Design, Synthesis and Preliminary Pharmacological Evaluation of New Non-Steroidal Anti-Inflammatory Agents Having a 4-(Methylsulfonyl) Aniline Pharmacophore

Abstract
:
1. Introduction

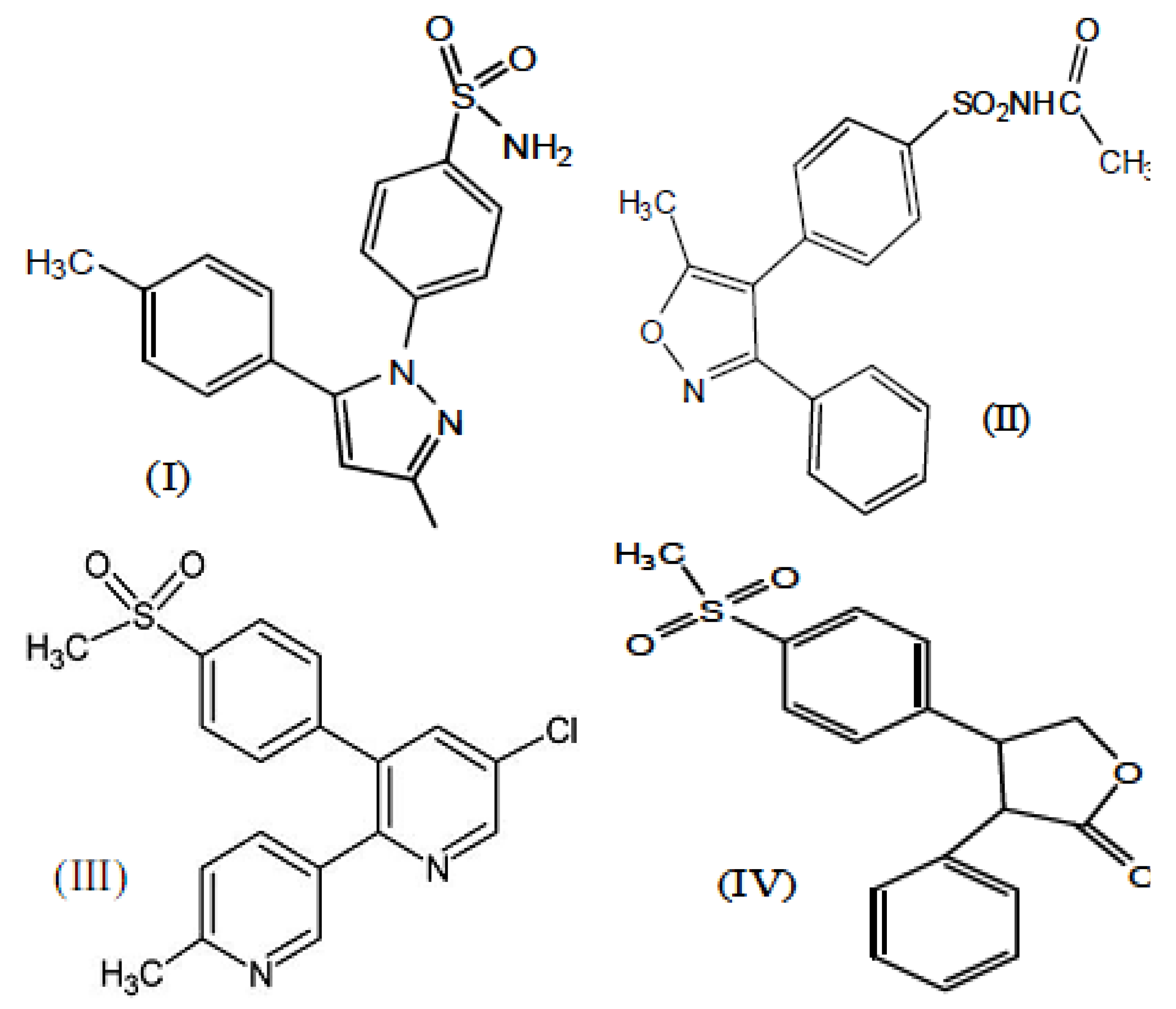
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
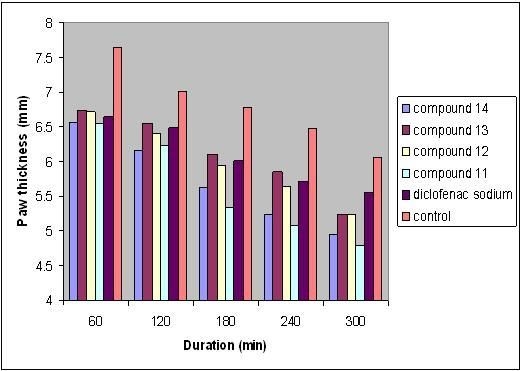
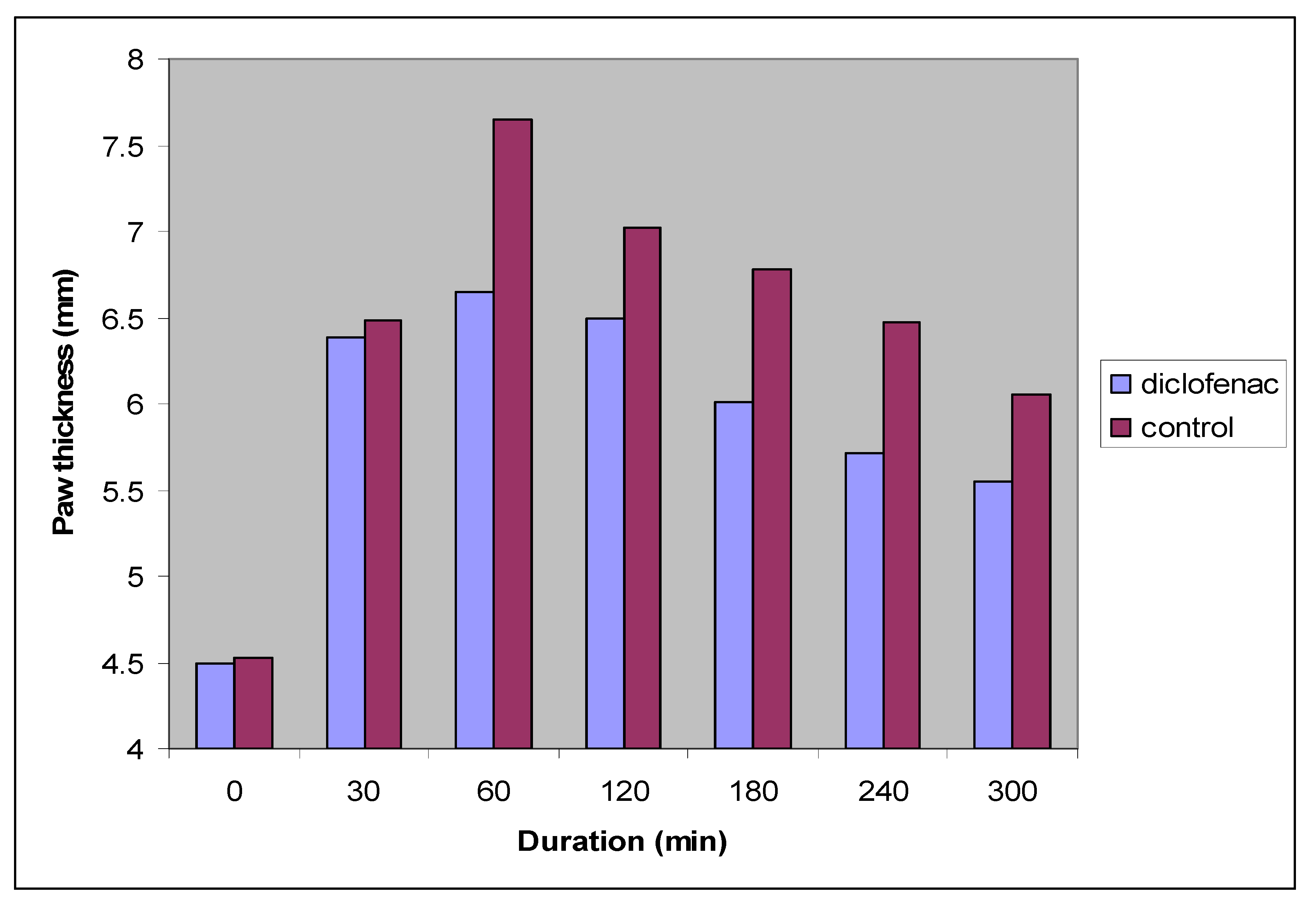
| Time (min) | Control (n = 6) | Diclofenac sodium (n = 6) | |
|---|---|---|---|
| Paw thickness (mm) | 0 | 4.53 ± 0.19 | 4.50 ± 0.08 |
| 30 | 6.48 ± 0.11 | 6.38 ± 0.14 | |
| 60 | 7.65 ± 0.16 | 6.65 ± 0.18 * | |
| 120 | 7.02 ± 0.18 | 6.49 ± 0.04 * | |
| 180 | 6.78 ± 0.04 | 6.01 ± 0.11 * | |
| 240 | 6.47 ± 0.11 | 5.71 ± 0.12 * | |
| 300 | 6.06 ± 0.03 | 5.55 ± 0.03 * |
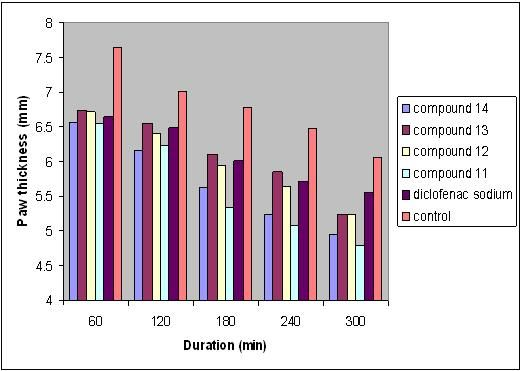
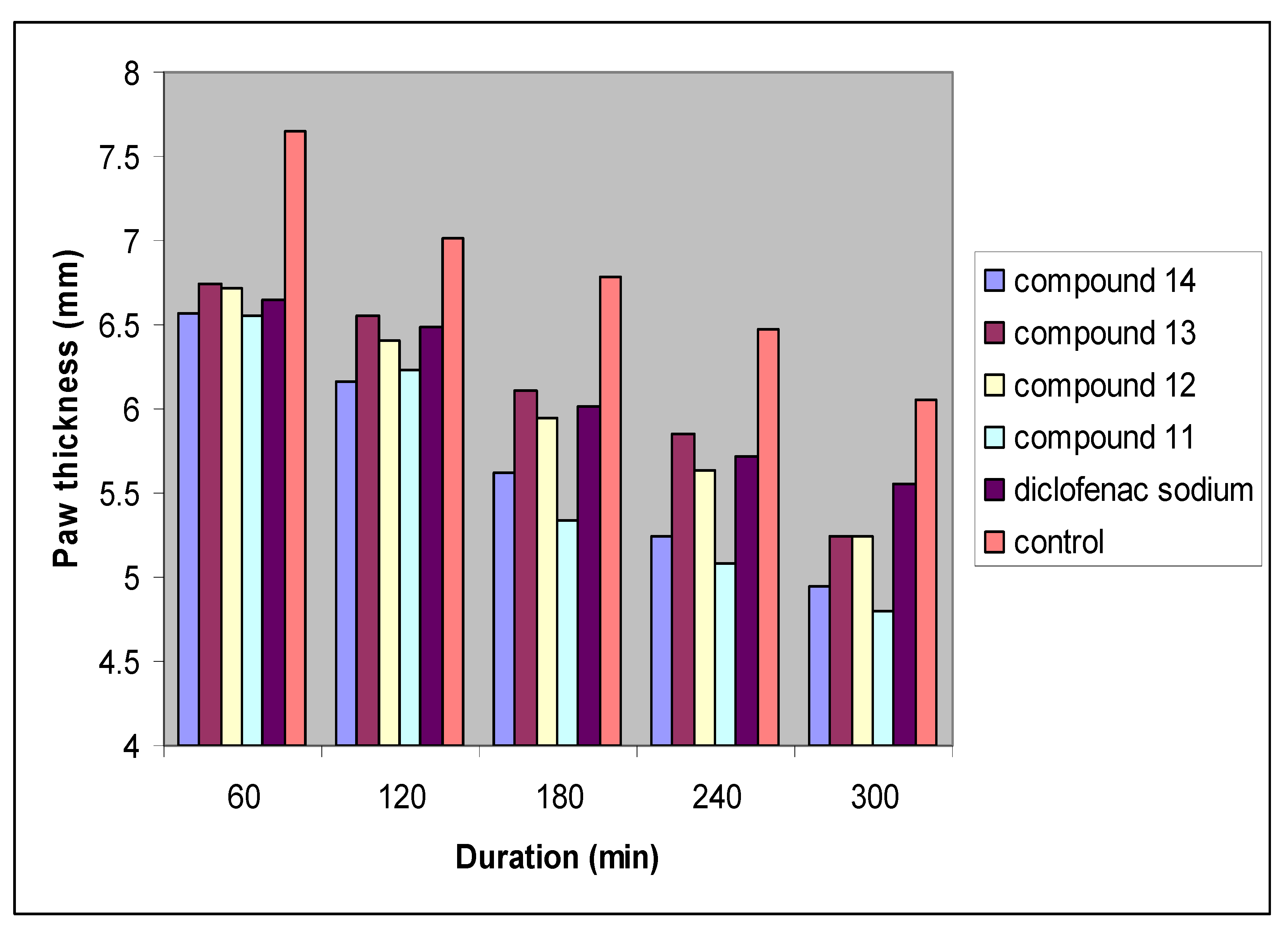
| Treatment groups | |||||||
|---|---|---|---|---|---|---|---|
| Time (min) | Control (n = 6) | Diclofenac sodium (n = 6) | Compound 11 (n = 6) | Compound 12 (n = 6) | Compound 13 (n = 6) | Compound 14 (n = 6) | |
| Paw thickness (mm) | 0 | 4.53 ± 0.19 | 4.50 ± 0.08 | 4.55 ± 0.11 | 4.49 ± 0.06 | 4.45 ± 0.08 | 4.43 ± 0.06 * |
| 30 | 6.48 ± 0.11 | 6.38 ± 0.14 | 6.33 ± 0.04 | 6.40 ± 0.11 | 6.39 ± 0.06 | 6.35 ± 0.14 | |
