A New Class of Heterocycles: 1,4,3,5-Oxathiadiazepane 4,4-dioxides

Abstract
:1. Introduction

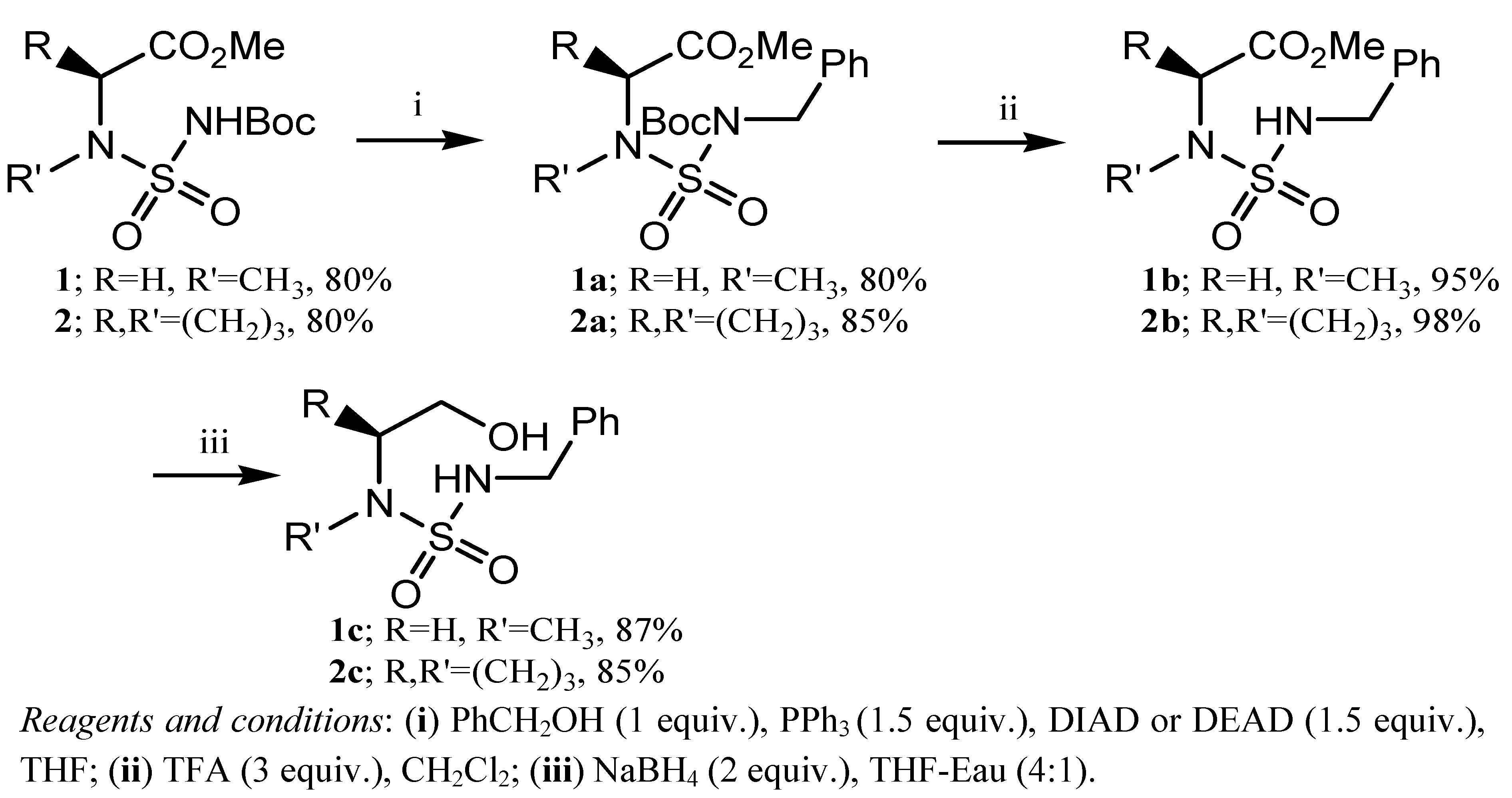
2. Results and Discussion



{kind=link}
{kind=link}
| Compounds | Yield (%) |
|---|---|
| 1d | 50 |
| 2d | 50 |
| 3d | 43 |
| 4d | 41 |
| 1e | 55 |
| 2e | 45 |
| 3e | 40 |
| 4e | 42 |
3. Experimental
3.1. General
3.2. General Synthetic Procedure for Carbamoylation-Sulfamoylation: Preparation of 1 and 2
3.3. General Procedure for the Synthesis of N-Boc, N′-(benzyl)sulfamides 1a and 2a
3.4. General Deprotection Procedure: Preparation of 1b and 2b
3.5. General Reduction Procedure; Preparation of 1c and 2c
3.6. General Procedure for the Preparation of 1,4,3,5-Oxathiadiazepanes 4,4-dioxides 1d–4d, 1e–4e
4. Conclusions
Acknowledgments
References and Notes
- Drews, J. Drug Discovery: A Historical Perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C.T. Update on the Development of HIV Entry Inhibitors. Curr. Med. Chem. 2003, 10, 925–953. [Google Scholar] [CrossRef]
- Silvestri, R.; Marfe, G.; Artico, M.; la Regina, G.; Lavecchia, A.; Novellino, E.; Morgante, M.; di Stefano, C.; Catalano, G.; Filomeni, G.; Abruzzese, E.; Ciriolo, M.R.; Russo, M.A.; Amadori, S.; Cirilli, R.; la Torre, F.; Salimei, P.S. Pyrrolo[1,2-b][1,2,5]benzothiadiazepines (PBTDs): A New Class of Agents with High Apoptotic Activity in Chronic Myelogenous Leukemia K562 Cells and in Cells from Patients at Onset and Who Were Imatinib-Resistant. J. Med. Chem. 2006, 49, 5840–5844. [Google Scholar]
- Lebegue, N.; Gallet, S.; Flouquet, N.; Carato, P.; Pfeiffer, B.; Renard, P.; Le´once, S.; Pierre, A.; Chavatte, P.; Berthelot, P. Novel Benzopyridothiadiazepines as Potential Active Antitumor Agents. J. Med. Chem. 2005, 48, 7363–7373. [Google Scholar] [CrossRef]
- Brzozowski, F.; Saczewski, F.; Neamati, N. Synthesis and anti-HIV-1 activity of a novel series of 1,4,2-benzodithiazine-dioxides. Bioorg.Med. Chem. Lett. 2006, 16, 5298–5302. [Google Scholar] [CrossRef]
- Iyenger, B.S.; Dorr, R.T.; Alberts, D.S.; Solyom, A.M.; Krutzsch, M.; Remers, W.A. 1,4-Disubstituted Anthracene Antitumor Agents. J. Med. Chem. 1997, 40, 3734–3738. [Google Scholar] [CrossRef]
