3.1. General
IR spectra were recorded on a Nicolet Magna-FTIR-750 spectrophotometer. 1H- and 13C-NMR spectra were recorded in CDCl3 on Varian Mercury-300 or Varian Mercury-400 instruments. The ESI-MS were carried out on Thermo Finnigan LCQDECAXP and the low-resolution EI-MS was measured on a MAT-95 spectrometer and HREI-MS on a MAT-77 spectrometer. Purity was recorded on Gilson high-performance liquid chromatography (HPLC) (306 pump, UV/Vis-156 Detector, 215 liquid handle). TLC was carried out with glass pre-coated silica gel GF254 plates. Spots were visualized under UV light. All the solvents and reagents were used directly as obtained commercially unless otherwise noted.
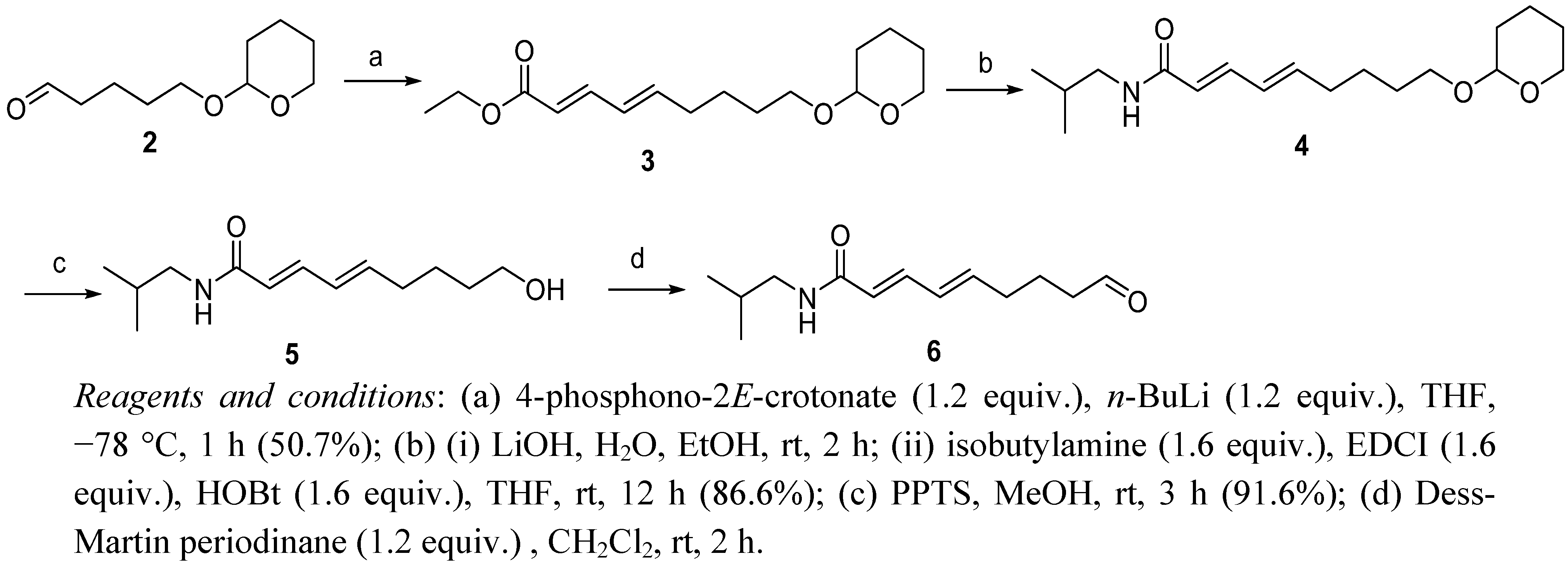
Ethyl 9-(tetrahydropyran-2-yloxy)-2E
,4E
-nonadienoate (
3). To a solution of triethyl 4-phosphono-2
E-crotonate (4.3 mL, 19.4 mmol) in anhydrous THF (50 mL) at −78 °C under Ar was added
n-BuLi (12.1 mL of a 1.6 M solution in hexane, 19.4 mmol). The reaction mixture was stirred at this temperature for 30 min, and then
2 [
8,
9] (3.0 g, 16.1 mmol) in anhydrous THF (20 mL) was added at −78 °C. The reaction mixture was stirred for an additional 1 h at this temperature, then stirred for 1 h at room temperature. The reaction was quenched with saturated NH
4Cl (30 mL). Then the mixture was extracted with CH
2Cl
2 (3 × 30 mL). The combined organic phases were washed with brine (2 × 50 mL), dried (Na
2SO
4), and evaporated. The residue was flash chromatographed (10:1 petroleum ether-EtOAc) to afford
3 as a colorless liquid (2.3 g, 50.7%):
1H-NMR (CDCl
3)
δ 1.27 (t, 3H,
J = 7.1 Hz), 1.48–1.81 (m, 10H), 2.18 (m, 2H), 3.34–3.49 (m, 2H), 3.70–3.84 (m, 2H), 4.17 (m, 2H,
J = 7.1 Hz), 4.55 (m, 1H), 5.75 (d, 1H,
J = 15.3 Hz), 6.15 (m, 2H), 7.24 (dd, 1H,
J = 15.4 Hz,
J = 14.0 Hz);
13C-NMR (CDCl
3)
