Total Synthesis of Flocoumafen via Knoevenagel Condensation and Intramolecular Ring Cyclization: General Access to Natural Products

Abstract
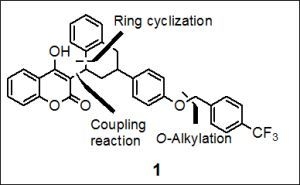
:
1. Introduction

2. Results and Discussion
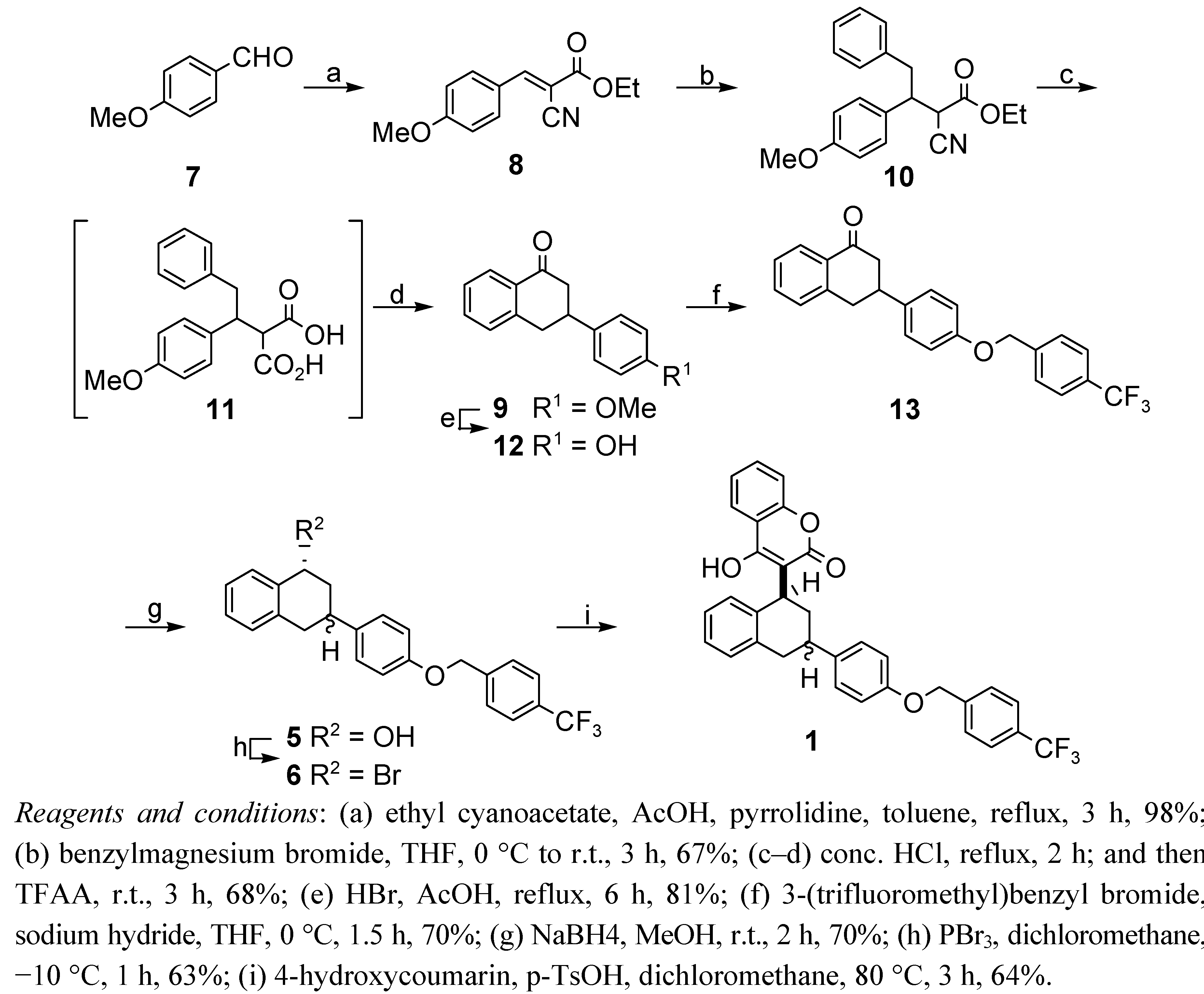
2.1. Total Synthesis


2.2. Structure Determination

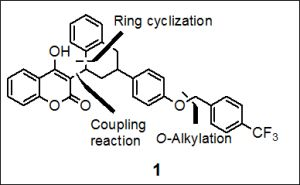{kind=link}
{kind=link}
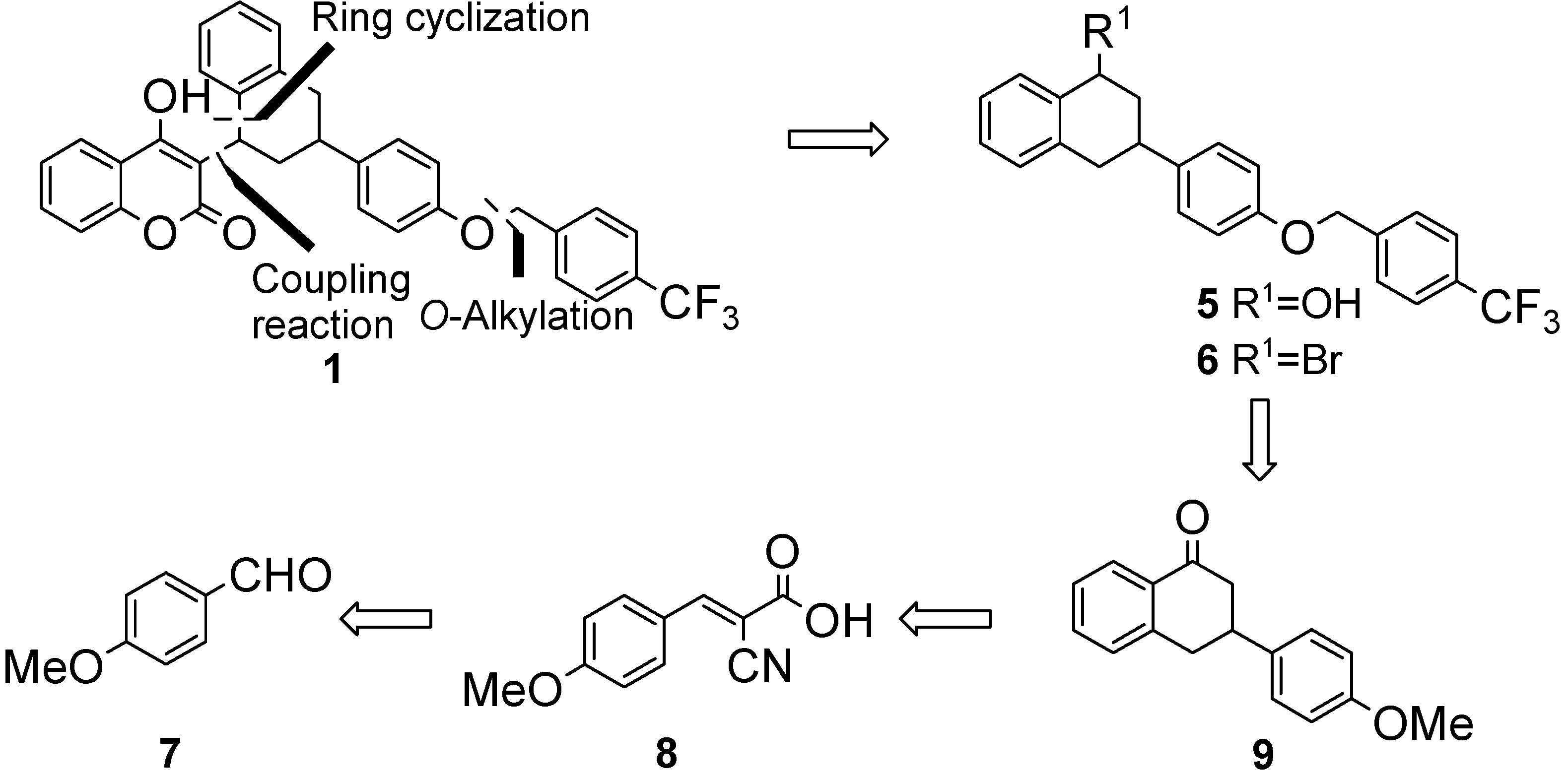{kind=link}
{kind=link}
{kind=link}
| Position | 1H (multi a, J in Hz) | 13C b | HMBC c | NOESY d |
|---|---|---|---|---|
| 5 | 7.72 (d, 7.5) | 123.1 | 4, 7, 9, CH | H6s, H7m |
| 1' | 4.87 (dd,5.5, 5.5) | 37.5 | H2'm, H3'w | |
| 2' | 2.52–2.42 (m)1.95–1.80 (m) | 36.7 | 3', 4', CH2 | H1'm, H3'm, H2"w, H6"w |
| 3' | 3.13–3.02 (m) | 9.8 | 2", 6", 4', 10', CH | H2"s, H6"s, H2'm, H1'w |
| 4' | 3.13–3.02 (m) | 38.6 | 5', 3', CH2 | H2's, H1'w, H2"w, H6"w |