- Thurston, D.E.; Subhas, B.D.; Howard, P.W.; Jen-kins, T.C.; Leoni, A.; Baraldi, P.G.; Guiotto, A.; Cacciari, B.; Kelland, L.R.; Follope, M.; Rault, S.J. Effect of A-Ring Modifications on the DNA-Binding Behavior and Cytotoxicity of Pyrrolo[2,1-c][1,4]benzodiazepines. J. Med. Chem. 1999, 42, 1951–1964. [Google Scholar] [CrossRef]
- Basolo, L.; Beccalli, E.M.; Borsini, E.; Broggini, G.; Khansaa, M.; Rigamonti, M. Access to a Novel Class of Tetracyclic 1,4-Benzodiazepin-5-ones Starting from α-Amino Acids by Pd-Catalyzed Amination/1,3-Dipolar Cycloaddition as the Key Steps. Eur. J. Org. Chem. 2010, 2010, 1694–1703. [Google Scholar]
- Mowbray, C.E.; Burt, C.; Corbau, R.; Gayton, S.; Hawes, M.; Perros, I.; Tran, D.A.; Price, F.J.; Quinton, M.D.; Selby, P.A.; Stupple, R.; Webster, A.W. Pyrazole NNRTIs 4: Selection of UK-453,061 (lersivirine) as a Development Candidate. Bioorg. Med. Chem. Lett. 2009, 19, 5857–5860. [Google Scholar]
- Broggini, G.; Marchi, I.; Martinelli, M.; Paladino, G.; Pilati, T.; Terraneo, A. Effective Synthesis of Enantiopure [1,2,3]Triazolo[1,5-a] and Pyrazolo[1,5a]pyrrolo[2,1c][1,4] benzodiazepines by Diastereoselective Intramolecular Azide and Nitrilimine Cycloadditions. Synthesis 2005, 2246–2252. [Google Scholar]
- Hurley, L.H.; Petrusek, R.L. Proposed structure of the anthramycin-DNA adduct. Nature 1979, 282, 529–531. [Google Scholar] [CrossRef]
- Cheatham, S.; Kook, A.; Hurley, L.H.; Barkley, M.D.; Remers, W.J. One and two dimensional proton NMR, fluorescence and molecular modeling studies on the tomaymycin-d(ATGCAT)2 adduct. Evidence for two covalent adducts with opposite orientations and stereochemistries at the covalent linkage site. J. Med. Chem. 1988, 31, 583–590. [Google Scholar] [CrossRef]
- Wang, J.J.; Hill, G.C.; Hurley, L.H. Template-directed design of a DNA-DNA crosslinker based upon a bis-tomaymycin-duplex adduct. J. Med. Chem. 1992, 35, 2995–3002. [Google Scholar] [CrossRef]
