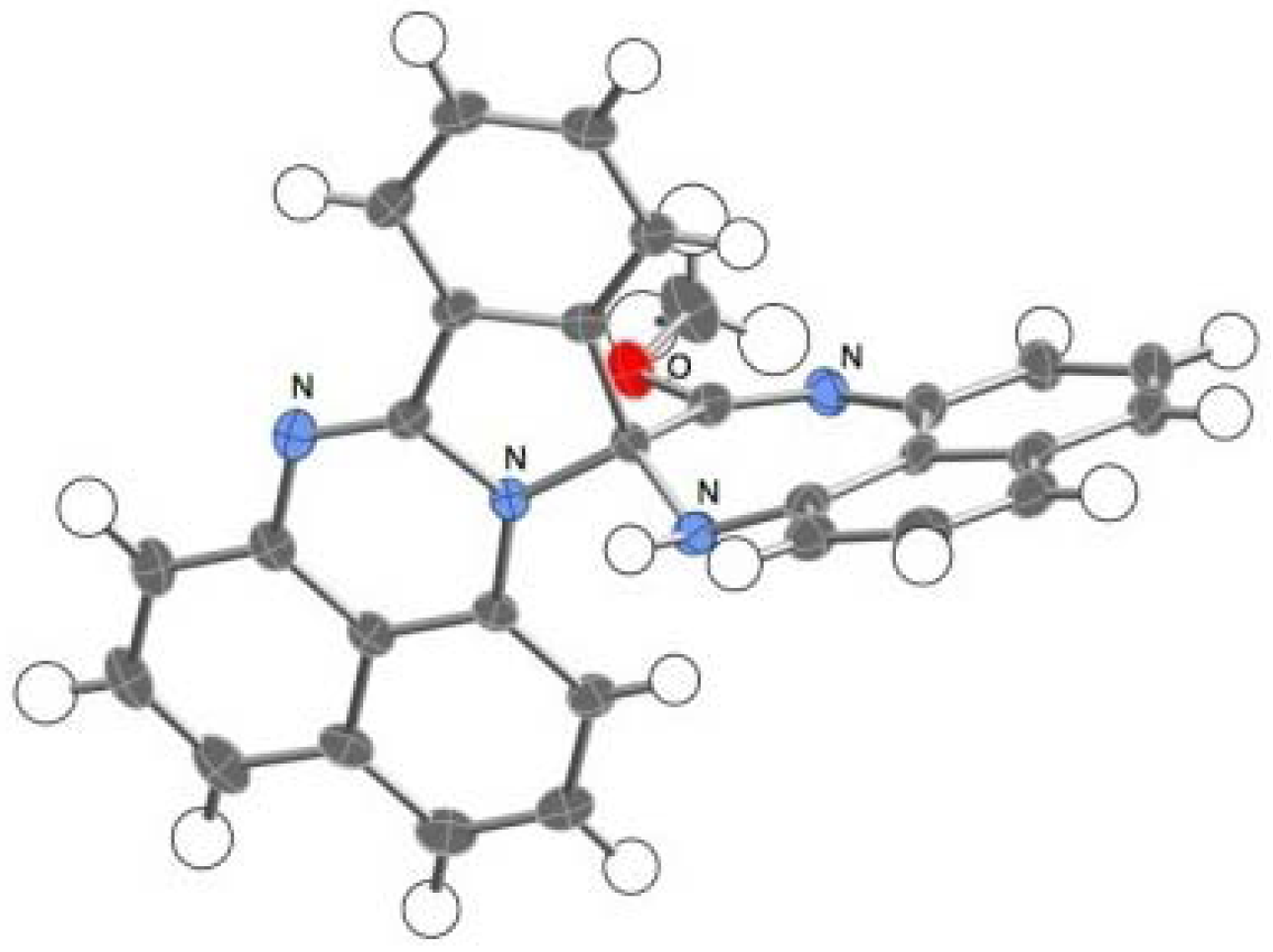
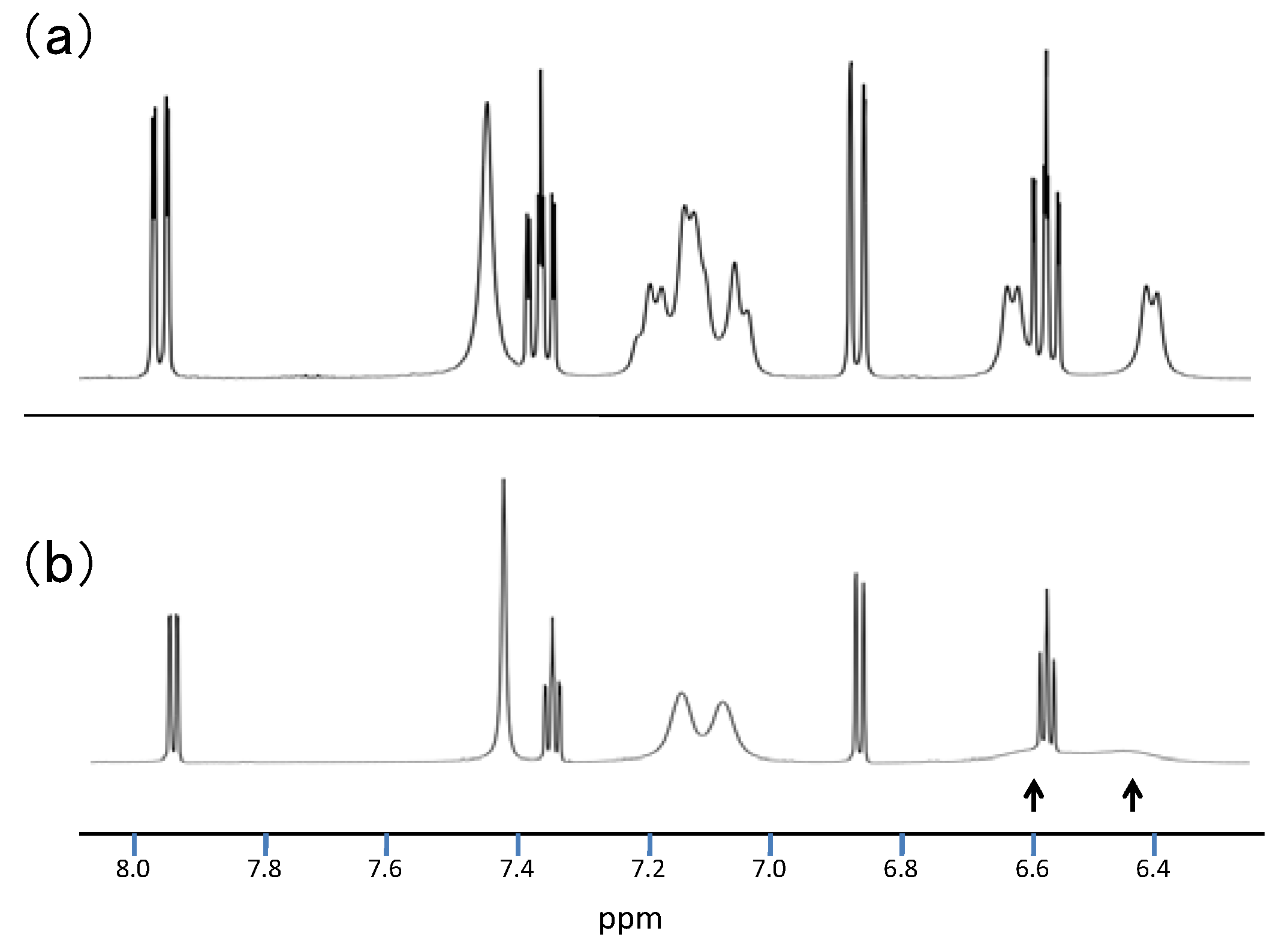
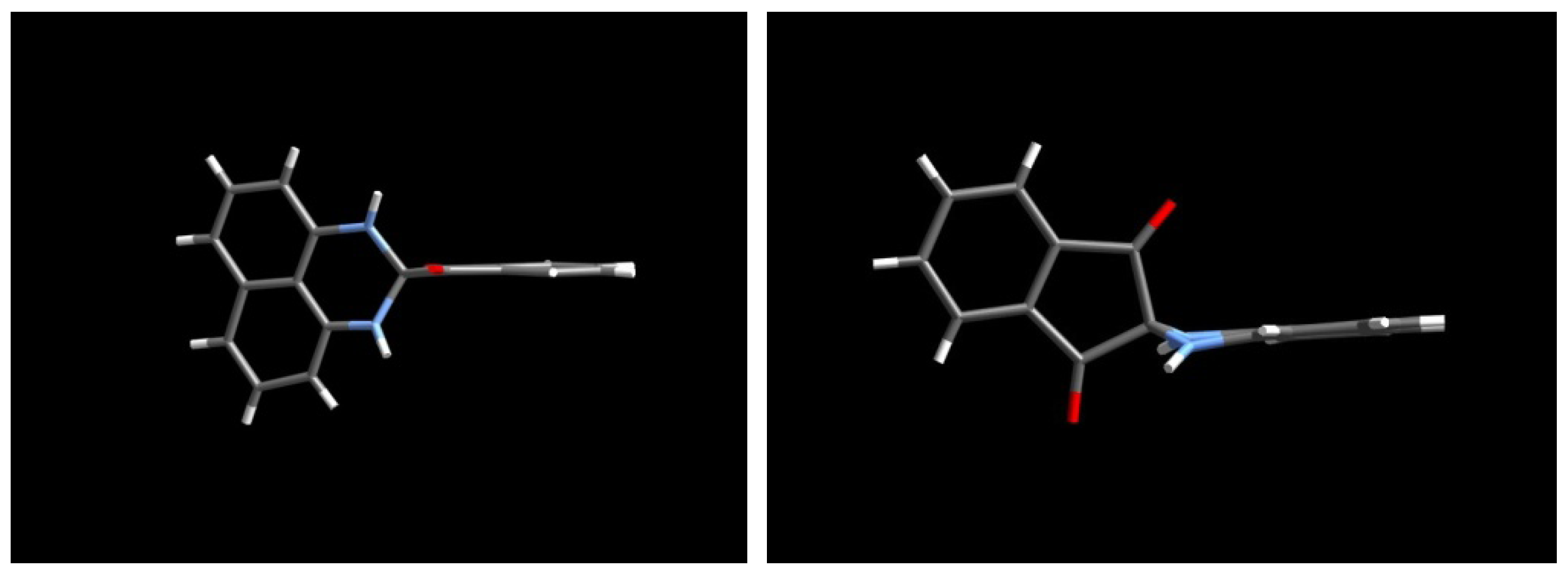