| 2" | 7.21 (d, 9.0) | 127.9 | H3"s | |
| 6" | 7.21 (d, 9.0) | 127.9 | H5"s |
| Position | 1H (multi a, J in Hz) | 13C b | HMBC c | NOESY d |
|---|---|---|---|---|
| 5 | 7.66 (dd, 1.5, 1.5) | 123.9 | 7, CH | H6s, H7m |
| 1' | 4.72 (t, 4.0) | 37.5 | 3', CH | H2'm, H8'w |
| 2' | 2.36-2.32 (m) | 35.9 | 4', CH2 | H1'm, H3'm, H4'w |
| 3' | 3.12-2.99 (m) | 36.5 | 2", 6", 4', CH | H2"s, H6"s, H2'm, H1'w |
| 4' | 3.23 (d, 12.0) | 39.8 | 1", 3', 5', 9', CH2 | H2's, H5'm, H2"m, H6"m, H1'w |
| 2" | 7.16 (d, 8.5) | 128.0 | H3"s, H2'w | |
| 6" | 7.16 (d, 8.5) | 128.0 | H5"s, H2'w |
| Parameter | cis-FCF | trans-FCF |
|---|---|---|
| Bond lengths (Å) | ||
| H1'-H2' | 2.37 | 2.39 |
| H1'-H3' | 2.60 | 3.83 |
| H5-C4 | 2.74 | 2.80 |
| H5-C7 | 3.41 | 3.39 |
| H5-C9 | 3.39 | 3.41 |
| Bond angle (deg) | ||
| C4'-C3'-C1" | 112.37 | 115.40 |
| Dihedral angles (deg) | ||
| H5-C5-C6-C4 | 0.05 | 1.93 |