- Montzouris, J.A.; Wang, J.J.; Thurston, D.E.; Hurley, L.H. Comparison of a DSB-120 DNA Interstrand Cross-Linked Adduct with the Corresponding Bis-tomaymycin Adduct: An Example of a Successful Template-Directed Approach to Drug Design Based upon the Monoalkylating Compound Tomaymycin. J. Med. Chem. 1994, 37, 3132–3140. [Google Scholar] [CrossRef]
- Boudjabi, S.; Dewynter, G.; Voyer, N.; Toupet, L.; Montero, J.L. Sulfahydantoins as Tripeptide Constraints: Synthesis and Structure of Chiral Substituted 3-Oxo-1,2,5-thiadiazolidine 1,1-Dioxides. Eur. J. Org. Chem. 1999, 2275–2283. [Google Scholar]
- Regainia, Z.; Abdaoui, M.; Aouf, N.E.; Dewynter, G.; Montero, J.L. Synthesis of 1,2,5-Thiadiazolidines 1,1-dioxides (Cyclosulfamides) Starting from Amino Acids and Chlorosulfonyl Isocyanate. Tetrahedron 2000, 56, 381–387. [Google Scholar] [CrossRef]
- Berredjem, M.; Djebbar, H.; Regainia, Z.; Aouf, N.E.; Dewynter, G.; Winum, J.Y.; Montero, J.L. Synthese et N-Acylation Regiospecifique de 1,2,5-Thiadiazolidines 1,1-Dioxydes Chirales. Phosphorus Sulfur 2003, 178, 693–705. [Google Scholar] [CrossRef]
- Regainia, Z.; Winum, J.Y.; Smain, F.Z.; Toupet, L.; Aouf, N.E.; Montero, J.L. General synthesis of n-membered cyclic sulfamides. Tetrahedron 2003, 59, 6051–6056. [Google Scholar] [CrossRef]
- Bendjeddou, A.; Djeribi, R.; Regainia, Z.; Aouf, N. N,N’-Substituted 1,2,5 Thiadiazolidine 1,1-Dioxides: Synthesis, Selected Chemical and Spectral Proprieties and Antimicrobial Evaluation. Molecules 2005, 10, 1387–1398. [Google Scholar] [CrossRef]
- Kumar, G.B.; Patel, H.V.; Shah, A.C.; Trenkle, M.; Cardin, C.J. Diastereoselective formation of 2-aryl-3-arenesulfonyl 4-ethyl-1,3-oxazolidines: An X-ray crystallographic and 1H NMR study. Tetrahedron-Asymmetry 1996, 7, 3391–3396. [Google Scholar]
- Wilken, J.; Wallbaum, S.; Saak, W.; Haase, D.; Pohl, S.; Laxmikant, N.; Patkar, N.; Arun, N.; Chittari, P.; Rajappa, S.; Martens, J. Utilization of Industrial Waste Materials, 6. Utilization of Derivatives of the Bicyclic Proline Analog (all-R)-Octahydrocyclopenta[b]pyrrol-2-carboxylic Acid in the Stereoselective Synthesis. Liebigs Ann. 1996, 927–934. [Google Scholar]