| 60 | 7.65 ± 0.16 | 6.65 ± 0.18 * | 6.55 ± 0.13 * | 6.72 ± 0.12 * | 6.75 ± 0.04 * | 6.57 ± 0.03 * | |
| 120 | 7.02 ± 0.18 | 6.49 ± 0.04 *a | 6.23 ± 0.06 *b | 6.41 ± 0.05 *a | 6.56 ± 0.06 *a | 6.16 ± 0.18 *b | |
| 180 | 6.78 ± 0.04 | 6.01 ± 0.11 *a | 5.34 ± 0.04 *b | 5.95 ± 0.03 *a | 6.11 ± 0.08 *a | 5.62 ± 0.04 *c | |
| 240 | 6.47 ± 0.11 | 5.71 ± 0.12 *a | 5.08 ± 0.10 *b | 5.64 ± 0.04 *a | 5.85 ± 0.16 *a | 5.24 ± 0.05 *b | |
| 300 | 6.06 ± 0.03 | 5.55 ± 0.03 *a | 4.80 ± 0.02 *b | 5.25 ± 0.06 *c | 5.52 ± 0.12 *a | 4.94 ± 0.12 *b | |

3. Experimental
3.1. General


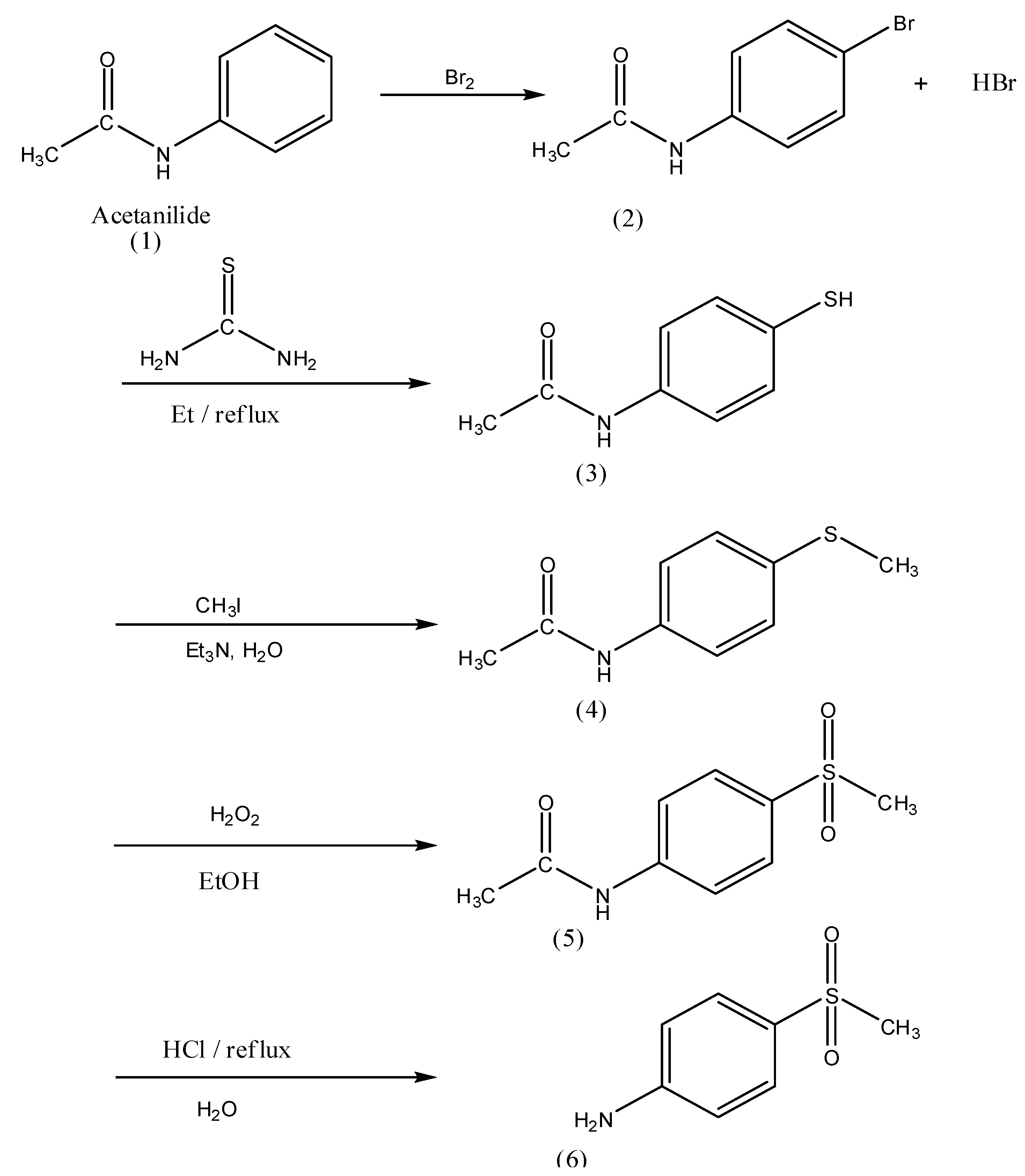
3.2. Synthesis of N-(4-Bromophenyl)acetamide (2)
3.3. Synthesis of N-(4-Mercaptophenyl)acetamide (3)
3.4. Synthesis of N-(4-(Methylthio)phenyl)acetamide (4)
3.5. Synthesis of N-(4-(Methylsulfonyl)phenyl)acetamide (5)
3.6. Synthesis of 4-(Methylsulfonyl)aniline (6)
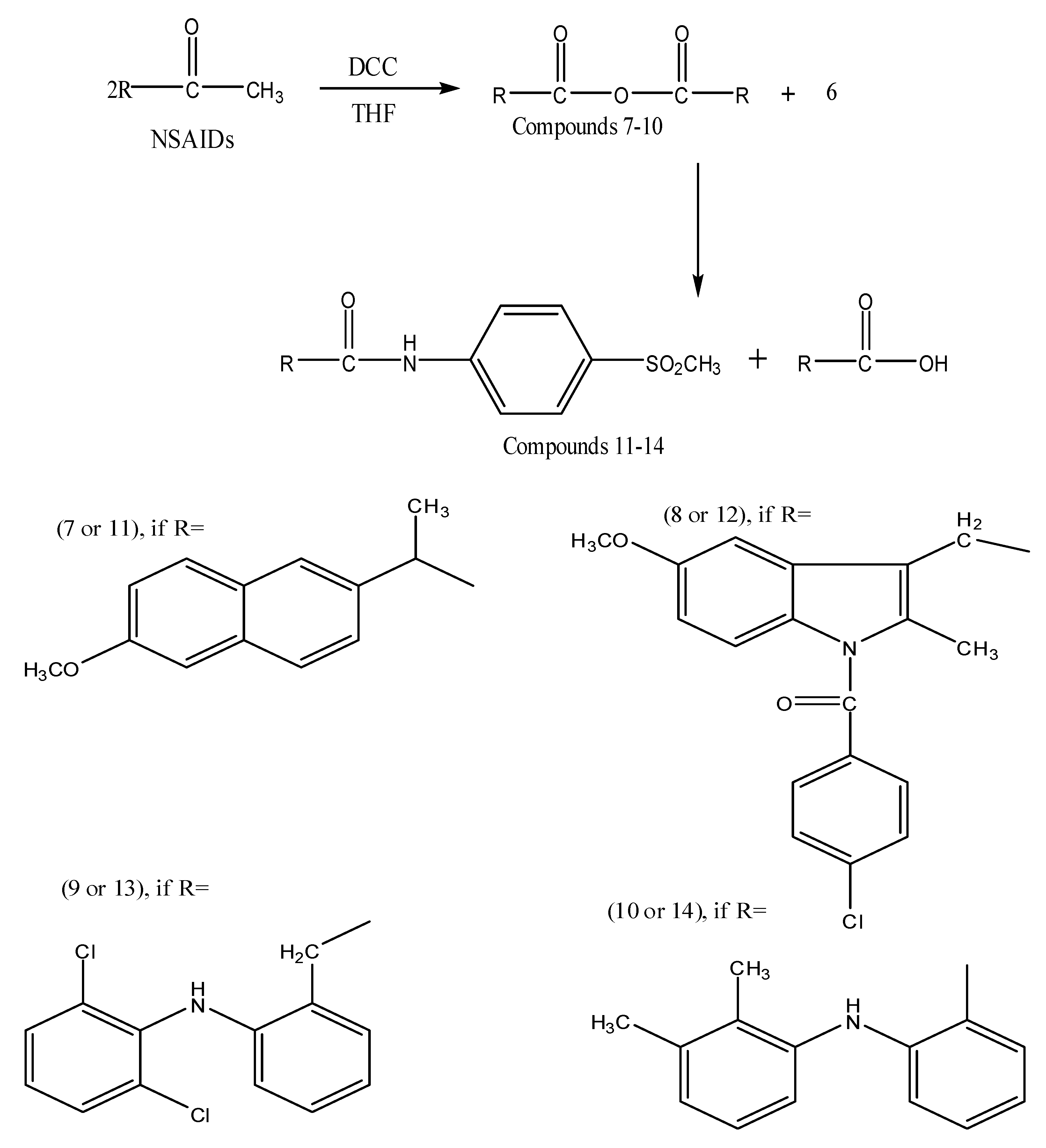
3.7. General Procedure for Synthesis of Acid Anhydride Derivatives of NSAIDs 7-10
3.8. General Procedure for Synthesis of the Final Compounds 11–14
3.9. Pharmacology
3.10. Anti-Inflammatory Activity
4. Conclusions
Acknowledgments
References and Notes