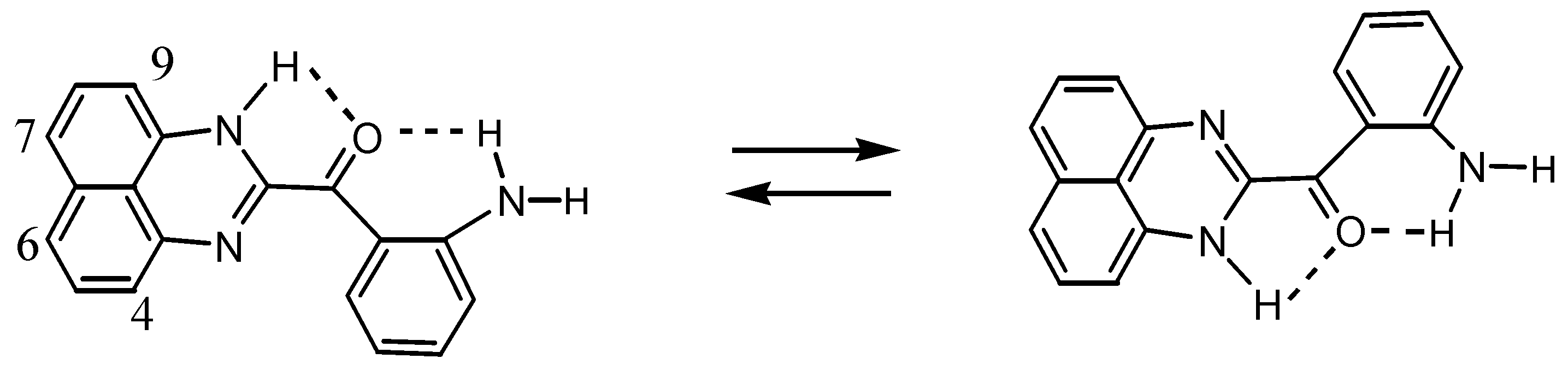
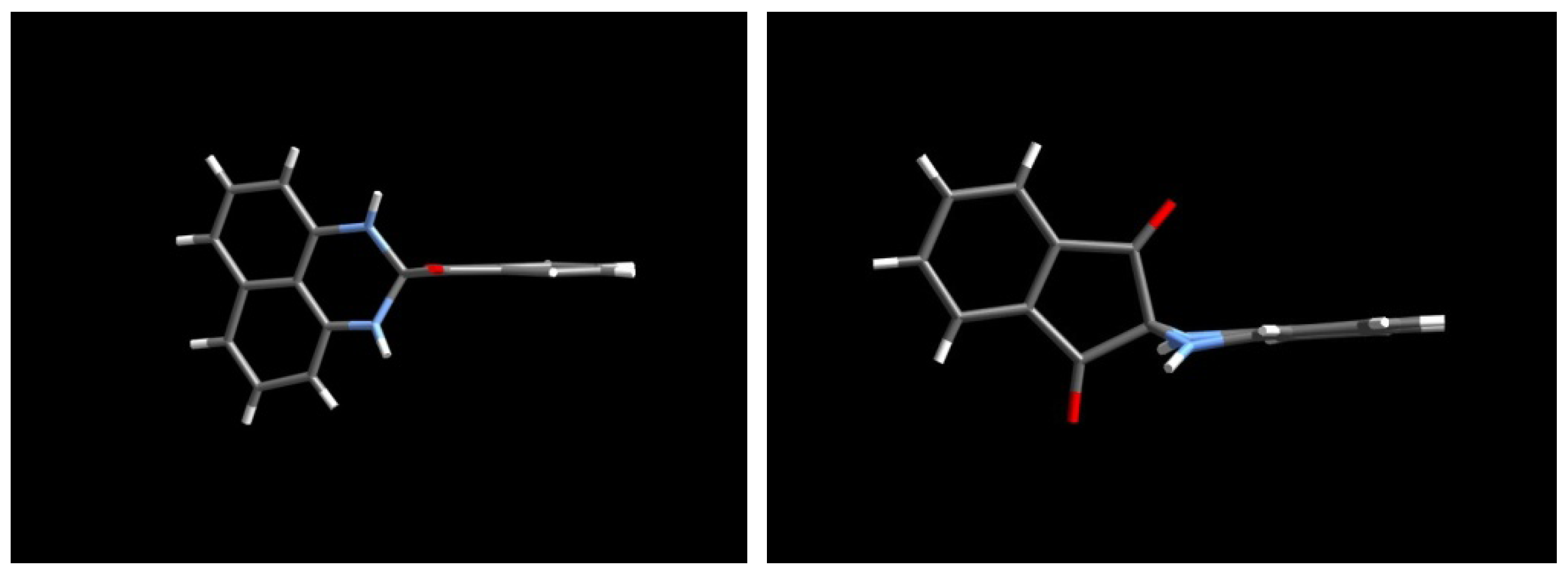
2.3.Structural Characteristics of the New Spiro-N,N-ketals

3. Experimental
General
3.1. Reaction with Ninhydrin
3.1.1. In Acetonitrile
3.1.2. In Methanol
3.1.3. In Methanol with p-Toluenesulfonic Acid
3.2. Reaction with Isatin
3.3. X-ray Crystal Structure Analysis
4. Conclusions
- Sample Availability: Contact the authors.
