Contribution of Cation-π Interactions in Iminium Catalysis

Department of Chemistry, Ochanomizu University, Otsuka, Bunkyo-ku, Tokyo, 112-8610, Japan
*
Author to whom correspondence should be addressed.
Molecules 2012, 17(2), 2161-2168; https://doi.org/10.3390/molecules17022161
Submission received: 12 January 2012
/
Revised: 9 February 2012
/
Accepted: 15 February 2012
/
Published: 21 February 2012
(This article belongs to the Special Issue Organocatalysis)
Abstract
:Ab initio calculations were carried out for a benzyl-substituted iminium cation derived from (E)-crotonaldehyde and a chiral imidazolidinone that was developed as an organocatalyst by MacMillan et al. At the MP2 level of theory it is predicted that the phenyl group is close to the iminium moiety in the most stable conformer, suggesting that the cation-π interaction contributes to the stabilization of this conformer. Energy decomposition analyses on model systems indicate that the electrostatic and polarization terms make significant contribution to the attractive interactions between the benzene ring and the iminium cation.
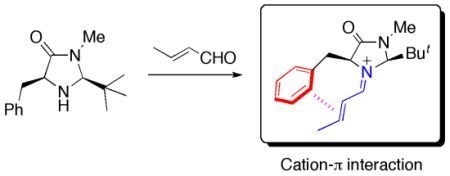
1. Introduction
In organocatalytic reactions, non-covalent interactions as well as steric hindrance are crucial for the achievement of high selectivity as they affect the conformations of the reaction intermediates and/or transition states. Such interactions, for example, π-π, CH-π and cation-π interactions, are rather weak and therefore, it may be difficult to predict the dominant conformers for the species involving the key steps. Information on the factors that control the conformation of the reactive species will be valuable in the design of new organocatalysts with high reactivity and selectivity.
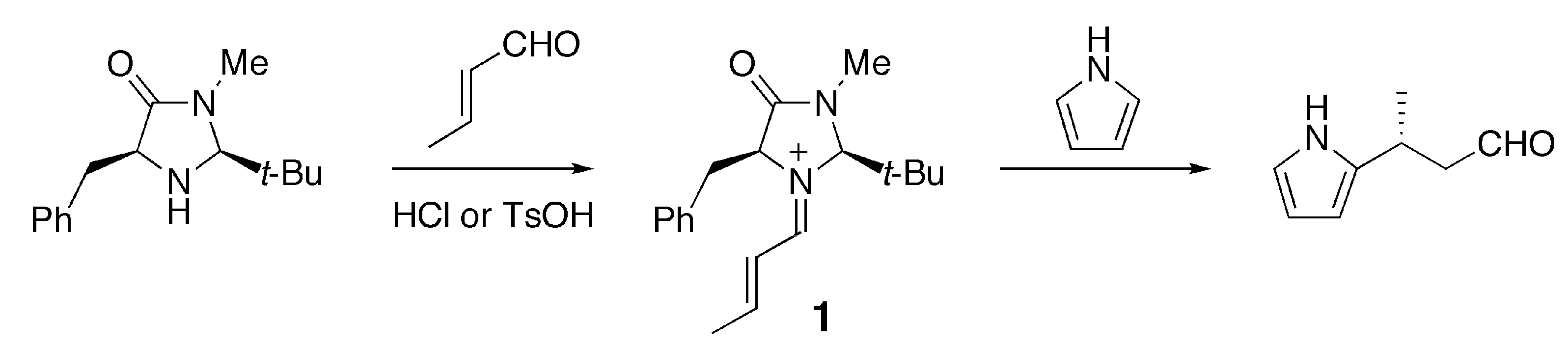
In the present paper, we focus on a benzyl-substituted iminium cation 1 [1] and demonstrate that the cation-π interaction has a significant influence on the conformational preference for this reaction intermediate. Iminium cation 1 is formed from (E)-crotonaldehyde and a chiral imidazolidinone, which was developed by MacMillan and coworkers as an enantioselective organocatalyst (Scheme 1) [2].
Scheme 1.
Iminium intermediate 1 derived from (E)-crotonaldehyde and the imidazolidinone developed by MacMillan et al [2].
Scheme 1.
Iminium intermediate 1 derived from (E)-crotonaldehyde and the imidazolidinone developed by MacMillan et al [2].