| H5-C5-C8-C7 | 179.83 | 178.18 |
| H5-C5-C6-C9 | 0.17 | 2.72 |
| H2'-C2'-C3'-C4' | 4.28 | 2.45 |

3. Experimental
3.1. General
3.2. General Procedure for the Knoevenagel Reaction: Synthesis of (E)-Ethyl 2-cyano-4-methoxy-cinnamte (8) [18]
3.3. Synthesis of Compounds 10, 9, 12, 13, and 5
3.3.1. 3-(4-Methoxyphenyl)-l-tetralone (9)
3.3.2. 3-(4-Hydroxyphenyl)-l-tetralone (12)
3.3.3. 3-[1,2,3,4-Tetrahydro-3-[4-(4-trifluoromethylbenzyloxy)phenyl]-1-naphthalen-1-one (13)
3.3.4. 3-[1,2,3,4-Tetrahydro-3-[4-(4-trifluoromethylbenzyloxy)phenyl]-1-naphthalen-1-ol (5)
3.4. Synthesis of Flocoumafen (1)
 -MS (ESI+) m/z 565.1 [M+Na]+, HRMS calcd for C33H26F3O4 [M+H]+ 543.1783, found m/z 543.1772; trans-Flocoumafen: mp 107.3 °C. IR (neat, NaCl) νmax 3401 (OH), 1665 (C=O), 1612 (C=C), 1080 (C-O), 831 cm-1; LCMS (ESI+) m/z 565.3 [M+Na]+; HRMS calcd for C33H26F3O4 [M+H]+ 543.1783, found m/z 543.1772.
