Synthesis of Azanucleosides through Regioselective Ring-Opening of Epoxides Catalyzed by Sulphated Zirconia under Microwave and Solvent-Free Conditions

 ,
, 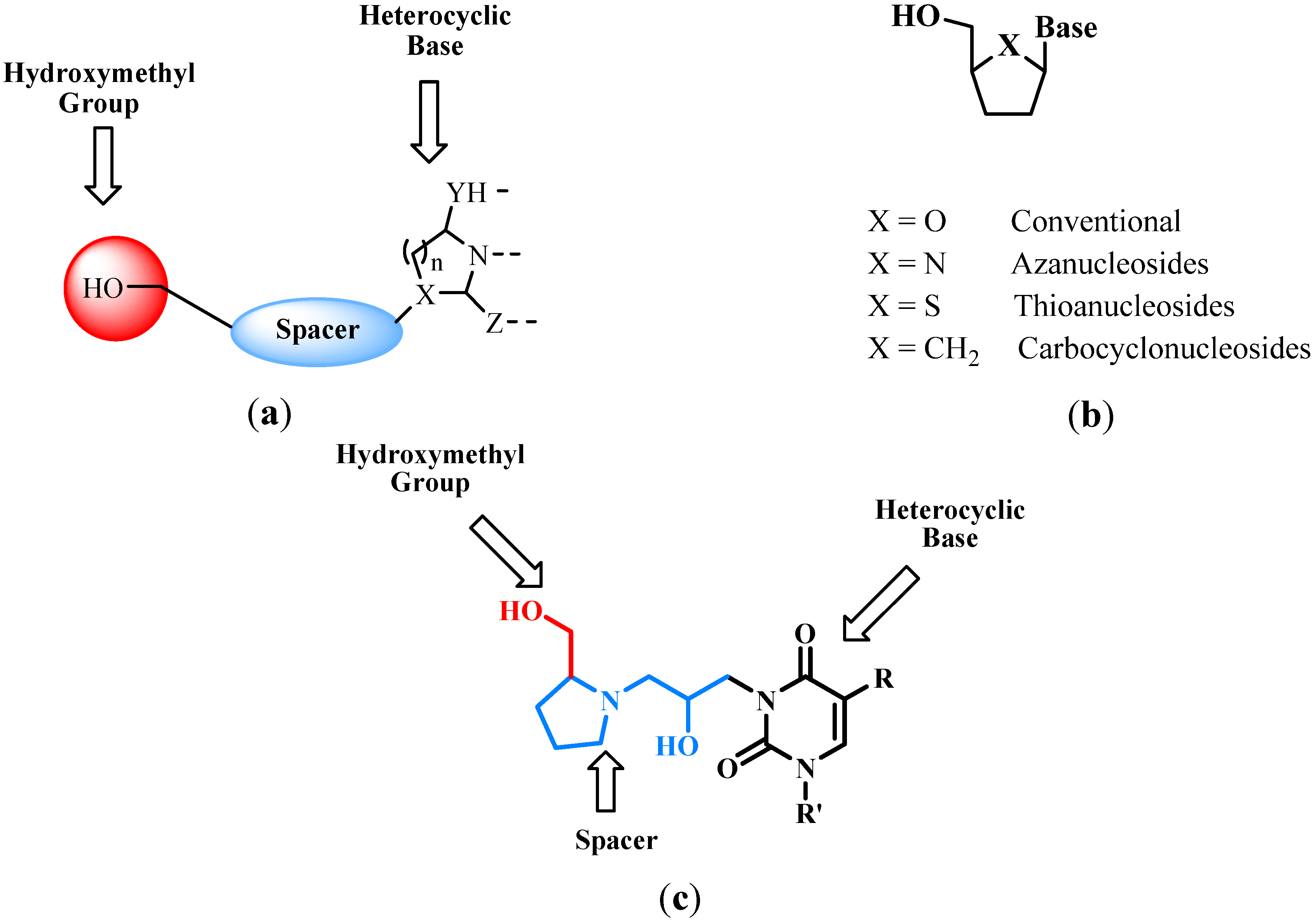{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
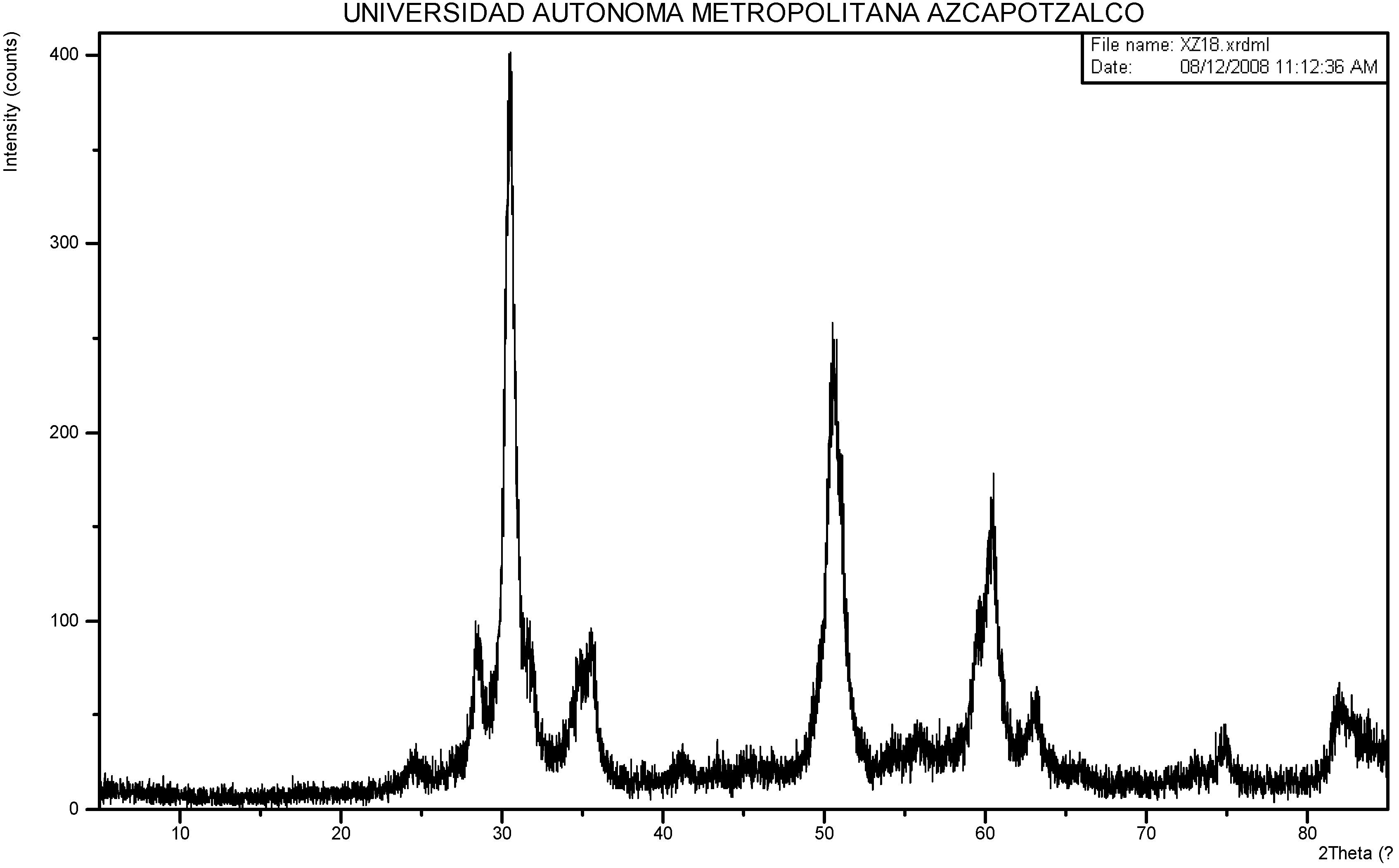
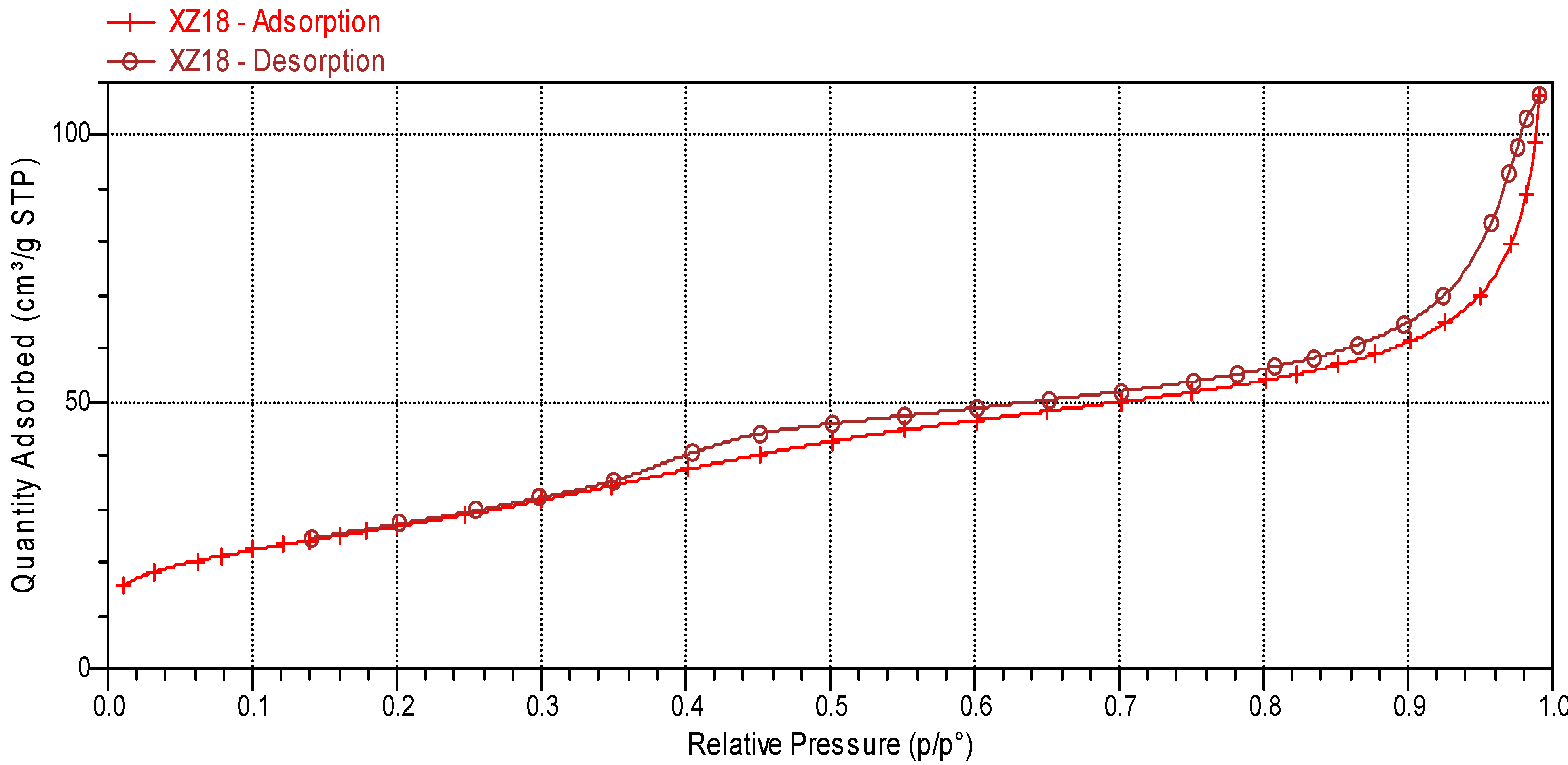
2.1. Sulphated Zirconia Characterization.



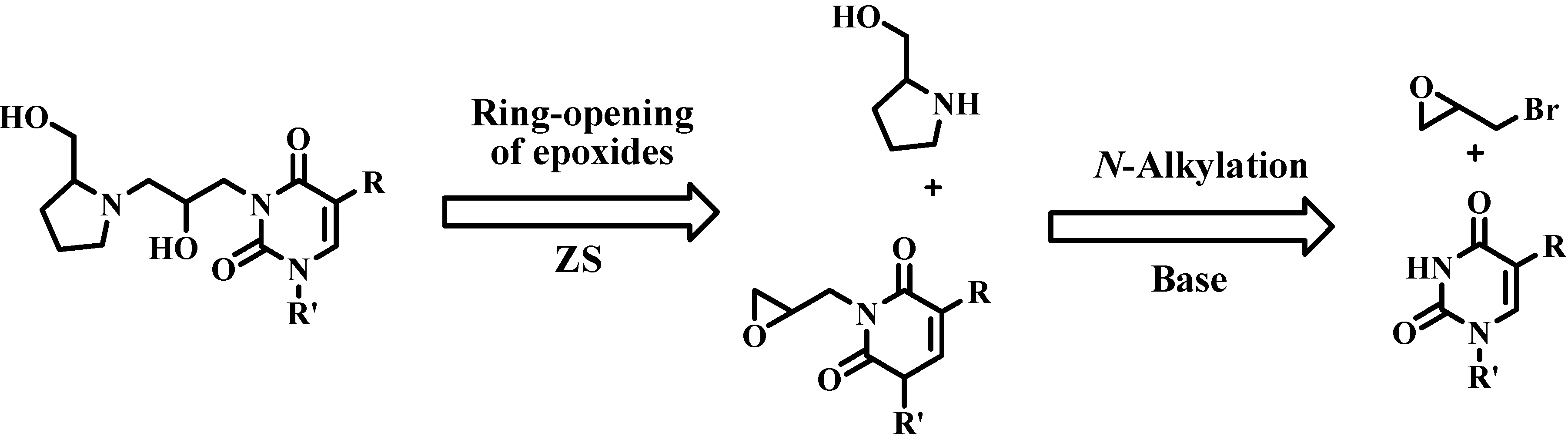
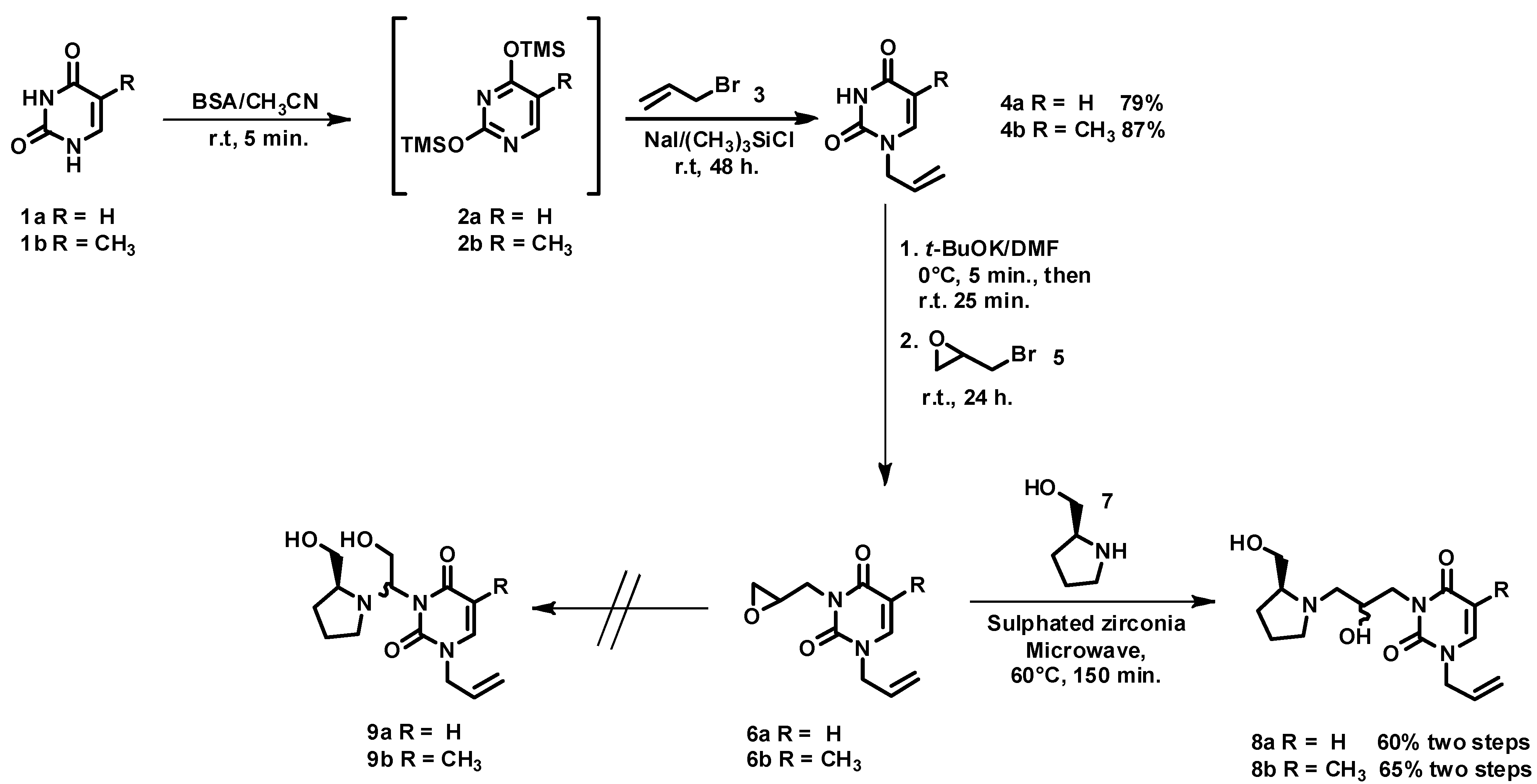
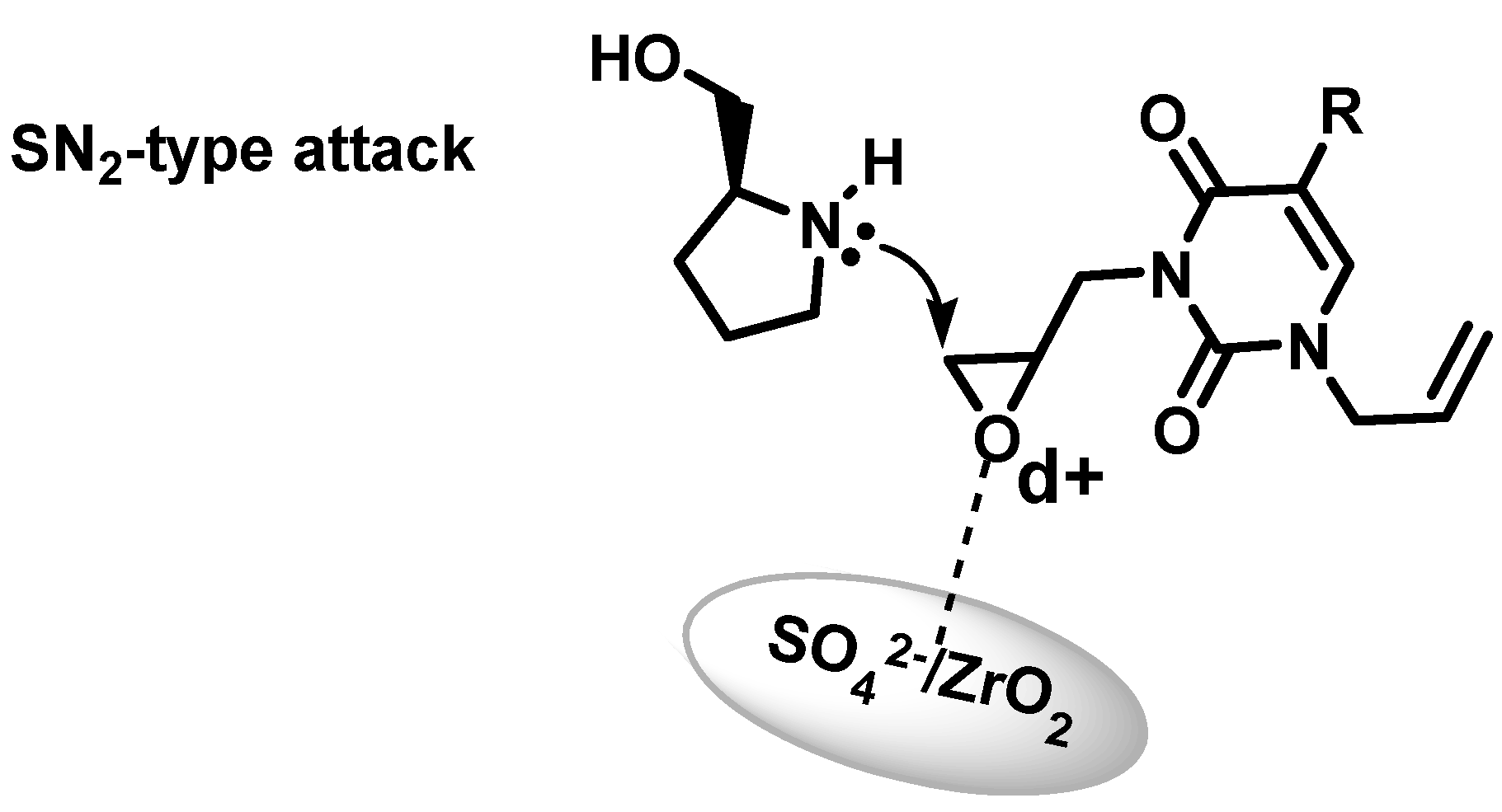
2.2. Azanucleosides Synthesis


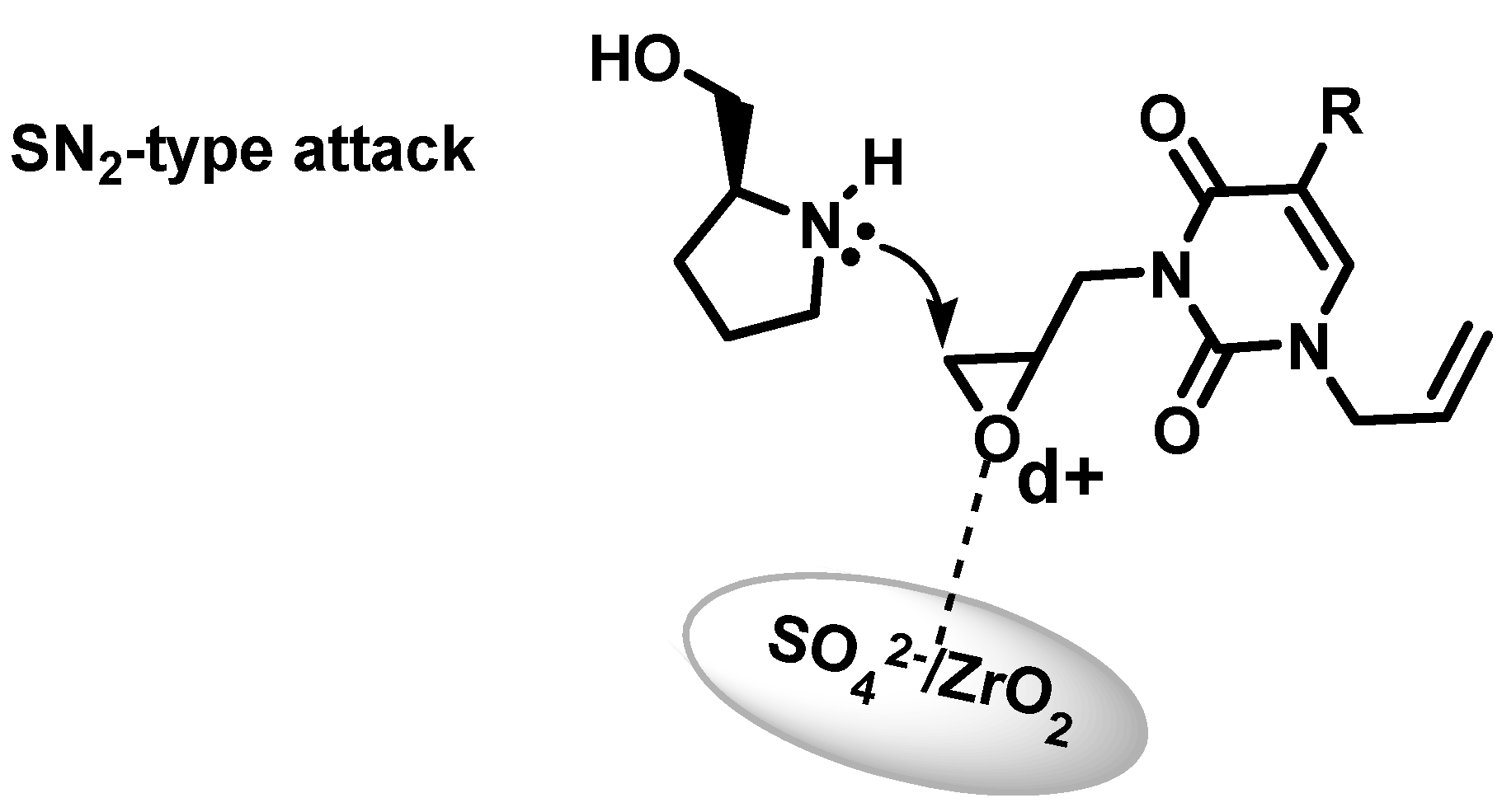
3. Experimental
3.1. General
3.2. Catalyst Synthesis [5]
3.3. Synthesis of Azanucleosides
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds 3a, 3b, 6a and 6b are available from the authors.
References and Notes
- Zane, F.; Melada, S.; Signorretto, M.; Pinna, F. Active and recyclable sulphated zirconia catalysts for the acylation of aromatic compounds. Appl. Catal. AGen. 2006, 299, 137–144. [Google Scholar] [CrossRef]
- Pérez-Luna, M.; Cosultchi, A.; Toledo-Antonio, J.A.; Cortés-Jácome, M.A. n-Pentane isomerization over Pt- and Ni-Pt-promoted sulfated zirconia catalysts supported on alumina. Catal. Lett. 2009, 131, 285–293. [Google Scholar] [CrossRef]