References
- Sinibaldi, M.-E.; Canet, I. Synthetic approachs to spiroaminals. Eur. J. Org. Chem. 2008, 2008, 4391–4399. [Google Scholar] [CrossRef]
- Schönberg, A.; Singer, E.; Osch, M.; Hoyer, G.-A. Å°ber die Reaktion von N,N'-Disubstituierten Äthylendiamin mit Ninhydrin. Ein 1,4-Diaza-spiro[4,4]nonan-system mit Ungewöhnlichen Eigenschaften. Tetrahedron Lett. 1975, 37, 3217–3220. [Google Scholar]
- Schönberg, A.; Singer, E.; Eschenhof, B.; Hoyer, G.-A. Å°ber Reaktionen des Ninhydrins bzw. 1,2,3-Indantrions mit 1,2- und 1,3-bifunktionellen Verbindungen. Ein Beitrag zur Bildung von Spiroverbindungen aus Ninhydrin. Chem. Ber. 1978, 111, 3058–3067. [Google Scholar] [CrossRef]
- Schönberg, A.; Singer, E.; Eckert, P. Å°ber die photochemische Epoxidierung einer Carbonylgruppe mit Methanol. Chem. Ber. 1980, 113, 3094–3097. [Google Scholar] [CrossRef]
- Simmons, H.E.; Fukunaga, T. Spiroconjugation. J. Am. Chem. Soc. 1967, 89, 5208–5215. [Google Scholar] [CrossRef]
- Minkin, V. Photo-, Thermo-, Solvato-, and electrochromic spiroheterocyclic compounds. Chem. Rev. 2004, 104, 2751–2776. [Google Scholar] [CrossRef]
- Maslak, P.; Chopra, A.; Moylan, C.R.; Wortmann, R.; Lebus, S.; Rheingold, A.L.; Yap, G.P.A. Optical properties of spiroconjugated charge-transfer dyes. J. Am. Chem. Soc. 1996, 118, 1471–1481. [Google Scholar]
- Kaupp, G.; Naimi-Jamal, M.R.; Schmeyers, J. Quantitative reaction cascades of ninhydrin in the solid state. Chem. Eur. J. 2002, 8, 594–600. [Google Scholar] [CrossRef]
- Deady, L.W.; Desneves, J.; Ross, A.C. Synthesis of some 11H-indeno[1,2-b]quinoxalin-11-one. Tetrahedron 1993, 49, 9823. [Google Scholar] [CrossRef]
- Israel, M.; Jones, L.C.; Modest, E.J. 6H-Indeno[1,2-b]pyrido[3,2-e]pyrazines. A new heterocyclic ring system. J. Heterocycl. Chem. 1972, 9, 255–258. [Google Scholar] [CrossRef]
- Zeller, K.-P. The Chemistry of the Quinonoid Compounds; Patai, S., Ed.; Wiley: London, UK, 1974; Volume 1, p. 236. [Google Scholar]
- Mosher, W.A.; Banks, T.E. Reaction of 2-acyl-1,3-indandiones with 1,8-naphthalenediamine. A new route to 2-substituted perimidines. J. Org. Chem. 1971, 36, 1477–1480. [Google Scholar] [CrossRef]
- Schank, K.; Leider, R.; Click, C.; Glock, R. Chemie freier cyclischer vicinaler Tricarbonyl-verbindungen. Helvetica. Chem. Acta 2004, 87, 869–924. [Google Scholar] [CrossRef]
- Popp, F.D. Synthesis of potential antineoplastic agents. XXI. Compounds related to ellipticine. J. Heterocycl. Chem. 1972, 9, 1399–1401. [Google Scholar] [CrossRef]
- Popp, F.D. Reaction of isatine with aromatic o-diamines. J. Heterocycl. Chem. 1969, 6, 125–127. [Google Scholar] [CrossRef]
- Bergman, J.; Staalhandske, C.; Vallberg, H. Studies of the reaction between indole-2,3-diones (isatins) and secondary aliphatic amines. Acta Chem. Scand. 1997, 51, 753–759. [Google Scholar] [CrossRef]
- Bergman, J.; Engqvist, R.; Stälhandske, C.; Wallberg, H. Studies of the reactions between indole-2,3-diones (isatins) and 2-aminobenzylamine. Tetrahedron 2003, 59, 1033–1048. [Google Scholar] [CrossRef]