-MS (ESI+) m/z 565.1 [M+Na]+, HRMS calcd for C33H26F3O4 [M+H]+ 543.1783, found m/z 543.1772; trans-Flocoumafen: mp 107.3 °C. IR (neat, NaCl) νmax 3401 (OH), 1665 (C=O), 1612 (C=C), 1080 (C-O), 831 cm-1; LCMS (ESI+) m/z 565.3 [M+Na]+; HRMS calcd for C33H26F3O4 [M+H]+ 543.1783, found m/z 543.1772.4. Conclusions
Supplementary Materials
Acknowledgments
- Sample Availability: Samples of the compounds in mg scale are available from the authors.
References and Notes
- Au, N.; Rettie, A.E. Pharmacogenomics of 4-hydroxycoumarin anticoagulants. Drug Metab. Rev. 2008, 40, 355–375. [Google Scholar] [CrossRef]
- Ghate, M.; Kusanur, R.A.; Kulkarni, M.V. Synthesis and in vivo analgesic and anti-inflammatory activity of some bi heterocyclic coumarin derivatives. Eur. J. Med. Chem. 2005, 40, 882–887. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Mazzon, E.; Bevilaqua, C.; Costantino, G.; Britti, D.; Mazzullo, G.; De Sarro, A.; Caputi, A.P. Cloricromene, a coumarine derivative, protects against collagen-induced arthritis in Lewis rat. Br. J. Pharmacol. 2000, 131, 1399–1407. [Google Scholar] [CrossRef]
- Kontogiorgis, C.A.; Hadjipavlou-Litina, D.J. Synthesis and antiinflammatory activity of coumarin derivatives. J. Med. Chem. 2005, 48, 6400–6408. [Google Scholar] [CrossRef]
- Kurokawa, M.; Kumeda, C.A.; Yamamura, J.I.; Kamiyama, T.; Shiraki, K. Antipyretic activity of cinnamyl derivatives and related compounds in influenza virus-infected mice. Eur. J. Pharmacol. 1998, 348, 45–51. [Google Scholar] [CrossRef]
- Nawrot-Modranka, J.; Nawrot, E.; Graczyk, J. In vivo antitumor, in vitro antibacterial activity and alkylating properties of phosphorohydrazine derivatives of coumarin and chromon. Eur. J. Med. Chem. 2006, 41, 1301–1309. [Google Scholar] [CrossRef]
- Al-Soud, Y.A.; Al-Sa'doni, H.H.; Amajaour, H.A.S.; Salih, K.S.M.; Mubarak, M.S.; Al-Masoudi, N.A.; Jaber, I.H. Synthesis, characterization and anti-HIV and Antitumor activities of new coumarin derivatives. Z. Naturforsch. B. 2008, 63, 83–89. [Google Scholar]
- Nolan, K.A.; Zhao, H.; Faulder, P.F.; Frenkel, A.D.; Timson, D.J.; Siegel, D.; Ross, D., Jr.; Burke, T.R.; Stratford, I.J.; Bryce, R.A. Coumarin-based inhibitors of human NAD(P)H:quinone oxidoreductase-1. Identification, structure-activity, off-target effects and in vitro human pancreatic cancer toxicit. J. Med. Chem. 2007, 50, 6316–6325. [Google Scholar]
- Ito, Y. Comparison of acceptability of three anticoagulant rodenticides by the root rat, Rattus rattus. Med. Entomol. Zool. 2003, 54, 337–341. [Google Scholar]
- Stanchev, S.; Momekov, G.; Jensen, F.; Manolov, I. Synthesis, computational study and cytotoxic activity of new 4-hydroxycoumarin derivatives. Eur. J. Med. Chem. 2008, 43, 694–706. [Google Scholar] [CrossRef]
- Lawley, W.J.; Charlton, A.J.A.; Hughson, E.J.; Grundy, H.H.; Brown, P.M.; Jones, A. Development of a cell culture/ELISA assay to detect anticoagulant rodenticides and its application to analysis of rodenticide treated grain. J. Agric. Food Chem. 2006, 54, 1588–1593. [Google Scholar] [CrossRef]