- Zhou, Y.; John, F.H.; Lenard, M.L. The Nonsteroidal Anti-Inflammatory Drug Indomethacin Induces Heterogeneity in Lipid Membranes: Potential Implication for Its Diverse Biological Action. PLoS One 2010, 5, 1–8. [Google Scholar]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The Role of Cyclooxygenase-2 in Cell Proliferation and Cell Death in Human Malignancies. Int. J. Cell Biol. 2010, 2010. [Google Scholar]
- Hiroki, F.; Masafumi, K.; Hiromi, F.; Tsukasa, M.; Yuichiro, Y.; Zhonghua, Q.; Matthew, D.B. Effect of selective cyclooxygenase-2 (COX-2) inhibitor treatment on glucose-stimulated insulin secretion in C57BL/6 mice. Biochem. Biophys. Res. Commun. 2007, 363, 37–43. [Google Scholar]
- Laura, G.; Ennio, O.; Gary, W. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: old and new mechanisms of action. J. Neurochem. 2004, 91, 521–536. [Google Scholar] [CrossRef]
- Carol, A.R.; Lawrence, J.M. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 29–33. [Google Scholar]
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Isakson, P. Distribution of COX-1 and COX-2 in normal and inflamed tissues. Adv. Exp. Med. Biol. 1997, 400A, 167–170. [Google Scholar]
- Paloucek, F.P.; Rynn, K.O. Nonsteroidal Anti-Inflammatory Drugs. In Clinical Toxicology; Ford, M.D., Ed.; WB Saunders: Philadelphia, PA, USA, 2001; pp. 281–284. [Google Scholar]