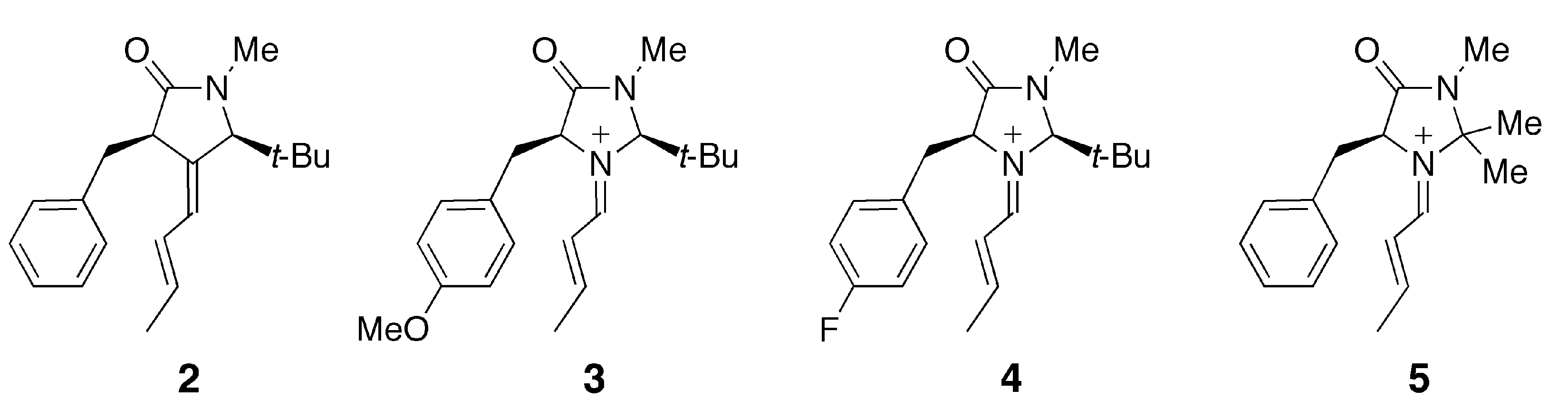
This intermediate undergoes various types of C-C bond formation reactions including Friedel-Crafts alkylation [2,3,4], Diels-Alder cycloaddition [1,5] and 1,3-dipolar additions [6]. MacMillan et al. attempted to explain the observed enantioselectivity of the products based on the conformation of the iminium intermediate predicted by MM3 calculations [2,3]. Houk et al. later estimated the relative energies of plausible conformers for the iminium intermediate and transition state in the Friedel-Crafts alkylation of pyrrole or N-methylindole by DFT calculations [7,8]. They suggested that the E/Z configuration of the C=N bond and the conformation of the benzyl group significantly affect the stereoselectivity of the product. Although the results of the calculations by Houk et al. can account for the experimental observations, they do not explain the origin of the conformational preference. One of our group recently proposed the cation-π interaction as one of the factors determining the conformational preference of 1 [9]. According to calculations at the B3LYP/6-31G(d) level, the most stable conformation of 1 displays a short distance between the iminium and phenyl moieties. In the case of the carbon-analogue 2, such a conformer is slightly less stable than another conformer in which the phenyl group is more distant from the iminium moiety. Density functional theory, particularly the B3LYP hybrid functional, has been widely used for exploring organic reaction mechanisms [10]. In general, DFT methods may be inadequate for evaluating the magnitudes of non-covalent interactions such as dispersion interactions and charge-transfer interactions. In the present study, we have carried out quantum chemical calculations at the MP2 level of theory for different conformers of iminium 1 and its related species 2–4 (Figure 1).
Figure 1.
Structural formulas of the species related to the iminium intermediate 1.

Furthermore, energy decomposition analyses have been applied to interacting pair models consisting of a truncated iminium cation and benzene in order to characterize the non-covalent interactions between the phenyl and iminium moieties. The results of these calculations provide a better understanding of cation-π interactions and suggest that such interactions may be utilized to improve the selectivities of organocatalysts through stabilization of the desired conformers.