- Van Heerden, P.S.; Bezuidenhoudt, B.C.B.; Ferreira, D. Improved synthesis of the rodenticides diphenacoum and brodifacoum. J. Chem. Soc. Perkin Trans. 1 1997, 8, 1141–1146. [Google Scholar]
- Jung, J.C.; Lee, J.H.; Oh, S.; Lee, J.G.; Park, O.S. Synthesis and antitumor activity of 4-hydroxycoumarin derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 5527–5531. [Google Scholar] [CrossRef]
- Benfatti, F.; Cardillo, G.; Gentilucci, L.; Mosconi, E.; Tolomelli, A. Enzymatic resolution of ethyl 3-hydroxy-2-(1'-substituted-methylidene)butyrate by Pseudomonas cepacia lipase catalyzed acetylatio. Tetrahedron: Asymmetry 2007, 18, 2227–2232. [Google Scholar]
- Huang, Y.Z.; Zhou, Z.L.; Shi, L.L. Synthetic application of elementoorganic compounds of 15th and 16th groups. 102. Reactions of diazo compounds with carbonyl compounds mediated by diorganyl telluride and catalytic amount of cuprous iodide compounds: Conversion of aldehydes to alkenes via highly stabilized and stabilized telluronium ylide. Tetrahedron 1993, 49, 6821–6830. [Google Scholar]
- Fang, J.; Ren, J.; Wang, Z. Sc(OTf)3-catalyzed smooth tandem [3+2] cycloaddition/ring opening of donor-acceptor cyclopropane 1,1-diesters with enol silyl ethers. Tetrahedron Lett. 2008, 49, 6659–6662. [Google Scholar] [CrossRef]
- Lee, J.; Gauthier, D.; Rivero, R.A. Solid-phase synthesis of 3,4,5-substituted 1,5-benzodiazepin-2-ones. J. Org. Chem. 1999, 64, 3060–3065. [Google Scholar] [CrossRef]
- Novoa de Armas, H.; Blaton, N.M.; Peeters, O.M.; De Ranter, C.J.; Suarez, M.; Ochoa, E.; Verdecia, Y.; Salfran, E. Synthesis, crystal structure and molecular modeling (AM1) of two 5-arylidene derivatives of Meldrum's acid. J. Chem. Crystallogr. 2000, 30, 189–194. [Google Scholar] [CrossRef]
- Kohn, W.; Becks, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar]
- Spartan’06; Wavefunction Inc.: Irvine, CA, USA, 2006.
- Armarego, W.L.F.; Perrin, D.D. Butterworth-Heinemann, Purification of Laboratory Chemicals, 4th ed; Pergamon Press: Oxford, UK, 1997. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jung, J.-C.; Lim, E.; Lee, Y.; Min, D.; Ricci, J.; Park, O.-S.; Jung, M. Total Synthesis of Flocoumafen via Knoevenagel Condensation and Intramolecular Ring Cyclization: General Access to Natural Products. Molecules 2012, 17, 2091-2102. https://doi.org/10.3390/molecules17022091
Jung J-C, Lim E, Lee Y, Min D, Ricci J, Park O-S, Jung M. Total Synthesis of Flocoumafen via Knoevenagel Condensation and Intramolecular Ring Cyclization: General Access to Natural Products. Molecules. 2012; 17(2):2091-2102. https://doi.org/10.3390/molecules17022091
Chicago/Turabian StyleJung, Jae-Chul, Eunyoung Lim, Yongnam Lee, Dongguk Min, Jeremy Ricci, Oee-Sook Park, and Mankil Jung. 2012. "Total Synthesis of Flocoumafen via Knoevenagel Condensation and Intramolecular Ring Cyclization: General Access to Natural Products" Molecules 17, no. 2: 2091-2102. https://doi.org/10.3390/molecules17022091