- Reddy, B.M.; Patil, M.K. Organic syntheses and transformations catalyzed by sulfated zirconia. Chem. Rev. 2009, 109, 2185–2208. [Google Scholar] [CrossRef]
- Arata, K. Organic syntheses catalyzed by superacidic metal oxides: Sulfated zirconia and related compounds. Green Chem. 2009, 11, 1719–1728. [Google Scholar] [CrossRef]
- Negrón, G.; Angeles, D.; Lomas, L.; Martínez, Á.; Ramírez, M.; Martínez, R. An efficient synthesis of 6,6-dimethyl-2-(4-nitrophenyl)-1-(R-penyl)-4,5,6,7-tetrahydro-1H-4-indolones using a solid sulphated zirconia as catalyst. Heterocycles 2004, 63, 367–372. [Google Scholar]
- Negrón, G.; Palacios, L.; Angeles, D.; Lomas, L.; Gaviño, R. A mild and efficient method for the chemoselective synthesis of acylals from aromatic aldehydes and their deprotections catalyzed by sulfated zirconia. J. Braz. Chem. Soc. 2005, 16, 490–494. [Google Scholar] [CrossRef]
- Palacios-Grijalva, L.N.; Cruz-González, D.Y.; Lomas-Romero, L.; González-Zamora, E.; Ulibarri, G.; Negrón-Silva, G.E. Sulphated zirconia as an eco-friendly catalyst in acylal preparation under solvent-free conditions, acylal deprotection assisted by microwaves, and the synthesis of anhydro-dimers of o-hydroxybenzaldehyde. Molecules 2009, 14, 4065–4078. [Google Scholar] [CrossRef]
