Cytotoxic Phenylpropanoids and a New Triterpene, Turformosinic Acid, from Turpinia formosana Nakai

Abstract
:1. Introduction

2. Results and Discussion
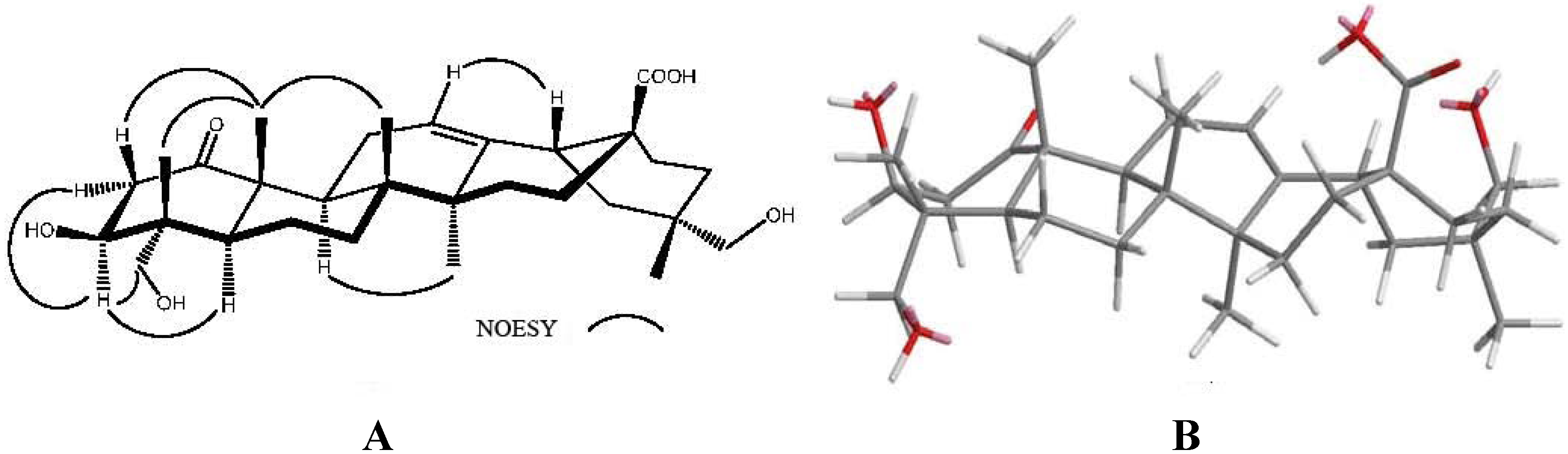
2.1. Structure Determination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
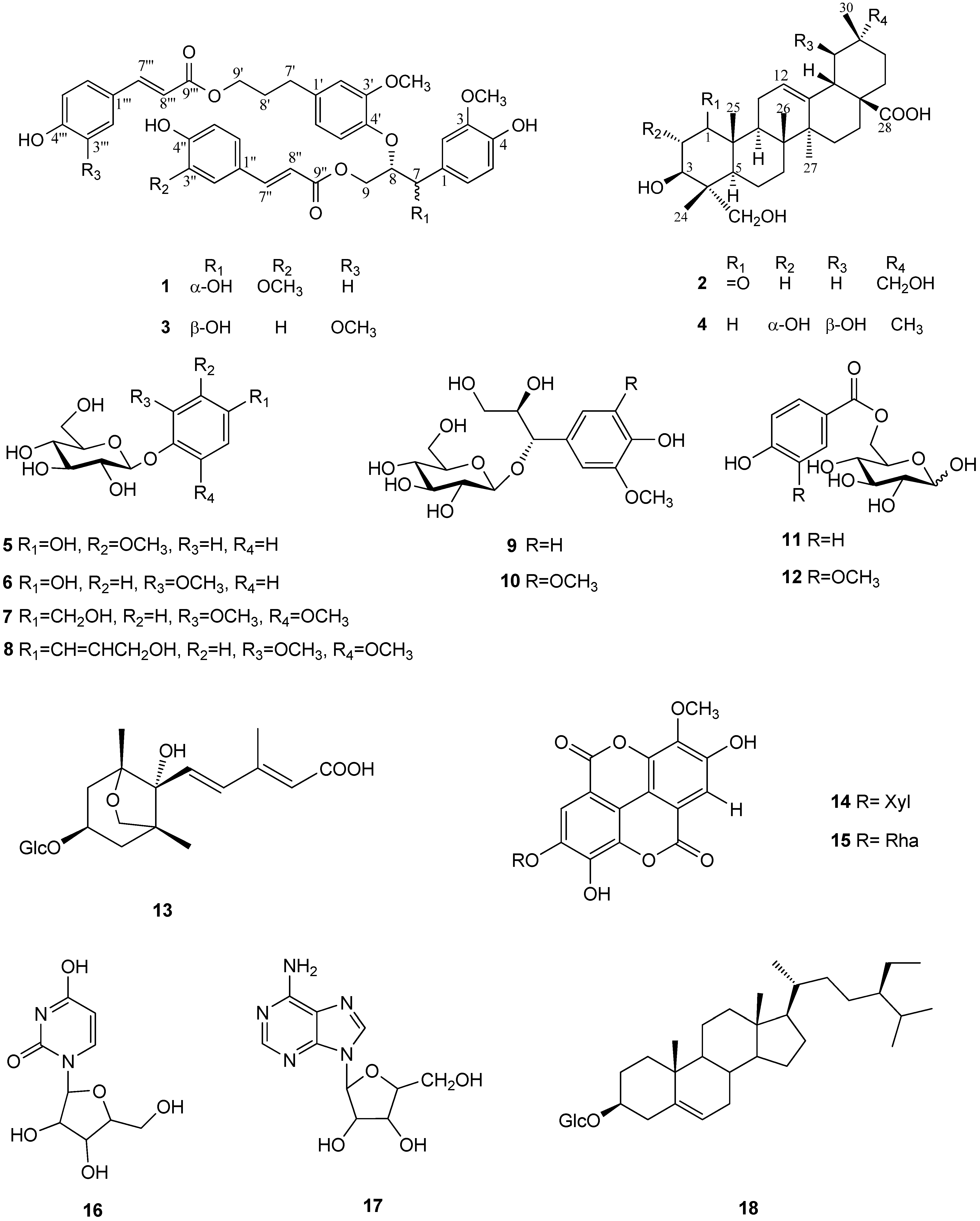
| 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Position | δH a | δC b | δC c | Position | δH a | δC b | δC c | |
| 1 | - | 133.8 | 131.0 | 1'' | - | 127.6 | 126.7 | |
| 2 | 7.07 (s) | 111.6 | 108.9 | 2'' | 7.13 (s) | 111.7 | 109.4 | |
| 3 | - | 148.7 | 146.8 | 3'' | - | 150.5 | 146.6 | |
| 4 | - | 146.9 | 145.1 | 4'' | - | 149.2 | 148.1 | |
| 5 | 6.76 (d, J = 8.0) | 115.8 | 114.2 | 5'' | 6.80 (d, J = 8.0) | 116.4 | 115.0 | |
| 6 | 6.88 (d, J = 8.0) | 120.8 | 119.3 | 6'' | 7.02 (d, J = 8.0) | 124.1 | 123.1 | |
| 7 | 4.92 (d, J = 5.5) | 74.1 | 72.3 | 7'' | 7.56 (d, J = 16.0) | 146.6 | 145.2 | |
| 8 | 4.60 (m) | 83.7 | 84.2 | 8'' | 6.32 (d, J = 16.0) | 115.5 | 115.1 | |
| 9 | 4.45 (m) | 64.8 | 62.8 | 9'' | - | 169.3 | 167.6 | |
| MeO-3 | 3.79 (s) | 56.4 | 55.8 | MeO-3'' | 3.84 (s) | 56.4 | 55.9 | |
| 1' | - | 137.5 | 137.2 | 1''' | - | 127.0 | 126.5 | |
| 2' | 6.73 (s) | 113.9 | 112.4 | 2''' | 7.32 (d, J = 8.0) | 131.2 | 130.0 | |
| 3' | - | 151.9 | 151.1 | 3''' | 6.78 (m) | 116.8 | 115.9 | |
| 4' | - | 147.2 | 145.0 | 4''' | - | 161.1 | 158.4 | |
| 5' | 6.64 (d, J = 7.5) | 121.7 | 120.4 | 5''' | 6.78 (m) | 116.8 | 115.9 | |
| 6' | 6.84 (d, J = 7.5) | 119.7 | 121.1 | 6''' | 7.32 (d, J = 8.0) | 131.2 | 130.0 | |
| 7' | 2.59 (dd, J = 7.5, 7.0) | 32.8 | 31.9 | 7''' | 7.36 (d, J = 16.0) | 146.7 | 145.2 | |