- Heydenreich, M.; Koch, A.; Lazar, L.; Szatmari, I.; Sillanpaa, R.; Kleinpeter, E.; Fulop, F. Synthesis and stereochemical studies of 1- and 2-phenyl-substituted 1,3-oxazino[4,3-a]isoquinoline derivatives. Tetrahedron 2003, 59, 1951–1959. [Google Scholar] [CrossRef]
- Perlmutter, P.; Tabone, M. A Simple Route to .alpha.-Substituted-.beta.-Amino Ester Precursors of Carbapenem Antibiotics. J. Org. Chem. 1995, 60, 6515–6522. [Google Scholar]
- Arnecke, R.; Bohmer, V.; Friebe, S.; Gebauer, S.; Krauss, G.J.; Thondorf, I.; Vgt, W. Regio- and diastereoselective condensation of resorcarenes with primary amines and formaldehyde. Tetrahedron Lett. 1995, 36, 6221–6224. [Google Scholar]
- Mitsunobu, O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis 1981, 1, 1–29. [Google Scholar] [CrossRef]
- Hughes, D.L. The Mitsunobu Reaction. Org. Reactions 1992, 42, 335–380. [Google Scholar]
- Takahashi, H.; Morimoto, I.; Higashiyama, K. Highly Diastereoselective Reaction of (3aS,6R)-5-Oxa-7,8a-diazaperhydroazulen-8-ones with Diethylzinc. Chem. Pharm. Bull. 1990, 38, 2627–2631. [Google Scholar] [CrossRef]
- Takahashi, H.; Morimoto, I.; Higashiyama, K. Asymmetirc Induction by Chiral Heterocyclic Compounds: Highly Diastereoselective Reaction of 5-Oxa-7,8a-diazaperhydroazulen-8-ones with Organometallic Reagents. Heterocycles 1990, 30, 287–290. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1d–4d and 1e–4e are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bendjeddou, A.; Abbaz, T.; Regainia, Z.; Aouf, N.-E. A New Class of Heterocycles: 1,4,3,5-Oxathiadiazepane 4,4-dioxides. Molecules 2012, 17, 1890-1899. https://doi.org/10.3390/molecules17021890
Bendjeddou A, Abbaz T, Regainia Z, Aouf N-E. A New Class of Heterocycles: 1,4,3,5-Oxathiadiazepane 4,4-dioxides. Molecules. 2012; 17(2):1890-1899. https://doi.org/10.3390/molecules17021890
Chicago/Turabian StyleBendjeddou, Amel, Tahar Abbaz, Zine Regainia, and Nour-Eddine Aouf. 2012. "A New Class of Heterocycles: 1,4,3,5-Oxathiadiazepane 4,4-dioxides" Molecules 17, no. 2: 1890-1899. https://doi.org/10.3390/molecules17021890