- Angeles-Beltrán, D.; Lomas-Romero, L.; Lara-Corona, V.H.; González-Zamora, E.; Negrón-Silva, G. Sulfated zirconia-catalyzed synthesis of 3,4-dihydropyrimidin-2(1H)-ones (DHPMs) under solventless conditions: Competitive multicomponent Biginelli vs. Hantzsch reactions. Molecules 2006, 11, 731–738. [Google Scholar] [CrossRef]
- Negrón-Silva, G.; Hernández-Reyes, C.X.; Angeles-Beltrán, D.; Lomas-Romero, L.; González-Zamora, E.; Méndez-Vivar, J. Comparative study of regioselective synthesis of β-aminoalcohols under solventless conditions catalyzed by sulfated zirconia and SZ/MCM-41. Molecules 2007, 12, 2515–2532. [Google Scholar] [CrossRef]
- Negrón-Silva, G.; Hernández-Reyes, C.X.; Angeles-Beltrán, D.; Lomas-Romero, L.; González-Zamora, E. Microwave-enhanced sulphated zirconia and SZ/MCM-41 catalyzed regioselective synthesis of β-amino alcohols under solvent-free conditions. Molecules 2008, 13, 977–985. [Google Scholar] [CrossRef]
- Guerra-Navarro, N.A.; Palacios-Grijalva, L.N.; Angeles-Beltrán, D.; Negrón-Silva, G.E.; Lomas-Romero, L.; González-Zamora, E.; Gaviño-Ramírez, R.; Navarrete-Bolaños, J. Synthesis of new pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-dione (PCU) cyanosilylated derivatives using sulphated zirconia and hydrotalcite as catalysts in microwave-assisted reactions under solvent free conditions. Molecules 2011, 16, 6561–6576. [Google Scholar]