- Dabiri, M.; Mohammadi, A.A.; Qaraat, H. An efficient and convenient protocol for the synthesis of novel 1H’-spiro[isoindoline-1,2'-quinazoline]-3,4'(3'H)-dione derivatives. Monatsh Chem. 2009, 140, 401–404. [Google Scholar] [CrossRef]
- Niume, K.; Kurosawa, S.; Toda, F.; Hasegawa, M.; Iwakura, Y. The condensation of isatin with o-phenylenediamine. Bull. Chem. Soc. Jpn. 1982, 55, 2293. [Google Scholar] [CrossRef]
- Henseke, G.; Lemke, W. Heterocyclic compounds. II. Higher-condensed quinoxalines. Chem. Ber. 1958, 91, 101–112. [Google Scholar] [CrossRef]
- Phadtare, S.B.; Vijayraghavan, R.; Shankarling, G.S.; MacFarlane, D.R. Efficient Synthesis of 2,3-Dihydro-1H-Perimidine Derivatives Using HBOB as a Novel Solid Acid Catalyst. Aust. J. Chem. 2012, 65, 86–90. [Google Scholar] [CrossRef]
- Yasaei, Z.; Mirzaei, P.; Bazgir, A. InCl3-catalyzed efficient synthesis of spiro-perimidine derivatives. Compt. Rend. Chim. 2010, 13, 1308–1312. [Google Scholar]
- Arya, K.; Dandia, A. Regioselective Synthesis of Biologically Important Scaffold Spiro [Indole-Perimidines]: An Antitumor Agents. Lett. Org. Chem. 2007, 4, 378–383. [Google Scholar] [CrossRef]
- Beam, C.F.; Heindel, N.D.; Chun, L.; Stefanski, A. The preparation of heterocyclic materials from 2-isocyanatobenzoyl chloride and difunctional nucleophiles. J. Heterocycl. Chem. 1976, 13, 421–411. [Google Scholar] [CrossRef]
- Yavari, I.; Adib, M.; Jahani-Moghaddam, F.; Bijanzadeh, H.R. Vinylphosphonium salt mediated simple synthesis of 7-oxo-7H-pyrido[1,2,3-cd]perimidine derivatives. Dynamic NMR spectroscopic study of prototropic tautomerism in ethyl 1H-perimidine-2-carboxylate. Tetrahedron 2002, 58, 6901–6906. [Google Scholar] [CrossRef]
- Mukano, Y.; Momochi, M.; Takanashi, Y.; Suzuki, M.; Wakabayashi, H.; Tetamae, H.; Kobayashi, K. Imino-enamine tautomerism and dynamic prototropy in 1-imino-3-amino-1H-indens. Tetrahedron 2010, 66, 605–611. [Google Scholar]
- The melting point of 6 is reported to be 244-246 °C in literature 22.
- CrystalStructure 3.8: Crystal Structure Analysis Package. Rigaku and Rigaku/MSC 2000-2006. 9009 New Trails Dr., The Woodlands, TX, USA.
- CRYSTALS Issue 11: Carruthers, J.R.; Rollett, J.S.; Betteridge, P.W., Kinna, D.; Pearce, L.; Larsen, A.; Gabe, E. Chemcal Crystallography Laboratory, Oxford, UK, 1999
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Akita, M.; Seto, H.; Aoyama, R.; Kimura, J.; Kobayashi, K. Novel Rearrangements in the Reactions Directed Toward Preparation of Spiro-N,N-ketals: Reactions of Naphthalene-1,8-diamine with Ninhydrin and Isatin. Molecules 2012, 17, 13879-13890. https://doi.org/10.3390/molecules171213879
Akita M, Seto H, Aoyama R, Kimura J, Kobayashi K. Novel Rearrangements in the Reactions Directed Toward Preparation of Spiro-N,N-ketals: Reactions of Naphthalene-1,8-diamine with Ninhydrin and Isatin. Molecules. 2012; 17(12):13879-13890. https://doi.org/10.3390/molecules171213879
Chicago/Turabian StyleAkita, Motoko, Hideyuki Seto, Reiko Aoyama, Junko Kimura, and Keiji Kobayashi. 2012. "Novel Rearrangements in the Reactions Directed Toward Preparation of Spiro-N,N-ketals: Reactions of Naphthalene-1,8-diamine with Ninhydrin and Isatin" Molecules 17, no. 12: 13879-13890. https://doi.org/10.3390/molecules171213879