| 8' | 1.89 (t, J = 7.0) | 31.4 | 30.3 | 8''' | 6.14 (d, J = 16.0) | 114.9 | 114.5 | |
| 9' | 4.09 (t, J = 6.0) | 64.8 | 63.8 | 9''' | - | 168.9 | 164.4 | |
| OMe-3' | 3.72 (s) | 56.4 | 55.8 | |||||
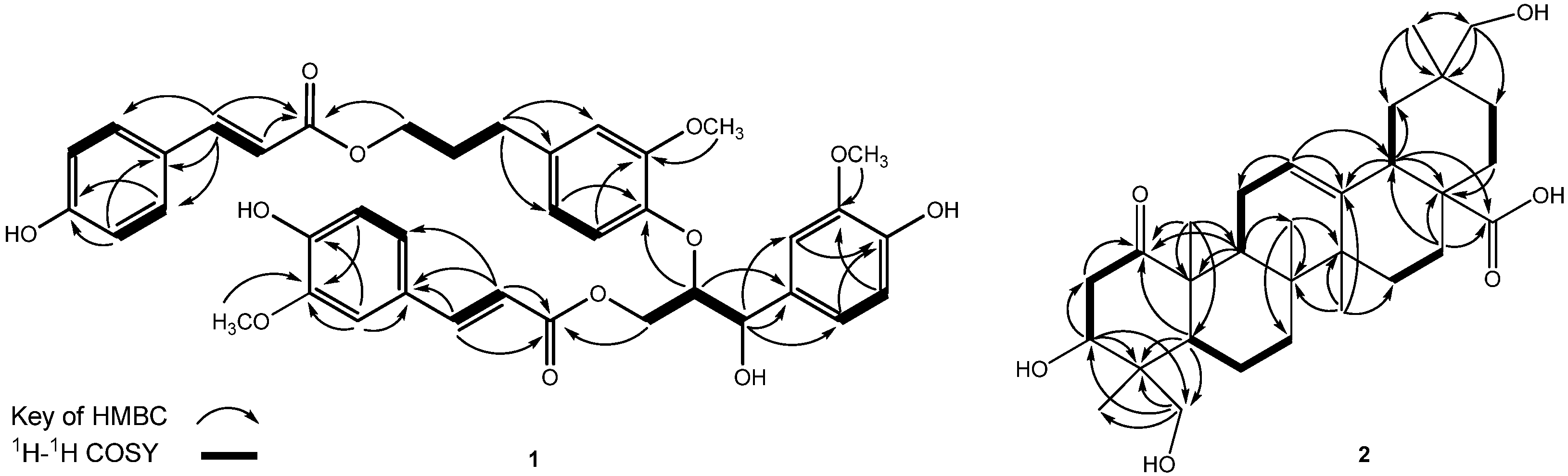
| 2 | ||||||
|---|---|---|---|---|---|---|
| Position | δH b | δC c | Position | δH b | δC c | |
| 1 | - | 215.0 | 16 | 1.99 (dd, J = 13.2, 3.2)1.60 (m) | 24.0 | |
| 2 | 2.29 (dd, J = 12.0, 5.2)3.10 (t, J = 12.0) | 44.6 | 17 | - | 48.0 | |
| 3 | 3.81 (dd, J = 12.0, 4.8) | 73.3 | 18 | 2.88 (dd, J = 13.6, 4.0) | 42.1 | |
| 4 | - | 44.0 | 19 | 1.80 (m), 1.09 (m) | 41.0 | |
| 5 | 1.35 (m) | 47.6 | 20 | - | 36.7 | |
| 6 | 1.56 (m) | 18.4 | 21 | 1.25 (d, J = 8.8)1.76 (m) | 33.0 | |
| 7 | 1.27 (dd, J = 10.0, 3.2)1.58 m | 33.2 | 22 | 1.14 (m)1.47 (m) | 29.2 | |
| 8 | - | 40.3 | 23 | 3.48 (d, J = 11.2)3.33 (d, J = 11.2) | 65.8 | |
| 9 | 2.25 (dd, J = 10.4, 5.6) | 40.2 | 24 | 0.86 (s) | 13.2 | |
| 10 | - | 53.2 | 25 | 1.33 (s) | 15.9 | |
| 11 | 2.27 (m), 1.83 (m) | 26.2 | 26 | 0.87 (s) | 18.3 | |
| 12 | 5.24 (t-like, J = 4.0) | 124.0 | 27 | 1.19 (s) | 26.3 | |
| 13 | - | 144.4 | 28 | - | 181.7 | |
| 14 | - | 43.1 | 29 | 3.19 (br s) | 74.3 | |
| 15 | 1.74 (m), 1.06 (m) | 28.7 | 30 | 0.92 (s) | 19.5 | |

2.2. Cytotoxicity and DHHP Radical Scavenging Activity
| Compound | Anti-oxidative a | Cytotoxicity b | |||||
| DPPH test | MCF-7 | Daoy | WiDr | Hep2 | |||
| ED50 (μg/mL) | ED50 (μg/mL) | ||||||
| 1 | 26.9 | - c | 11.46 | 12.07 | 13.22 | ||
| 3 | 28.4 | 13.69 | 6.07 | 4.45 | 3.60 | ||
| 5 | 23.6 | - | - | - | - | ||
| 6 | 30.8 | - | - | - | - | ||
| Positive control | 12.25 d | 0.15 e | 0.08 e | 0.15 e | 0.16 e | ||