- Peterson, M.L.; Vince, R. Synthesis and biological evaluation of 4-purinylpyrrolidine nucleosides. J. Med. Chem. 1991, 34, 2787–2797. [Google Scholar] [CrossRef]
- Ng, K.E.; Orgel, L.E. Replacement of the 3'-CH group by nitrogen in the carbocyclic analogue of thymidine. J. Med. Chem. 1989, 32, 1754–1757. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Santos, F.P.S.; Garcia-Manero, G. Therapy with azanucleosides for myelodysplastic syndromes. Nat. Rev. Clin. Oncol. 2010, 7, 433–444. [Google Scholar] [CrossRef]
- Crimmins, M.T. New developments in the enantioselective synthesis of cyclopentyl carbocyclic nucleosides. Tetrahedron 1998, 54, 9229–9272. [Google Scholar] [CrossRef]
- Zhu, X.F. The latest progress in the synthesis of carbocyclic nucleosides. Nucleosides Nucleotides Nucleic Acids 2000, 19, 651–690. [Google Scholar] [CrossRef]
- Piperno, A.; Chiacchio, M.A.; Iannazzo, D.; Romeo, R. Synthesis and biological activity of phosphonated nucleosides: Part 1. Furanose, carbocyclic and heterocyclic analogues. Curr. Med. Chem. 2006, 13, 3675–3695. [Google Scholar]