2. Results and Discussion
2.1. Conformational Preferences in Iminium Intermediate 1 and Related Species 2–5
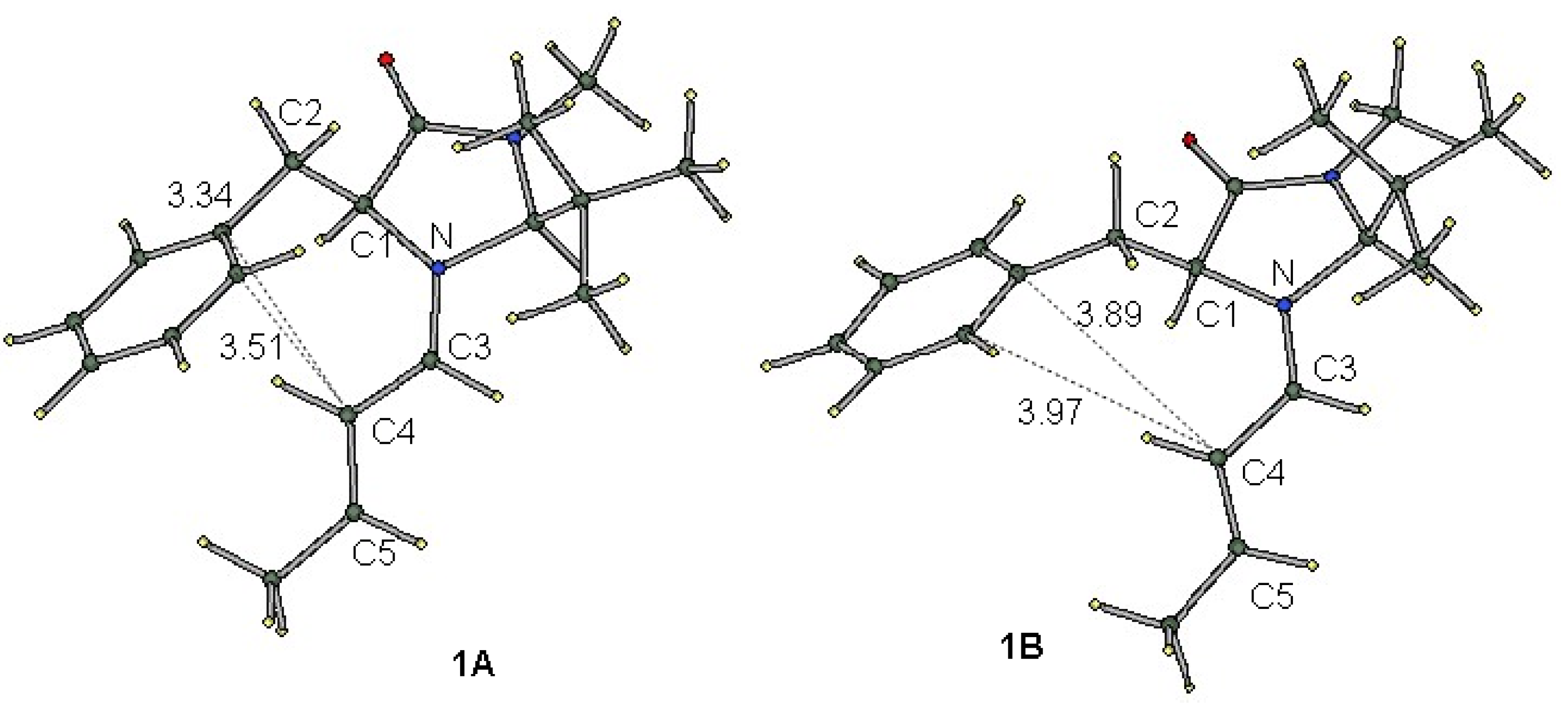
Figure 2 shows the B3LYP-optimized structure of the most stable conformers for iminium intermediate 1. These conformers are different from each other in the orientation of the phenyl group; the dihedral angles of N−C1−C2−Ph are −72.8° and −134.5° for 1A and 1B, respectively. As a result, the distance between the phenyl and iminium moieties is shorter in 1A than in 1B. Short contacts of less than 3.6 Å are seen between the iminium α-carbon (C4) and the phenyl group in conformer 1A. The interatomic distances C4−Cipso and C4−Cortho are 3.34 and 3.51 Å, respectively, while the corresponding distances in 1B are much longer (3.89 and 3.97 Å, see Table 1). As is seen in Figure 2, the N=C3−C4=C5 π-system and the benzene ring are not parallel in either 1A or 1B.