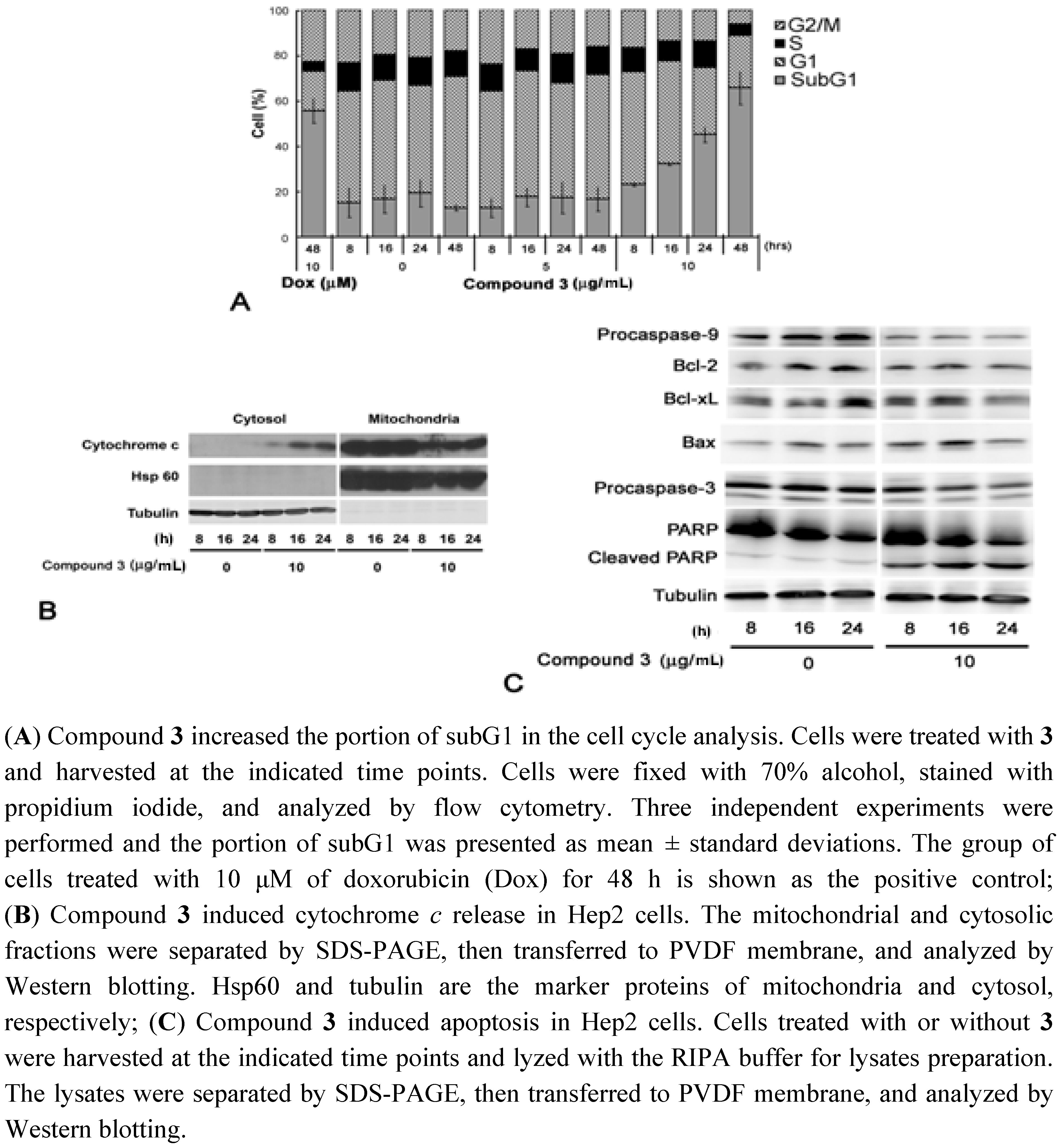
2.3. Cell Cycle Analysis and Immunobloting
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectral Data
3.5. Cytotoxicity Assay
3.6. DPPH Radical Scavenging Activity
3.7. Cell Cycle and DNA Content Analysis
3.8. Preparation of Mitochondrial and Cytosolic Fractions
3.9. Immunoblotting
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds 1 and 2 are available from the authors.
References and Notes
- The Catalogue of Medicinal Plant Resources in Taiwan; Hanasty Printing Co. Ltd.: Taipei, Taiwan, 2003; p. 298.
- Li, H.L. Flora of Taiwan, 2nd ed; Editorial Committee of the Flora of Taiwan: Taipei, Taiwan, 2003; Volume 3, p. 663. [Google Scholar]
- Horgen, F.D.; Edrada, R.A.; de los Reyes, G.; Agcaoili, F.; Madulid, D.A.; Wongpanich, V.; Angerhofer, C.K.; Pezzuto, J.M.; Soejarto, D.D.; Farnsworth, N.R. Biological screening of rain forest plot trees from Palawan Island (Philippines). Phytomedicine 2001, 8, 71–81. [Google Scholar] [CrossRef]
- Matthew, S.; Kao, K.C.; Chang, Y.S.; Abreu, P. Ellagic acid glycosides from Turpinia ternata. Nat. Prod. Res. 2007, 21, 83–88. [Google Scholar] [CrossRef]
- Jantova, S.; Nagy, M.; Ruzeková, L.; Grancai, D. Antibacterial activity of plant extracts from the families Fabaceae, Oleaceae, Philadelphaceae, Rosaceae and Staphyleace. Phytother. Res. 2000, 14, 601–603. [Google Scholar] [CrossRef]
- Jantova, S.; Nagy, M.; Ruzeková, L.; Grancai, D. Cytotoxic effects of plant extracts from the families Fabaceae, Oleaceae, Philadelphaceae, Rosaceae and Staphyleace. Phytother. Res. 2001, 15, 22–25. [Google Scholar] [CrossRef]