- Katzung, B.G. Basic and Clinical Pharmacology, 9th ed; McGraw-Hill: New York, NY, USA, 2004; p. 298. [Google Scholar]
- Claus, S.; William, E.B.; Alan, R.B. Human cyclo-oxygenase-1 and an alternative splice variant: Contrasts in expression of mRNA, protein and catalytic activities. Biochem. J. 2005, 385, 57–64. [Google Scholar] [CrossRef]
- Nina, Z.; Katarina, O.; Damjan, G.; Maja, J. Cyclooxygenase in normal human tissue-is cox-1 really a constitutive isoform, and cox-2 an inducible isoform? J. Cell. Mol. Med. 2009, 13, 3753–3763. [Google Scholar] [CrossRef]
- Min, Y.; Crawford, M.A. Essential Fatty Acids. In The Eicosanoid; Curtis-Prior, P., Ed.; John Wiley and Sons Ltd.: West Sussex, UK, 2004; pp. 257–276. [Google Scholar]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; van de Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar]
- Domenico, P.; Jean-Michel, D. Selective Cyclooxygenase-2 Inhibitors Development in Cardiovascular Medicine. Circulation 2005, 112, 1073–1079. [Google Scholar] [CrossRef]
- DeWitt, D.L.; Meade, E.A.; Smith, W.L. PGH synthase isoenzyme selectivity: The potential for safer nonsteroidal antiinflammatory drugs. Am. J. Med. 1993, 95 Suppl. 1, S40–S44. [Google Scholar]