Figure 2.
Optimized structures of conformers 1A (Left) and 1B (Right) with atom numbering. The figures indicate the interatomic distances in Å.
Figure 2.
Optimized structures of conformers 1A (Left) and 1B (Right) with atom numbering. The figures indicate the interatomic distances in Å.

The relative electronic energies of these conformers for iminium intermediate 1 and the related species 2–5 are summarized in Table 1. As was previously predicted by the DFT calculations [9], conformer 1A is more stable than 1B. For all the iminium cations 1, 3, 4 and 5, conformer A is more stable than conformer B according to either DFT or MP2 calculations. On the other hand, the two conformers of the carbon analogue, 2A and 2B, were predicted to have almost the same energy. The difference in the conformational preference between the iminium cations and the non-charged carbon analogue suggests that the cation-π interaction plays an important role in the stabilization of conformer A.
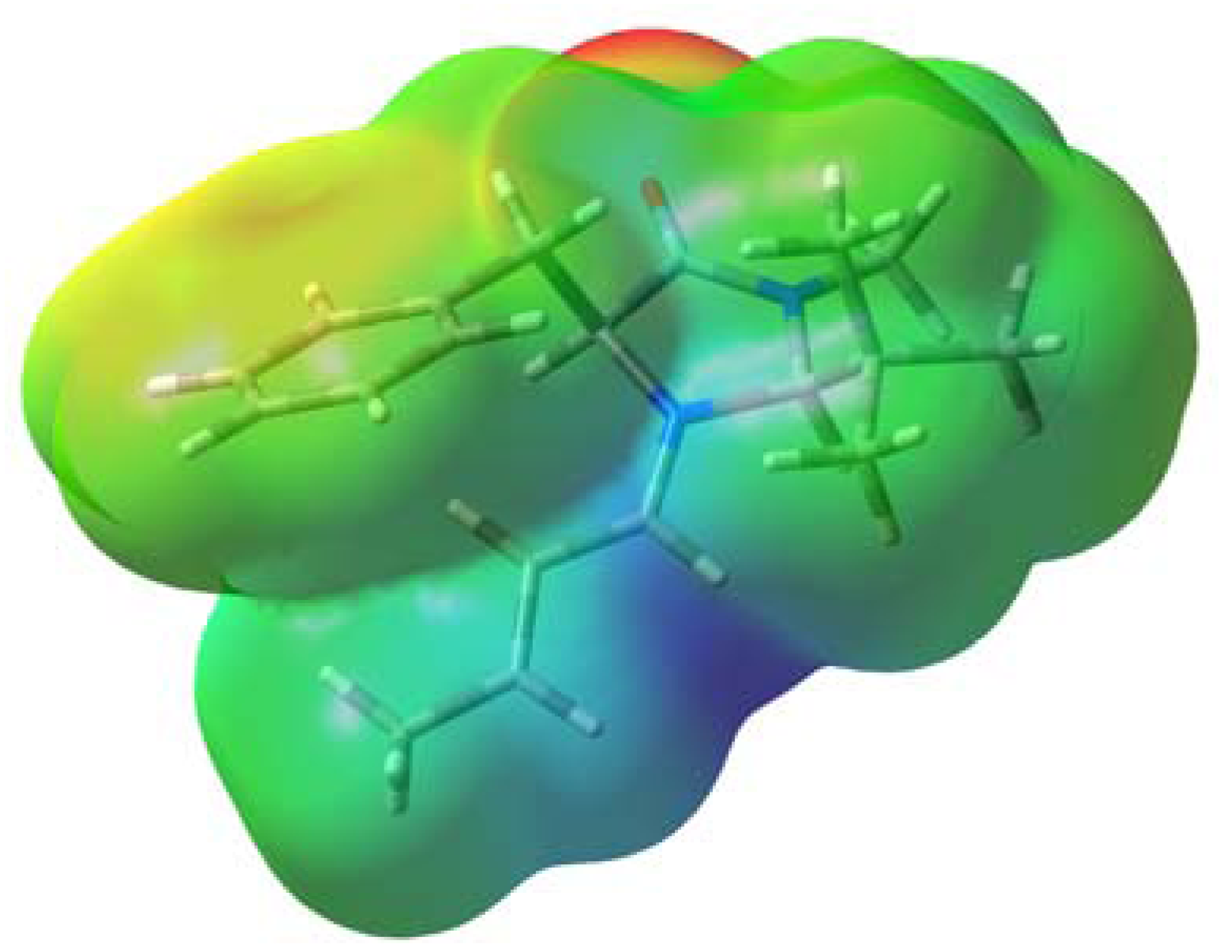
Figure 3 shows the electrostatic potential for conformer 1A. The iminium moiety possesses high positive charge while the phenyl group is only slightly positive. Thus, the polarisable π-electrons in the phenyl group are likely to be attracted by the positive charge in the iminium moiety. It is expected that such an interaction would be enhanced with increases in the electron density of the π-system. We carried out calculation for the corresponding p-methoxy- and p-fluoro derivatives, 3 and 4, at the same level of theory in order to examine the effects of the electron-donating and electron-withdrawing substituents. The predicted energy difference between conformer A and B increased in the order 4 (X = F) < 1 (X = H) < 3 (X = OMe) as shown in Table 1. This result supports the assumption that the cation-π interaction contributes to the predominance of conformer A over B.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Relative electronic energies (kcal/mol) and non-bonded interatomic distances (Å) for conformers A and B of 1–5.