- Takeda, Y.; Okada, Y.; Masuda, T.; Hirata, E.; Shinzato, T.; Takushi, A.; Yu, Q.; Otsuka, H. New megastigmane and tetraketide from the leaves of Euscaphis japonica. Chem. Pharm. Bull. 2000, 48, 752–754. [Google Scholar]
- Yu, Q.; Otsuka, H.; Hirata, E.; Shinzato, T.; Takeda, Y. Turpinionosides A–E: Megastigmane glucosides from leaves of Turpinia ternate Nakai. Chem. Pharm. Bull. 2002, 50, 640–644. [Google Scholar] [CrossRef]
- Yu, Q.; Matsunami, K.; Otsuka, H.; Takeda, Y. Staphylionosides A–K: Megastigmane glucosides from the leaves of Staphylea bumalda DC. Chem. Pharm. Bull. 2005, 53, 800–807. [Google Scholar] [CrossRef]
- Nakanish, T.; Iida, N.; Inatomi, Y.; Murata, H.; Inada, A.; Murata, J.; Lang, F.A.; Iinuma, M.; Tanaka, T.; Sakagami, Y. A monoterpene glucoside and three megastigmane glycosides from Juniperus communis var. depressa. Chem. Pharm. Bull. 2005, 53, 783–787. [Google Scholar] [CrossRef]
- Takeda, Y.; Okada, Y.; Masuda, T.; Hirata, E.; Takushi, A.; Otsuka, H. Euscapholide and its glucoside from leaves of Euscaphis japonica. Phytochemistry 1998, 49, 2565–2658. [Google Scholar]
- Konishi, T.; Otani, T.; Kiyosawa, S.; Fujiwara, Y. Constituents of the capsules of Euscaphis japonica (THUNB.) KANTIZ. Chem. Pharm. Bull. 1996, 44, 863–864. [Google Scholar] [CrossRef]
- Rudiyansyah; Lambert, L.K.; Garson, M.J. Lignans and triterpenes from the bark of Durio carinatus and Durio oxleyanus. J. Nat. Prod. 2010, 73, 1649–1654. [Google Scholar] [CrossRef]
- Wu, P.L.; Chuang, T.H.; He, C.X.; Wu, T.S. Bioassay-guided isolation and cytotoxicity of phenylpropanoid esters from the stems of Hibiscus taiwanensis. Bioorg. Med. Chem. 2004, 12, 2193–2197. [Google Scholar]