- Piperno, A.; Giofrè, S.V.; Iannazzo, D.; Romeo, R.; Romeo, G.; Chiacchio, U.; Rescifina, A.; Piotrowska, D.G. Synthesis of C 40 truncated phosphonated carbocyclic 20-Oxa-30-azanucleosides as antiviral agents. J. Org. Chem. 2010, 75, 2798–2805. [Google Scholar] [CrossRef]
- Merino, P. Heterocyclic nucleosides. Chemical synthesis and biological properties. Curr. Med. Chem. Anti Infective Agents 2002, 1, 389–411. [Google Scholar] [CrossRef]
- Wu, Q.; Simons, C. Synthetic methodologies for C-nucleosides. Synthesis 2004, 10, 1533–1553. [Google Scholar]
- Amblard, F.; Nolan, S.P.; Agrofoglio, L.A. Metathesis strategy in nucleoside chemistry. Tetrahedron 2005, 61, 7067–7080. [Google Scholar] [CrossRef]
- de Clercq, E.; Holy, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef]
- Simons, C.; Wu, Q.; Htar, T.T. Recent advances in antiviral nucleoside and nucleotide therapeutics. Curr. Top. Med. Chem. 2005, 5, 1191–1203. [Google Scholar] [CrossRef]
- Zhou, X.X.; Lettler, E. Nucleoside analogs as anti-HBV agents. Curr. Top. Med. Chem. 2006, 6, 851–865. [Google Scholar] [CrossRef]
- Len, C.; Postel, D. Synthesis of 2',3'-didehydro-2',3'-dideoxynucleosides via nucleoside route. Curr. Org. Synth. 2006, 3, 261–281. [Google Scholar] [CrossRef]