δ 14.2, 19.6, 25.4, 29.2, 30.7, 32.7, 60.1, 62.3, 67.2, 98.8, 119.2, 128.5, 144.2, 144.9, 167.2; IR (KBr)
vmax 3117, 2941, 1714, 1643, 1618, 1404, 1259, 1138, 1034, 870 cm
−1.
N-Isobutyl-9-(tetrahydropyran-2-yloxy)-2E,4E-nonadienamide (4). To an ice-cooled solution of 3 (1.0 g, 3.55 mmol) in EtOH (20 mL) was added 1 N LiOH (17.8 mL, 17.8 mmol) and the solution was stirred at room temperature for 12 h. The reaction mixture was diluted with H2O (20 mL) and washed with EtOAc (2 × 20 mL). The aqueous layer was acidified (pH 2.0) with 2 N HCl and extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with brine (2 × 50 mL), dried (Na2SO4), and evaporated to give 9-(tetrahydropyran-2-yloxy)-2E,4E-nonadienoic acid as a yellow solid. The acid was used directly in the next step. The acid, EDCI (1.1 g, 5.68 mmol), HOBt (0.77 g, 5.68 mmol) and isobutylamine (0.56 mL, 5.68 mmol) were dissolved in anhydrous THF (30 mL), and the reaction mixture was stirred at room temperature for 12 h. The reaction mixture was diluted with H2O (30 mL) and extracted with CH2Cl2 (2 × 30 mL). The combined organic phases were washed with brine (2 × 50 mL), dried (Na2SO4), and evaporated. The residue was flash chromatographed (5:1 petroleum ether-EtOAc) to afford 4 as a colorless oil (0.95 g, two step total yield 86.6%); 1H-NMR (CDCl3) δ 0.92 (d, 6H, J = 6.7 Hz), 1.51–1.84 (m, 11H), 2.18 (m, 2H), 3.16 (t, 2H, J = 6.5 Hz), 3.47 (m, 2H), 3.75 (m, 2H), 4.56 (m, 1H), 5.53 (brs, 1H), 5.74 (d, 1H, J = 14.9 Hz), 6.09 (m, 2H), 7.19 (dd, 1H, J = 14.8 Hz, J = 14.8 Hz); 13C-NMR (CDCl3) δ 19.6, 20.1, 25.4, 25.4, 28.5, 29.2, 30.7, 32.7, 46.8, 62.3, 67.2, 98.8, 121.9, 128.4, 141.0, 142.5, 166.3; IR (KBr) vmax 3282, 2953, 2870, 1659, 1630, 1551, 1352, 1261, 1121, 1034, 999 cm−1; ESIMS m/z 332 [M + Na]+, 310 [M + H]+; HREIMS: calcd for C18H31NO3Na [M + Na]+, 332.2202; found, 332.2201.