- Shimomura, H.; Sashida, Y.; Oohara, M. Lignans from Machilus thunbergii. Phytochemistry 1987, 26, 1513–1515. [Google Scholar]
- Seca, A.M.; Silva, A.M.; Silvestre, A.J.; Cavaleiro, J.A.; Domingues, F.M.; Pascoal-Neto, C. Phenolic constituents from the core of Kenaf (Hibiscus cannabinus). Phytochemistry 2001, 56, 759–767. [Google Scholar]
- Jossang, A.; Seuleiman, M.; Maidou, E.; Bodo, B. Pentacyclic triterpenes from Combretum nigricans. Phytochemistry 1996, 41, 591–594. [Google Scholar]
- Okada, Y.; Omae, A.; Okuyama, T. A new triterpenoid isolated from Lagerstronemia speciosa (L.) Pers. Chem. Pharm. Bull. 2003, 51, 452–454. [Google Scholar] [CrossRef]
- Wangensteen, H.; Miron, A.; Alamgir, M.; Rajia, S.; Samuelsen, A.B.; Malterud, K.E. Antioxidant and 15-lipoxygenase inhibitory activity of rotenoids, isoflavones and phenolic glycosides from Sarcolobus globosus. Fitoterapia 2006, 77, 290–295. [Google Scholar] [CrossRef]
- Inoshiri, S.; Sasaki, M.; Kohda, H.; Otsuka, H.; Yamasaki, K. Aromatic glycosides from Berchemia racemosa. Phytochemistry 1987, 26, 2811–2814. [Google Scholar]
- Park, K.M.; Yang, M.C.; Lee, K.H.; Kim, K.R.; Choi, S.U.; Lee, K.R. Cytotoxic phenolic constituents of Acer tegmentosum Maxim. Arch. Pharm. Res. 2006, 29, 1086–1090. [Google Scholar] [CrossRef]
- Dübeler, A.; Voltmer, G.; Gora, V.; Lunderstädt, J.; Zeeck, A. Phenols from Fagus sylvatica and their role in defence against Cryptococcus fagisuga. Phytochemistry 1997, 45, 51–57. [Google Scholar]
- Sugiyama, M.; Kikuchi, M. Studies on the constituents of Osmanthus Species. X. Structures of phenolic glucosides from the leaves of Osmanthus asiaticus Nakai. Chem. Pharm. Bull. 1992, 40, 325–326. [Google Scholar] [CrossRef]