| B3LYP | MP2 | HF | C4 a -- C ipso | C4 a -- C ortho | |
|---|---|---|---|---|---|
| 1A | 0.00 b | 0.00 | 0.00 | 3.34 | 3.51 |
| 1B | 0.91 b | 2.00 | 0.32 | 3.89 | 3.97 |
| 2A | 0.00 b | 0.00 | 0.00 | 3.47 | 3.57 |
| 2B | −0.38 b | 0.86 | −0.64 | 4.11 | 4.23 |
| 3A | 0.00 | 0.00 | 0.00 | 3.33 | 3.56 |
| 3B | 1.09 | 2.33 | 0.53 | 3.82 | 3.93 |
| 4A | 0.00 | 0.00 | 0.00 | 3.35 | 3.52 |
| 4B | 0.74 | 1.75 | 0.00 | 3.89 | 3.99 |
| 5Ac | 0.00 | − | − | 3.29 | 3.48 |
| 5Bc | 1.03 | − | − | 3.77 | 3.83 |
Figure 3.
Electrostatic potential map for conformer 1A plotted onto an isodensity surface (0.004 e au−3) obtained from the density calculated at the B3LYP/6-31G(d) level. The colour code corresponds to the charge from 0.03 (red) to 0.16 (dark blue).
Figure 3.
Electrostatic potential map for conformer 1A plotted onto an isodensity surface (0.004 e au−3) obtained from the density calculated at the B3LYP/6-31G(d) level. The colour code corresponds to the charge from 0.03 (red) to 0.16 (dark blue).

It should be noted that the cation-π interaction is not the only factor controlling the relative stabilities of the conformers for this type of iminium intermediates. Houk et al. predicted that the most stable conformer for a similar iminium cation 5 is not 5A, but rather a different conformer in which the benzene ring faces one of the methyl groups at position C2 in the imidazolidine ring [7,8]. According to their calculations at the B3LYP/6-311+G(d,p)//B3LYP/6-31G(d) level, the electronic energy of conformer 5A is only 0.2 kcal/mol higher than that of the most stable conformer. The energy difference between 5A and 5B (1.03 kcal/mol) is quite similar to that between 1A and 1B (0.91 kcal/mol) at the B3LYP/6-31G(d) level. It is likely that the cation-π interaction stabilizes conformer 5A relative to 5B as in the case of iminium 1. Thus, a subtle balance of the non-bonding interactions governs the relative stabilities of conformers for the iminium intermediates.