N-Isobutyl-9-hydroxy-2E,4E-nonadienamide (5). To a solution of 4 (0.60 g, 1.94 mmol) in MeOH (20 mL) was added PPTS (0.1 g), and the reaction mixture was stirred at room temperature for 3 h. The solvent was evaporated and then H2O (20 mL) and CH2Cl2 (20 mL) were added. The phases were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 30 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), and evaporated to give 5 as a colorless oil (0.40 g, 91.6%); 1H-NMR (CDCl3) δ 0.92 (d, 6H, J = 6.6 Hz), 1.47–1.63 (m, 4H), 1.77 (m, 1H), 2.18 (m, 2H), 3.16 (m, 2H), 3.65 (t, 2H, J = 6.3 Hz), 5.54 (brs, 1H), 5.74 (d, 1H, J = 14.9 Hz), 6.09 (m, 2H), 7.22 (dd, 1H, J = 14.8 Hz, J = 14.9 Hz); 13C-NMR (CDCl3) δ 20.1, 24.9, 28.5, 32.1, 32.6, 46.9, 62.5, 122.0, 128.5, 141.0, 142.4, 166.5; IR (KBr) vmax 3290, 2931, 2870, 1659, 1630, 1551, 1404, 1265, 1161, 1061, 999 cm−1; ESIMS m/z 451 [2M + H]+, 226 [M + H]+; HREIMS: calcd for C13H23NO2Na [M + Na]+, 248.1626; found, 248.1624.
N-Isobutyl-9-oxo-2E,4E-nonadienamide (6). Compound 5 (0.40 g, 1.78 mmol) in CH2Cl2 (10 mL) was added to the solution of Dess-Martin periodinane (0.90 g, 2.13 mmol) in CH2Cl2 (20 mL) at 0 °C, and the mixture was stirred at room temperature for 2 h. Saturated Na2S2O3 aqueous solution (20 mL) and CH2Cl2 (20 mL) were added. The phases were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 30 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), and evaporated to give 6 as a yellow solid. The crude aldehyde was used directly in the next step.
5-(2-(3,4-Methylenedioxyphenyl)ethylsulfonyl)-1-phenyltetrazole (8a). To an ice-cooled solution of 7a (2.2 g, 13.3 mmol), 1-phenyl-5-thioltetrazole (2.6 g, 14.6 mmol) and triphenylphosphine (3.8 g, 15 mmol) in anhydrous THF (150 mL) under Ar was added DIAD (2.9 mL, 14.6 mmol) in anhydrous THF (30 mL) over 20 min. The reaction mixture was stirred at room temperature for 1 h and the solvent was evaporated. H2O (50 mL) and Et2O (50 mL) were added to the residue. The phases were separated and the aqueous layer was extracted with Et2O (2 × 30 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), and evaporated. The residue was flash chromatographed (5:1 petroleum ether-EtOAc) to afford crude 5-(2-(3,4-methylenedioxyphenyl)-ethylthio)-1-phenyltetrazole as a white solid.
To an ice-cooled solution of the thioether (4.1 g, 12.6 mmol) in EtOH (15 mL) under Ar was added hexaammonium heptamolybdate tetrahydrate (4.7 g, 3.8 mmol) in H2O2 (5 mL) over 10 min. The reaction mixture was stirred at room temperature for 48 h. H2O (50 mL) and Et2O (50 mL) were added to the mixture. The phases were separated and the aqueous layer was extracted with Et2O (2 × 30 mL). The combined organic phases were washed with brine (50 mL), dried (Na2SO4), and evaporated. The residue was flash chromatographed (5:1 petroleum ether-EtOAc) to afford 8a as a white solid (4.1 g, 94.6%); m.p. 95–97 °C; 1H-NMR (CDCl3) δ 3.19 (t, 2H, J = 8.1 Hz), 3.96 (t, 2H, J = 8.1 Hz), 5.96 (s, 2H), 6.72–6.75 (m, 3H), 7.61–7.71 (m, 5H); 13C-NMR (CDCl3) δ 28.3, 57.4, 101.1, 108.6, 108.8, 121.6, 125.0, 129.7, 129.8, 131.5, 132.9, 146.8, 148.0, 153.3; IR (KBr) vmax 2926, 1593, 1504, 1450, 1354, 1248, 1148, 1038, 918, 766, 642, 523 cm−1; EIMS m/z 358 [M]+, 148 (100%); HREIMS: calcd for C16H14N4O4S, 358.0736; found, 358.0742.