- Ouyang, M.A.; Kuo, Y.H. Water-soluble constituents from aerial roots of Ficus microcarpa. J. Asian Nat. Prod. 2006, 8, 625–630. [Google Scholar] [CrossRef]
- Kaneko, T.; Ohtani, K.; Kasai, R.; Yamasaki, K.; Duc, N.M. n-Alkyl glycosides and p-hydroxybenzoyloxy glucose from fruits of Crescentia cujete. Phytochemistry 1998, 47, 259–263. [Google Scholar]
- Ding, H.Y.; Wu, Y.C.; Lin, H.C.; Chan, Y.Y.; Wu, P.L.; Wu, T.S. Glycosides from Paeonia suffruticosa. Chem. Pharm. Bull. 1999, 47, 652–655. [Google Scholar] [CrossRef]
- Champavier, Y.; Comte, G.; Vercauteren, J.; Allais, D.P.; Chulia, A.J. Norterpenoid and sesquiterpenoid glucosides from Juniperus phynicea and Galega officinalis. Phytochemistry 1999, 50, 1219–1223. [Google Scholar]
- Khallouki, F.; Haubner, R.; Huall, W.E.; Erben, G.; Spiegelhalder, B.; Bartsch, H.; Owen, R.W. Isolation, purification and identification of ellagic acid derivatives, catechins, and procyanidins from the root bark of Anisophyllea dichostyla R. Br. Food. Chem. Toxic 2007, 45, 472–485. [Google Scholar] [CrossRef]
- Hillis, W.E.; Yazaki, Y. Properties of some methylellagic acids and their glycosides. Phytochemistry 1973, 12, 2963–2968. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chang, F.R.; Teng, C.M.; Wu, Y.C. Cheritamine, a new N-fatty acyl tryamine and other constituents from the stems of Annona cherimola. J. Chin. Chem. Soc. 1999, 46, 77–86. [Google Scholar]
- Pretsch, E.; Clerc, T.; Seki, J.; Simon, W. Tables of Spectral Data for Structure Determination of Organic Compounds; Springer-Verlag: Berlin, Germany, 1989. [Google Scholar]