2.2. Origin of Interaction between the Iminium and Phenyl Moieties
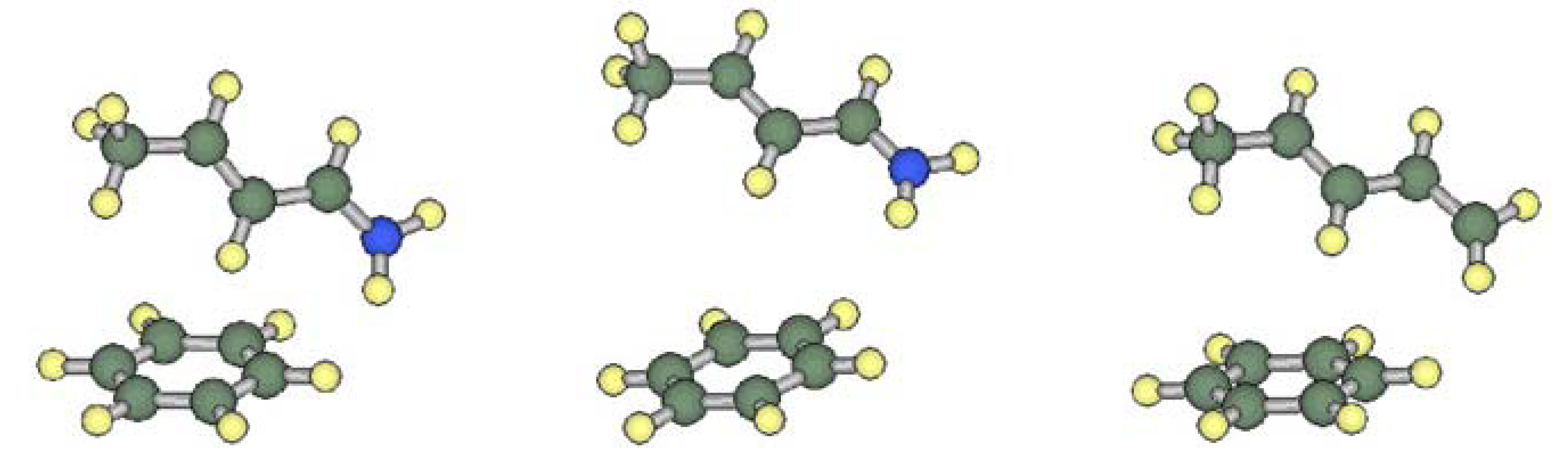
In order to characterize the interaction between the iminium and phenyl moieties in 1A, we calculated the interaction energy for a hypothetical interacting pair model M1A, which consists of a (E)-but-2-eniminium cation and a benzene molecule (see Figure 4). The mutual orientation of the components was fixed to be the same as that in 1A. The models corresponding to 1B, 2A, 3A and 4A were also examined for comparison. The interaction energies corrected for basis set superposition error (BSSE) are listed in Table 2. The MP2 calculation predicted that significant attractive interactions work between the iminium cation and the arene in models M1A, M3A and M4A. In model M1B, this interaction is weaker as the separation distance of the components is larger. In contrast, the interaction energy between the diene and benzene in M2A is close to zero.
Figure 4.
Interacting pair models M1A (left), M1B (middle) and M2A (right).

To clarify the origin of the attractive interactions, the total interaction energies, E(MP2), were decomposed into the individual contributions from several terms. The correlation energies, defined as E(correlation) = E(MP2)−E(HF), are similar to each other for M1A, M2A, M3A and M4A as shown in Table 2. On the other hand, the stabilization energy within the Hartree-Fock approximation depends on the electronic nature of the components, and the order is M2A < M4A < M1A < M3A. That is, the attractive interaction is the largest between the iminium and the electron-rich arene. The E(HF) term was further decomposed into the electrostatic, polarization, charge-transfer, exchange-repulsion, and higher-order coupling terms by using the Morokuma-Kitaura energy decomposition scheme [11]. In the case of M1A, the magnitude of summed E(electrostatic) and E(polarization) is larger than the magnitude of E(correlation). The charge-transfer interaction also contributes to the stabilization of M1A although only small fractional charge (0.08e) is transferred from the iminium to benzene. The magnitude of any type of interaction is attenuated in M1B due to the larger separation between the two components.
Tsuzuki and Yamada investigated the cation-π interactions between benzene and pyridinium (or N-methylpyridinium) cation with high-level ab initio calculations up to the CCSD(T) level at the basis set limit [12]. According to their calculations, there are local minima in which the C-H bond is directed toward the benzene plane (edge-to-face complex) with total stabilization energies of ca. −8 kcal/mol. They applied distributed multipole analyses with MP2/cc-pVTZ wavefunctions and revealed that the contribution from the electrostatic and induction terms is larger than the magnitude of the correlation energies. For the present models M1-M4, the geometries were not optimized but fixed at positions identical to those in the real systems 1–4. At such non-optimal geometries, the total stabilization energies of M1A and M3A are rather large [13]. The dominance of electrostatic and polarization terms is also similar to the trends observed for the above benzene-pyridinium complexes, although we used an easier energy decomposition scheme with Hartree-Fock wavefunctions.