- Merino, P. Heterocyclic nucleosides: Chemical synthesis and biological properties. Curr. Med. Chem. 2006, 13, 539–545. [Google Scholar] [CrossRef]
- Romeo, G.; Chiacchio, U.; Corsaro, A.; Merino, P. Chemical synthesis of heterocyclic-sugar nucleoside analogues. Chem. Rev. 2010, 110, 3337–3370. [Google Scholar] [CrossRef]
- Yokoyama, M.; Momotake, A. Synthesis and biological activity of azanucleosides. Synthesis 1999, 9, 1541–1554. [Google Scholar] [CrossRef]
- Chiacchio, U.; Borrello, L.; Crispino, L.; Rescifina, A.; Merino, P.; Macchi, B.; Balestrieri, E.; Mastino, A.; Piperno, A.; Romeo, G. Stereoselective synthesis and biological evaluations of novel 3'-deoxy-4'-azaribonucleosides as inhibitors of hepatitis C virus RNA replication. J. Med. Chem. 2009, 52, 4054–4057. [Google Scholar] [CrossRef] [Green Version]
- Ganem, B.; Papandreou, G.J. Mimicking the glucosidase transition state: Shape/charge considerations. J. Am. Chem. Soc. 1991, 113, 8984–8985. [Google Scholar] [CrossRef]
- Yokoyama, M.; Toyoshima, H.; Shimizu, M.; Togo, H. Stereoselective coupling of riboses with metallic salts of aromatic heterocycles. J. Chem. Soc. Perkin Trans. 1997, 1, 29–34. [Google Scholar]
- Schramm, V.L. Enzymatic transition states and transition state analogue design. Annu. Rev. Biochem. 1998, 67, 693–720. [Google Scholar] [CrossRef]