- Lee, C.K.; Lee, P.H.; Kuo, Y.H. The chemical constituents from the aril of Cassia fistula L. J. Chin. Chem. Soc. 2001, 48, 1053–1058. [Google Scholar]
- Zhang, L.J.; Chiou, C.T.; Cheng, J.J.; Huang, H.C.; Kuo, L.M.; Liao, C.C.; Bastow, K.F.; Lee, K.H.; Kuo, Y.H. Cytotoxic polyisoprenyl benzophenonoids from Garcinia subelliptica. J. Nat. Prod. 2010, 73, 557–562. [Google Scholar] [CrossRef]
- Chang, C.L.; Zhang, L.J.; Chen, R.Y.; Kuo, L.M.Y.; Huang, J.P.; Huang, H.C.; Lee, K.H.; Wu, Y.C.; Kuo, Y.H. Antioxidant and anti-inflammatory phenylpropanoid derivatives from Calamus quiquesetinervius. J. Nat. Prod. 2010, 73, 1482–1488. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, H.-C.; Chiou, C.-T.; Hsiao, P.-C.; Liaw, C.-C.; Zhang, L.-J.; Chang, C.-L.; Chen, I.-S.; Chen, W.-C.; Lee, K.-H.; Kuo, Y.-H. Cytotoxic Phenylpropanoids and a New Triterpene, Turformosinic Acid, from Turpinia formosana Nakai. Molecules 2012, 17, 1837-1851. https://doi.org/10.3390/molecules17021837
Huang H-C, Chiou C-T, Hsiao P-C, Liaw C-C, Zhang L-J, Chang C-L, Chen I-S, Chen W-C, Lee K-H, Kuo Y-H. Cytotoxic Phenylpropanoids and a New Triterpene, Turformosinic Acid, from Turpinia formosana Nakai. Molecules. 2012; 17(2):1837-1851. https://doi.org/10.3390/molecules17021837
Chicago/Turabian StyleHuang, Hui-Chi, Chun-Tang Chiou, Ping-Chun Hsiao, Chia-Ching Liaw, Li-Jie Zhang, Chao-Lin Chang, Ih-Sheng Chen, Wen-Chi Chen, Kuo-Hsiung Lee, and Yao-Haur Kuo. 2012. "Cytotoxic Phenylpropanoids and a New Triterpene, Turformosinic Acid, from Turpinia formosana Nakai" Molecules 17, no. 2: 1837-1851. https://doi.org/10.3390/molecules17021837