In contrast to the iminium-benzene pairs, the correlation effect dominates over the electrostatic or polarization effects for the chargeless diene-benzene pair, M2A. In the real systems 1A, 1B, 3A and 4A, the electrostatic interaction will be somewhat diminished since the sum of the atomic charges in the iminium moiety amounts to 0.6−0.7 rather than unity. However, the significant difference between M1A and M2A clearly indicates that the attractive interaction between the iminium and phenyl moieties in 1A can be understood in terms of the cation-π interaction, in which the electrostatic attraction makes a sizable contribution. Thus, the cation-π interaction appears to be one of the factors that controlling conformational preference in the iminium intermediates for organocatalytic systems.
Table 2.
Interaction energies (kcal/mol) and transferred fractional charges for the interacting pair models.
| M1A | M1B | M2A | M3A | M4A | |
|---|---|---|---|---|---|
| E(MP2) a | −5.32 | −3.60 | −0.30 | −6.58 | −3.44 |
| E(HF) a | 0.08 | −1.08 | 4.81 | −0.47 | 2.32 |
| E(correlation) b | −5.40 | −2.52 | −5.10 | −6.10 | −5.76 |
| E(electrostatic) | −4.34 | −0.58 | −2.09 | − | − |
| E(polarization) | −3.05 | −2.50 | −0.35 | − | − |
| E(charge transfer) | −2.10 | −1.39 | −1.41 | − | − |
| E(exchange repulsion) | 7.86 | 2.36 | 7.49 | − | − |
| E(others) | 0.91 | 0.54 | 0.30 | − | − |
| BSSE | 0.81 | 0.48 | 0.87 | − | − |
| Transferred charge c | 0.08e | 0.02 | 0.01 | 0.09 | 0.08 |
a The basis set of 6-311G(d,p) was used. BSSE was corrected; b defined as E(MP2)−E(HF); c Based on the atomic charges calculated by electrostatic potential fitting at the MP2/6-311G(d,p) level.
3. Computational Details
The structures of iminium intermediate 1 and its carbon analogue 2 were taken from a previous work [9], in which they were optimized at the B3LYP/6-31G(d) level. The geometries of p-methoxy- or p-fluoro-substituted derivatives, 3 and 4 respectively, were optimized at the same level of theory. Single-point energy calculations were carried out at the MP2/6-31G(d,p) level to compare the electronic energies between conformers A and B for each species 1–4.
The interacting pair models, which consisted of benzene and but-2-eniminium cation or 1,3-pentadiene, were constructed from the optimized structures of 1 or 2 by removing the atoms not belonging to the phenyl or N(or C)=CHCH=CHCH3 moiety followed by addition of terminating H-atoms at a distance of 1.087 Å (Carom-H), 1.00 Å (N-H) or 1.10 Å (Colefin-H). The interaction energies calculated at the MP2/6-311G(d,p) level for the fixed geometries as well as basis set superposition errors (BSSE) were corrected by the counterpoise method. The energy decompositionanalyses were performed based on the Morokuma-Kitaura scheme [11] for the HF/6-311G(d,p) reference wavefunctions. The atomic charges were obtained by electrostatic potential fitting using the Merz-Singh-Kollman scheme [14,15]. All the calculations were carried out with Gaussian03 [16] and GAMESS [17] program packages.
4. Conclusions
The present study revealed that the cation-π interaction manifests itself in benzyl-substituted iminium 1, which is the key intermediate in the chiral imidazolidinone-catalyzed C-C bond formation developed by MacMillan et al. In the most stable conformer 1A, the phenyl group is located close to the iminium moiety. The electrostatic and polarization effects contribute to the stabilization of this conformer together with the correlation interaction. These findings will provide an insight into the design of new organocatalysts with high reactivity and selectivity. As demonstrated by the present study, quantum chemical calculations provide valuable information on non-covalent interactions affecting the conformation of key intermediates in organocatalytic reactions.