- Ubasawa, M.; Takashima, H.; Sekiya, K. A convenient one-pot synthesis of acycleonucleosides. Chem. Pharm. Bull. 1995, 43, 142–143. [Google Scholar] [CrossRef]
- Negrón, G.; Quiclet-Sire, B.; Diaz, Y.; Gaviño, R.; Cruz, R. A new synthesis of 2',3'-didehydro-3'-deoxy-3-alkylthymidine. Nucleosides Nucleotides 1995, 14, 1539–1543. [Google Scholar] [CrossRef]
- Reddy, B.M.; Patil, M.K.; Reddy, B.T.; Park, S.-E. Effcient synthesis of β-amino alcohols by regioselective ring-opening of epoxides with anilines catalyzed by sulfated zirconia under solvent-free conditions. Catal. Commun. 2008, 9, 950–954. [Google Scholar] [CrossRef]
- Das, B.; Thirupathi, P.; Kumar, R.A.; Reddy, K.R. Effcient synthesis of 3-alkyl indoles through regioselective ring opening of epoxides catalyzed by sulfated zirconia. Catal. Commun. 2008, 9, 635–638. [Google Scholar] [CrossRef]
- Paryzek, Z.; Tabaczka, B. The preparation of 1-allyluracil. N(1)-Alkylation of N(3)-protected uracil derivatives. Org. Prep. Proc. Int. 2001, 33, 400–405. [Google Scholar]