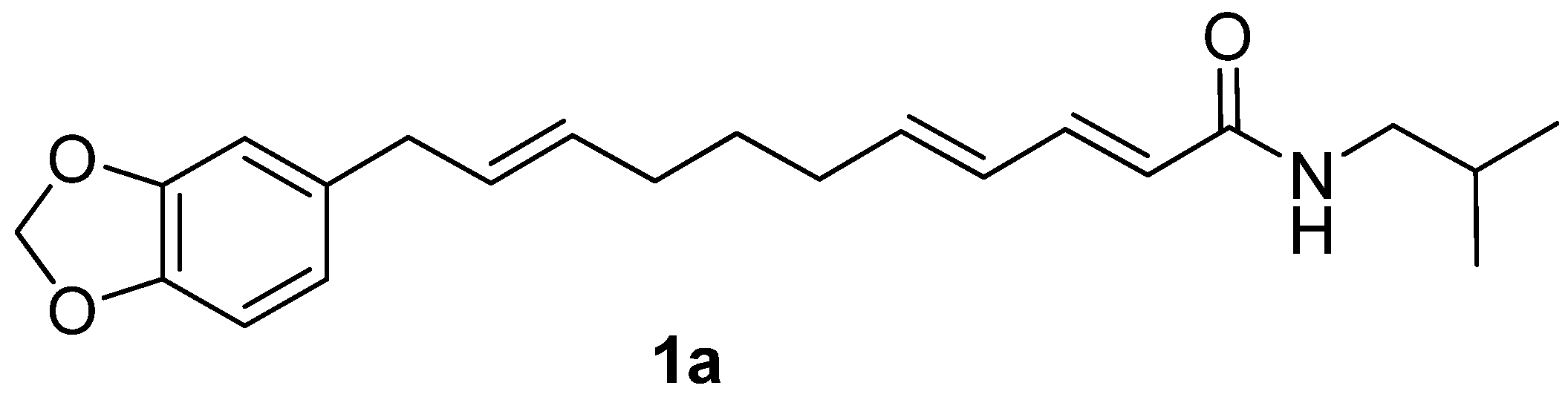
Laetispicine (
1a). To a solution of
8a (0.87 g, 2.44 mmol) in anhydrous DME (30 mL) at −60 °C under Ar was added KHMDS (2.7 mL of a 1 M solution in hexane, 2.70 mmol). The reaction mixture was stirred at this temperature for 1 h, and then
6 (0.50 g, 2.22 mmol) in anhydrous DME (10 mL) was added at −60 °C. The reaction mixture was stirred for an additional 2 h at this temperature, then stirred for 30 min at room temperature. The reaction was quenched with saturated NH
4Cl (30 mL). Then the mixture was extracted with Et
2O (3 × 30 mL). The combined organic phases were washed with brine (2 × 50 mL), dried (Na
2SO
4), and evaporated. The residue was flash chromatographed (3:1 petroleum ether-EtOAc) to afford
1a as a white solid (0.46 g, two step total yield 58.2%); m.p. 95–96 °C (lit. [
4] 93–94 °C);
1H-NMR (400 MHz, CDCl
3)
δ 0.92 (d, 6H,
J = 6.6 Hz), 1.50 (m, 2H), 1.81 (m, 1H), 2.03 (m, 2H), 2.15 (m, 2H), 3.15 (t, 2H,
J = 6.3 Hz), 3.23 (d, 2H,
J = 5.7 Hz), 5.52 (m, 2H,
J = 15.0 Hz,
J = 15.4 Hz), 5.74 (d, 1H,
J = 14.7 Hz), 5.91 (s, 2H), 6.06 (m, 2H,
J = 15.0 Hz,
J = 15.0 Hz), 6.60–6.74 (m, 3H), 7.17 (dd, 1H,
J = 15.0 Hz,
J = 9.9 Hz);
13C-NMR (CDCl
3)
δ 20.1, 28.4, 28.6, 31.8, 32.3, 38.7, 46.9, 100.7, 108.1, 108.9, 121.1, 121.9, 128.5, 129.6, 131.1, 134.7, 141.1, 142.5, 145.6, 147.5, 166.3; IR (KBr)
vmax 3425, 3302, 2955, 2922, 1655, 1628, 1614, 1551, 1506, 1487, 1250, 1001, 968 cm
−1; EIMS
m/z 355 [M]
+, 220 (100%); HREIMS: calcd for C
22H
29NO
3, 355.2147; found, 355.2155.