- Zarraga, I.G.E.; Schwarz, E.R. Coxibs and heart disease: What we have learned and what else we need to know. J. Am. Coll. Cardiol. 2007, 49, 1–14. [Google Scholar]
- Chen, Y.F.; Jobanputra, P.; Barton, P.; Bryan, S.; Fry-Smith, A.; Harris, G.; Taylor, R.S. Cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs (etodolac, meloxicam, celecoxib, rofecoxib, etoricoxib, valdecoxib andlumiracoxib) for osteoarthritis and rheumatoid arthritis: A systematic review and economic evaluation. Health Technol. Assess. 2008, 12, 1–278. [Google Scholar]
- Regina, B.; Samir, S.A. COX-3 and the mechanism of action of paracetamol/acetaminophen. Prostaglandins Leukotrienes Essent. Fatty Acids 2005, 72, 85–87. [Google Scholar] [CrossRef]
- Bruce, N.C. Cyclooxygenase-2-selective inhibitors: Translating pharmacology into clinical utility. Cleve. Clin. J. Med. 2002, 69, 13–19. [Google Scholar]
- Schnitzer, T.J.; Burmester, G.R.; Mysler, E.; Hochberg, M.C.; Doherty, M.; Ehrsam, E.; Gitton, X.; Krammer, G.; Mellein, B.; Matchaba, P.; Gimona, A.; Haekey, C.J. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: Randomized controlled trial. Lancet 2004, 364, 665–674. [Google Scholar]