Supplementary Materials
Supplementary File 1Acknowledgements
We thank Keiko Takano, Ochanomizu University, for kind providing the computational facilities.
References and Notes
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels-Alder reaction. J. Am. Chem. Soc. 2000, 122, 4243–2444. [Google Scholar]
- Paras, N.A.; MacMillan, D.W.C. New strategies in organic catalysis: The first enantioselective organocatalytic Friedel-Crafts alkylation. J. Am. Chem. Soc. 2001, 123, 4370–4371. [Google Scholar]
- Austin, J.F.; MacMillan, D.W.C. Enantioselective organocatalytic indole alkylations. Design of a new highly effective chiral amine for iminium catalysis. J. Am. Chem. Soc. 2002, 124, 1172–1173. [Google Scholar]
- Paras, N.A.; MacMillan, D.W.C. The enantioselective organocatalytic 1,4-addition of electron-rich benzenes to α, β-unsaturated aldehydes. J. Am. Chem. Soc. 2002, 124, 7894–7895. [Google Scholar]
- Wilson, R.A.; Jen, W.S.; MacMillan, D.W.C. Enantioselective organocatalytic intramolecular Diels-Alder reactions. The asymmetric synthesis of Solanapyrone D. J. Am. Chem. Soc. 2005, 127, 11616–11617. [Google Scholar] [CrossRef]
- Jen, W.S.; Wiener, J.J.M.; MacMillan, D.W.C. New strategies for organic catalysis: The first enantioselective organocatalytic 1,3-Dipolar cycloaddition. J. Am. Chem. Soc. 2000, 122, 9874–9875. [Google Scholar] [CrossRef]
- Gordillo, R.; Carter, J.; Houk, K.N. Theoretical explorations of enantioselective alkylation reactions of pyrroles and indoles organocatalyzed by chiral imidazolidinones. Adv. Synth. Catal. 2004, 346, 1175–1185. [Google Scholar] [CrossRef]
- Allemann, C.; Gordillo, R.; Clemente, F.R.; Cheong, P.H.; Houk, K.N. Theory of asymmetric organocatalysis of aldol and related reactions: Rationalizations and predictions. Acc. Chem. Res. 2004, 37, 558–569. [Google Scholar] [CrossRef]
- Yamada, S.; Fossey, J. Nitrogen cation-pi interactions in asymmetric organocatalytic synthesis. Org. Biomol. Chem. 2011, 9, 7275–7281. [Google Scholar] [CrossRef]
- Cheong, P.H.-Y.; Legault, C.Y.; Um, J.M.; Celebi-Ölcüm, N.; Houk, K.N. Quantum mechanical investigations of organocatalysis: Mechanisms, reactivities, and selectivities. Chem. Rev. 2011, 111, 5042–5137. [Google Scholar] [CrossRef]
- Morokuma, K.; Kitaura, K. A new energy decomposition scheme for molecular interactions within Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Mikami, M.; Yamada, S. Origin of attraction, magnitude, and directionality of interactions in benzene complexes with pyridiunium cations. 2007. [Google Scholar]
- According to Refereence [12], the MP2 method overestimates the correlation effects typically by ca. 30% compared with the more accurate CCSD(T) method at the basis set limit. On the other hand, use of small basis set underestimates the correlation energy compared with the value at the basis set limit. In the present calculations at the MP2 level with medium-size basis set, these errors could be partially cancelled out. Thus, the relative correlation energies in the present series are believed to be correct at least qualitatively.
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar]
- Besler, B.H.; Merz, K.M.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 111, 431–439. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A., Jr.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N.J.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, Ö.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian 03; Gaussian, Inc.: Wallingford CT, USA, 2003. [Google Scholar]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.J.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Mori, Y.; Yamada, S. Contribution of Cation-π Interactions in Iminium Catalysis. Molecules 2012, 17, 2161-2168. https://doi.org/10.3390/molecules17022161
AMA Style
Mori Y, Yamada S. Contribution of Cation-π Interactions in Iminium Catalysis. Molecules. 2012; 17(2):2161-2168. https://doi.org/10.3390/molecules17022161
Chicago/Turabian StyleMori, Yukie, and Shinji Yamada. 2012. "Contribution of Cation-π Interactions in Iminium Catalysis" Molecules 17, no. 2: 2161-2168. https://doi.org/10.3390/molecules17022161