5-(Phenethylsulfonyl)-1-phenyltetrazole (8b). Mitsunobu reaction of 7b (5.0 g, 0.041 mol) and then oxidation under similar conditions to those applied for 8a to afford 8b as a white solid (8.8 g, 68.3%): m.p. 90–92 °C; 1H-NMR (300 MHz, CDCl3): δ 3.27 (m, 2H), 4.01 (m, 2H), 7.25–7.34 (m, 5H), 7.61–7.72 (m, 5H); ESIMS m/z 315 [M + H]+.
5-(4-Methoxyphenethylsulfonyl)-1-phenyltetrazole (8c). Mitsunobu reaction of 7c (3.9 g, 0.026 mol) and then oxidation under similar conditions to those applied for 8a to afford 8c as a white solid (5.9 g, 67.3%); m.p. 99–101 °C; 1H-NMR (300 MHz, CDCl3) δ 3.21 (m, 2H), 3.80 (s, 3H), 3.97 (m, 2H), 6.86 (d, 2H, J = 8.4 Hz), 7.17 (d, 2H, J = 8.4 Hz), 7.61–7.69 (m, 5H); EIMS m/z 344 [M]+, 134 (100%).
5-(3,4-Dimethoxyphenethylsulfonyl)-1-phenyltetrazole (8d). Mitsunobu reaction of 7d (4.5 g, 0.025 mol) and then oxidation under similar conditions to those applied for 8a to afford 8d as a white solid (6.9 g, 75.1%); m.p. 155–157 °C; 1H-NMR (300 MHz, CDCl3) δ 3.21 (m, 2H), 3.87 (s, 3H), 3.88 (s, 3H), 3.98 (m, 2H), 6.75–6.81 (m, 3H), 7.60–7.71 (m, 5H); ESIMS m/z 375 [M + H]+.
5-(3,4,5-Trimethoxyphenethylsulfonyl)-1-phenyltetrazole (8e). Mitsunobu reaction of 7e (4.1 g, 0.019 mol) and then oxidation under similar conditions to those applied for 8a to afford 8e as a white solid (5.7 g, 72.6%); m.p. 133–135 °C; 1H-NMR (300 MHz, CDCl3) δ 3.21 (m, 2H), 3.83 (s, 3H), 3.86 (s, 6H), 4.00 (m, 2H), 6.45 (s, 2H), 7.61–7.71 (m, 5H); EIMS m/z 404 [M]+, 194 (100%).
5-(2-(2,3-Dihydrobenzofuran-5-yl)ethylsulfonyl)-1-phenyltetrazole (8f). Mitsunobu reaction of 7f (4.6 g, 0.028 mol) and then oxidation under similar conditions to those applied for 8a to afford 8f as a white solid (7.1 g, 71.5%); m.p. 114–115 °C; 1H-NMR (300 MHz, CDCl3) δ 3.17–3.22 (m, 4H), 3.95 (m, 2H), 4.57 (t, 2H, J = 6.6 Hz), 6.72 (d, 1H, J = 6.0 Hz), 6.97 (m, 1H), 7.09 (s, 1H), 7.58–7.70 (m, 5H); EIMS m/z 356 [M]+, 146 (100%).