- Bengt, S.; Ralf, M.; Per-Johah, J. Membrane prostaglandin E synthase-1: A novel therapeutic target. Pharmacol. Rev. 2007, 59, 207–224. [Google Scholar] [CrossRef]
- Mattia, C.; Coluzzy, F. COX-2 inhibitors: Pharmacological data and adverse effects. Minerva Anestesiol. 2005, 71, 461–470. [Google Scholar]
- Botting, R.M. Inhibitore of Cyclooxygenases: Mechanisms, Selectivity and Uses. J. physiol. Pharmacol. 2006, 57, 113–124. [Google Scholar]
- Francesca, C.; Leslie, J.C. Cyclooxygenase inhibition and thrombogenicity. Am. J. Med. 2001, 110, 28–32. [Google Scholar]
- Mukherjee, D.; Nissen, S.E.; Topol, E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA-J. Am. Med. Assoc. 2001, 286, 954–959. [Google Scholar]
- Giovanni, D.; Maria, B.D.; Chiara, C. Prevention of thrombosis and vascular inflammation: benefits and limitations of selective or combined COX-1, COX-2 and 5-LOX inhibitors. Trends Pharmacol. Sci. 2003, 24, 245–252. [Google Scholar] [CrossRef]
- Amresh, G.; Zeashan, H.; Rao, Ch.V.; Singh, P.N. Prostaglandin mediated anti-inflammatory and analgesic activity of Cissampelos pareira. Acta Pharm. Sci. 2007, 49, 153–160. [Google Scholar]