- Thibon, J.; Latxague, L.; Déléris, G. Synthesis of silicon analogues of acyclonucleotides incorporable in oligonucleotide solid-phase synthesis. J. Org. Chem. 1997, 62, 4635–4642. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hernández-Reyes, C.X.; Angeles-Beltrán, D.; Lomas-Romero, L.; González-Zamora, E.; Gaviño, R.; Cárdenas, J.; Morales-Serna, J.A.; Negrón-Silva, G.E. Synthesis of Azanucleosides through Regioselective Ring-Opening of Epoxides Catalyzed by Sulphated Zirconia under Microwave and Solvent-Free Conditions. Molecules 2012, 17, 3359-3369. https://doi.org/10.3390/molecules17033359
Hernández-Reyes CX, Angeles-Beltrán D, Lomas-Romero L, González-Zamora E, Gaviño R, Cárdenas J, Morales-Serna JA, Negrón-Silva GE. Synthesis of Azanucleosides through Regioselective Ring-Opening of Epoxides Catalyzed by Sulphated Zirconia under Microwave and Solvent-Free Conditions. Molecules. 2012; 17(3):3359-3369. https://doi.org/10.3390/molecules17033359
Chicago/Turabian StyleHernández-Reyes, Celia Xochitl, Deyanira Angeles-Beltrán, Leticia Lomas-Romero, Eduardo González-Zamora, Rubén Gaviño, Jorge Cárdenas, José Antonio Morales-Serna, and Guillermo E. Negrón-Silva. 2012. "Synthesis of Azanucleosides through Regioselective Ring-Opening of Epoxides Catalyzed by Sulphated Zirconia under Microwave and Solvent-Free Conditions" Molecules 17, no. 3: 3359-3369. https://doi.org/10.3390/molecules17033359