5-(2-(2,3-Dihydrobenzo [
1,
4]
dioxin-6-yl)ethylsulfonyl)-1-phenyltetrazole (
8g). Mitsunobu reaction of
7g (4.0 g, 0.022 mol) and then oxidation under similar conditions to those applied for
8a to afford
8g as a white solid (6.0 g, 72.3%); m.p. 113–115 °C;
1H-NMR (300 MHz, CDCl
3)
δ 3.15 (m, 2H), 3.95 (m, 2H), 4.24 (s, 4H), 6.69–6.83 (m, 3H), 7.60–7.71 (m, 5H); EIMS
m/z 372 [M]
+, 162 (100%).
5-(4-Chlorophenethylsulfonyl)-1-phenyltetrazole (8h). Mitsunobu reaction of 7h (4.5 g, 0.029 mol) and then oxidation under similar conditions to those applied for 8a to afford 8h as a white solid (7.5 g, 74.5%): m.p. 95–97 °C; 1H-NMR (300 MHz, CDCl3) δ 3.26 (m, 2H), 3.98 (m, 2H), 7.19–7.33 (m, 4H), 7.61–7.71 (m, 5H); EIMS m/z 349 [M]+, 138 (100%).
5-(4-Fluorophenethylsulfonyl)-1-phenyltetrazole (8i). Mitsunobu reaction of 7i (4.7 g, 0.034 mol) and then oxidation under similar conditions to those applied for 8a to afford 8i as a white solid (7.6 g, 67.3%); m.p. 99–100 °C; 1H-NMR (400 MHz, CDCl3) δ 3.25 (m, 2H), 3.99 (m, 2H), 7.02 (m, 2H), 7.24 (m, 2H), 7.59–7.70 (m, 5H); ESIMS m/z 333 [M + H]+.
N-Isobutyl-11-phenylundeca-2E,4E,9E-trienamide (1b). Julia reaction of 8b (0.15 g, 0.49 mmol) under similar conditions to those applied for latispicine (1a) afforded 1b as a white solid (72 mg, 51.2%); m.p. 80–82 °C; 1H-NMR (300 MHz, CDCl3) δ 0.92 (d, 6H, J = 6.6 Hz), 1.51 (m, 2H), 1.80 (m, 1H), 2.03 (m, 2H), 2.16 (m, 2H), 3.16 (t, 2H, J = 6.3 Hz), 3.33 (d, 2H, J = 6.0 Hz), 5.52 (m, 2H), 5.72 (d, 1H, J = 15.0 Hz), 6.08 (m, 2H), 7.14–7.31 (m, 6H); 13C-NMR (CDCl3) δ 20.1, 28.5, 28.6, 31.9, 32.4, 39.1, 47.0, 121.8, 125.9, 128.4, 128.5, 128.5, 129.5, 131.2, 140.9, 141.3, 142.7, 166.4; EIMS m/z 311 [M]+, 91 (100%); HREIMS: calcd for C21H29NO, 311.2249; found, 311.2258.
N-Isobutyl-11-(4-methoxyphenyl)undeca-2E,4E,9E-trienamide (1c). Julia reaction of 8c (0.20 g, 0.58 mmol) under similar conditions to those applied for latispicine (1a) afforded 1c as a white solid (90 mg, 50.1%); m.p. 65–67 °C; 1H-NMR (300 MHz, CDCl3) δ 0.92 (d, 6H, J = 6.6 Hz), 1.50 (m, 2H), 1.80 (m, 1H), 2.01 (m, 2H), 2.15 (m, 2H), 3.16 (t, 2H, J = 6.6 Hz), 3.26 (d, 2H, J = 6.3 Hz), 3.79 (s, 3H), 5.45–5.54 (m, 2H), 5.72 (d, 1H, J = 15.0 Hz), 6.04–6.10 (m, 2H), 6.83 (d, 2H, J = 8.7 Hz), 7.08 (d, 2H, J = 8.4 Hz), 7.18 (m, 1H); 13C-NMR (CDCl3) δ 20.2, 28.5, 28.6, 31.9, 32.3, 38.1, 46.9, 55.3, 113.8, 122.0, 128.5, 129.4, 129.9, 130.8, 133.0, 141.1, 142.6, 157.8, 166.4; EIMS m/z 341 [M]+, 121 (100%); HREIMS: calcd for C22H31NO2, 341.2355; found, 341.2360.