- Brain, S.F.; Antony, J.H.; Peter, W.G.; Austin, R.T. Vogel’s Textbook of Practical Organic Chemistry, 5th ed; Longman Scientific & Technical: London, UK, 1989; p. 918. [Google Scholar]
- Neslihan, D. Synthesis and Characterization of New Triheterocyclic Compounds Consisting of 1,2,4 Triazol-3-one,1,3,4-Thiadiazole and 1,3,4-Oxadiazole Rings. Turk. J. Chem. 2005, 29, 125–133. [Google Scholar]
- Azizi, N.; Khajeh, A.A.; Bolourtchian, M.; Saidi, M.R. A Green and Highly Efficient Alkylation of Thiols in Water. J. Iran. Chem. Soc. 2009, 6, 749–753. [Google Scholar] [CrossRef]
- Michael, B.S.; Jerry, M. Oxidation and Reduction. In March Advanced Organic Chemistry, 6th ed; John Wiley, Sons. Inc.: Hoboken, NJ, USA, 2007; pp. 425, 1411, 1780–1783. [Google Scholar]
- ChemSynthesis Chemical Database. Available online: http://www.chemsynthesis.com/ (accessed on 2 December 2011).
- Monther, F.M.; Abdul-Rassoul, W.; Samira, F. Synthesis and Preliminary Pharmacological Evaluation of amino benzene sulfonamide derivatives of diflunisal as anti-inflammation agents. Iraqi J. Pharm. 2008, 7-8, 42–49. [Google Scholar]
- Salem, B.S.; Mahdi, M.F.; Mohammed, M.H. Synthesis and Preliminary Pharmacological Study of Sulfonamide Conjugates with Ibuprofen and Indomethacin as New Anti-Inflammatory Agents. Iraqi J. Pharm. Sci. 2009, 18, 4. [Google Scholar]