N-Isobutyl-11-(3,4-dimethoxyphenyl)undeca-2E,4E,9E-trienamide (1d). Julia reaction of 8d (0.19 g, 0.51 mmol) under similar conditions to those applied for latispicine (1a) afforded 1d as a white solid (85 mg, 49.4%); m.p. 61–63 °C; 1H-NMR (300 MHz, CDCl3) δ 0.92 (d, 6H, J = 6.9 Hz), 1.50 (m, 2H), 1.78 (m, 1H), 2.03 (m, 2H), 2.16 (m, 2H), 3.16 (t, 2H, J = 6.3 Hz), 3.26 (d, 2H, J = 6.3 Hz), 3.86 (s, 3H), 3.86 (s, 3H), 5.47–5.53 (m, 2H), 5.70 (d, 1H, J = 14.7 Hz), 6.06–6.09 (m, 2H), 6.70–6.81 (m, 3H), 7.18 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ 20.1, 28.5, 28.6, 31.9, 32.3, 38.6, 46.9, 55.8, 56.0, 111.2, 111.8, 120.2, 121.9, 128.6, 129.8, 131.0, 133.6, 141.1, 142.5, 147.2, 148.8, 166.3; EIMS m/z 371 [M]+, 151 (100%); HREIMS: calcd for C23H33NO3, 371.2460; found, 371.2463.
N-Isobutyl-11-(3,4,5-trimethoxyphenyl)undeca-2E,4E,9E-trienamide (1e). Julia reaction of 8e (0.20 g, 0.50 mmol) under similar conditions to those applied for latispicine (1a) afforded 1e as a white solid (92 mg, 50.5%); m.p. 72–73 °C; 1H-NMR (300 MHz, CDCl3) δ 0.92 (d, 6H, J = 6.6 Hz), 1.52 (m, 2H), 1.79 (m, 1H), 2.05 (m, 2H), 2.17 (m, 2H), 3.16 (t, 2H, J = 6.6 Hz), 3.26 (d, 2H, J = 5.4 Hz), 3.82 (s, 3H), 3.84 (s, 6H), 5.52 (m, 2H), 5.68 (d, 1H, J = 14.7 Hz), 6.06 (m, 2H), 6.40 (s, 2H), 7.17 (m, 1H); 13C-NMR (CDCl3) δ 20.1, 28.4, 28.6, 31.8, 32.3, 39.4, 46.9, 56.0, 60.9, 105.3, 122.0, 128.7, 129.5, 131.3, 135.9, 136.8, 141.1, 142.4, 153.1, 166.4; EIMS m/z 401 [M]+, 181 (100%); HREIMS: calcd for C24H35NO4, 401.2566; found, 401.2567.
N-Isobutyl-11-(2,3-dihydrobenzofuran-5-yl)undeca-2E,4E,9E-trienamide (1f). Julia reaction of 8f (0.18 g, 0.51 mmol) under similar conditions to those applied for latispicine (1a) afforded 1f as a white solid (89 mg, 54.4%); m.p. 70–72 °C; 1H-NMR (300 MHz, CDCl3) δ 0.92 (d, 6H, J = 6.3 Hz), 1.50 (m, 2H), 1.79 (m, 1H), 2.02 (m, 2H), 2.15 (m, 2H), 3.14–3.25 (m, 6H), 4.54 (t, 2H, J = 8.7 Hz), 5.47–5.53 (m, 2H), 5.72 (d, 1H, J = 14.7 Hz), 6.08 (m, 2H), 6.70 (d, 1H, J = 8.1 Hz), 6.90 (d, 1H, J = 8.4 Hz), 7.00 (s, 1H), 7.17 (m, 1H); 13C-NMR (CDCl3)δ 20.1, 28.5, 28.6, 29.8, 31.9, 32.4, 38.5, 46.9, 71.2, 109.0, 121.9, 125.0, 127.0, 127.8, 128.5, 130.2, 130.7, 132.9, 141.2, 142.7, 158.3, 166.3; EIMS m/z 353 [M]+, 133 (100%); HREIMS: calcd for C23H31NO2, 353.2355; found, 353.2362.