- Chandrashekhar, S.P.; Naveen, K.J.; Amarjit, S.; Shinivas, K.K. Modulatory effect of COX inhibitors on sildenafil-induced antinociception. Pharmacology 2003, 69, 183–189. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compounds 2, 3, 4, 11, 12 and 14 are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mahdi, M.F.; Mohammed, M.H.; Jassim, A.A.K. Design, Synthesis and Preliminary Pharmacological Evaluation of New Non-Steroidal Anti-Inflammatory Agents Having a 4-(Methylsulfonyl) Aniline Pharmacophore. Molecules 2012, 17, 1751-1763. https://doi.org/10.3390/molecules17021751
Mahdi MF, Mohammed MH, Jassim AAK. Design, Synthesis and Preliminary Pharmacological Evaluation of New Non-Steroidal Anti-Inflammatory Agents Having a 4-(Methylsulfonyl) Aniline Pharmacophore. Molecules. 2012; 17(2):1751-1763. https://doi.org/10.3390/molecules17021751
Chicago/Turabian StyleMahdi, Monther Faisel, Mohamed Hassan Mohammed, and Akeel Abdul Kadhum Jassim. 2012. "Design, Synthesis and Preliminary Pharmacological Evaluation of New Non-Steroidal Anti-Inflammatory Agents Having a 4-(Methylsulfonyl) Aniline Pharmacophore" Molecules 17, no. 2: 1751-1763. https://doi.org/10.3390/molecules17021751