N
-Isobutyl-11-(2,3-dihydrobenzo [
1,
4]
dioxin-6-yl)undeca-2E
,4E
,9E
-trienamide (
1g). Julia reaction of
8g (0.15 g, 0.40 mmol) under similar conditions to those applied for latispicine (
1a) afforded
1g as a white solid (75 mg, 55.9%); m.p. 69–72 °C;
1H-NMR (300 MHz, CDCl
3)
δ 0.92 (d, 6H,
J = 6.6 Hz), 1.50 (m, 2H), 1.80 (m, 1H), 2.01 (m, 2H), 2.14 (m, 2H), 3.14–3.22 (m, 4H), 4.23 (s, 4H), 5.45–5.52 (m, 2H), 5.72 (d, 1H,
J = 14.7 Hz), 6.07 (m, 2H), 6.62–6.79 (m, 3H), 7.18 (m, 1H);
13C-NMR (CDCl
3)
δ 20.1, 28.5, 28.6, 31.9, 32.4, 38.3, 46.9, 64.3, 64.4, 117.0, 117.1, 121.3, 121.8, 128.5, 129.6, 131.0, 134.3, 141.2, 141.7, 142.7, 143.3, 166.4; EIMS
m/z 369 [M]
+, 149 (100%); HREIMS: calcd for C
23H
31NO
3, 369.2304; found, 369.2298.
N-Isobutyl-11-(4-chlorophenyl)undeca-2E,4E,9E-trienamide (1h). Julia reaction of 8h (0.17 g, 0.49 mmol) under similar conditions to those applied for latispicine (1a) afforded 1h as a white solid (82 mg, 52.8%); m.p. 70–73 °C; 1H-NMR (300 MHz, CDCl3) δ 0.93 (d, 6H, J = 6.6 Hz), 1.52 (m, 2H), 1.81 (m, 1H), 2.04 (m, 2H), 2.15 (m, 2H), 3.17 (t, 2H, J = 6.6 Hz), 3.30 (d, 2H, J = 5.7 Hz), 5.47–5.57 (m, 2H), 5.73 (d, 1H, J = 15.3 Hz), 6.08 (m, 2H), 7.10–7.28 (m, 5H); 13C-NMR (CDCl3) δ 20.1, 28.4, 28.6, 31.9, 32.3, 38.3, 46.9, 122.0, 128.4, 128.6, 129.0, 129.8, 131.6, 131.7, 139.4, 141.1, 142.5, 166.4; EIMS m/z 345 [M]+, 125 (100%); HREIMS: calcd for C21H28NOCl, 345.1859; found, 345.1856.
N-isobutyl-11-(4-fluorophenyl)undeca-2E,4E,9E-trienamide (1i). Julia reaction of 8i (0.19 g, 0.57 mmol) under similar conditions to those applied for latispicine (1a) afforded 1i as a white solid (98 mg, 57.5%); m.p. 75–77 °C; 1H-NMR (300 MHz, CDCl3) δ 0.92 (d, 6H, J = 6.8 Hz), 1.50 (m, 2H), 1.80 (m, 1H), 2.03 (m, 2H), 2.15 (m, 2H), 3.16 (t, 2H, J = 6.4 Hz), 3.29 (d, 2H, J = 6.4 Hz), 5.46–5.56 (m, 2H), 5.73 (d, 1H, J = 14.8 Hz), 6.08 (m, 2H), 6.95 (m, 2H), 7.10–7.26 (m, 3H); 13C-NMR (CDCl3) δ 20.1, 28.5, 28.6, 31.9, 32.4, 38.2, 46.9, 115.0, 115.2, 121.9, 128.5, 129.4, 129.8, 129.8, 131.4, 141.2, 142.6, 166.3; EIMS m/z 329 [M]+, 109 (100%); HREIMS: calcd for C21H28NOF, 329.2155; found, 329.2147.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









