3.3. Zn/CH2Br2/TiCl4 Reagent
TiCl4 (2.3 mL) was added dropwise over 10 min to a stirred suspension of zinc dust (5.75 g) in CH2Br2 (2 mL) and THF (50 mL, freshly distilled over sodium and benzophenone) at −40 °C. The mixture was allowed to warm to 5 °C and was then stirred at this temperature for 3 days to give a thick grey slurry of an active species. It was stored at −20 °C.
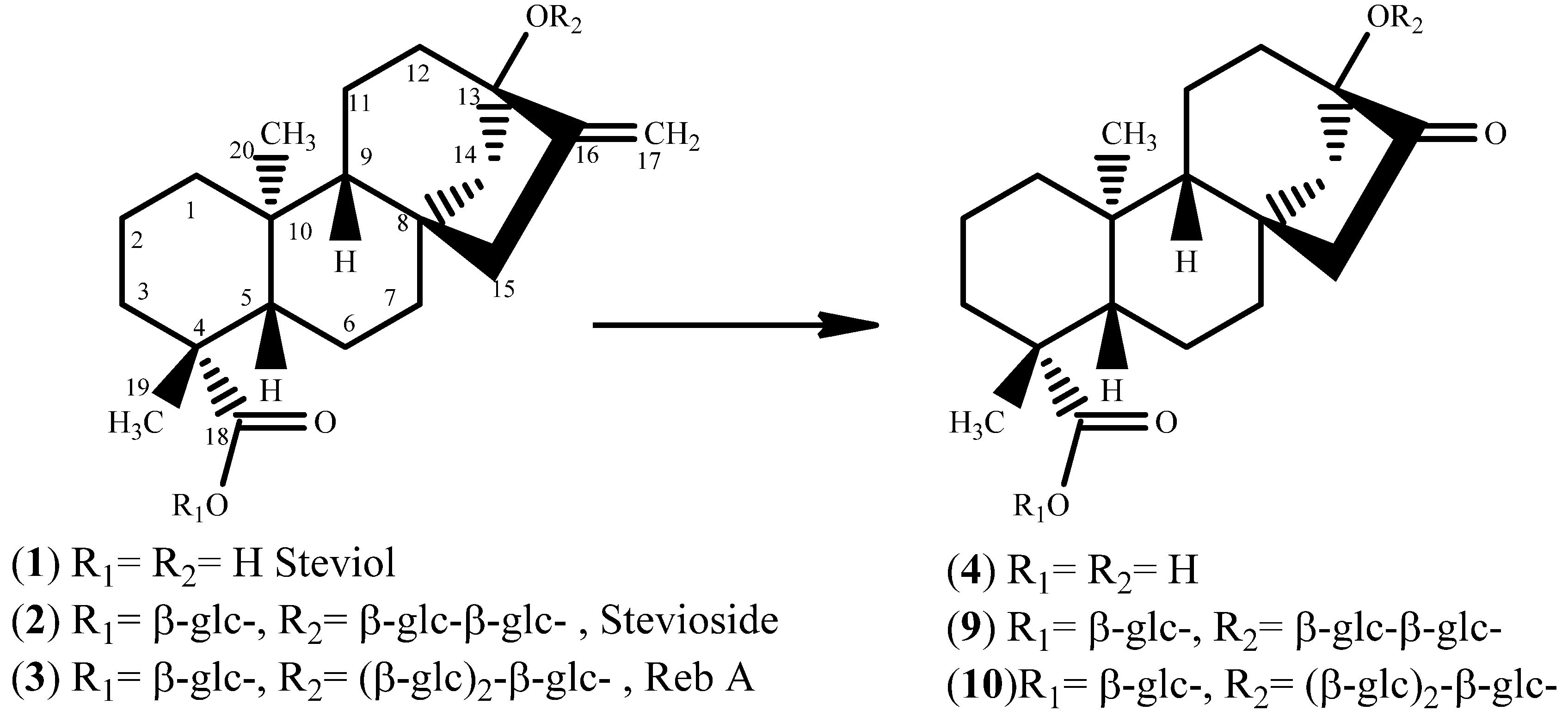
Steviol (
1) - (i)
Chemical Method [
6]: Stevioside (1.0 gm, 1.24 mmol) was suspended in water (75 mL) and treated with sodium periodate (1.5 g, 7.0 mmol) for 21 h at room temperature. The white frothy solution was then freeze-dried and was redissolved in 10% KOH solution (50 mL). The clear solution thus obtained was heated at 85 °C in an oil bath fitted with a reflux condenser. It was cooled and neutralized with 2% aqueous HCl to pH = 5.0. Heavy precipitation appeared at this time. The reaction mixture was then extracted with ethyl acetate three times (30 mL ea.). The combined ethyl acetate solution was dried over sodium sulfate and evaporated under reduced pressure to get 270 mg of crude steviol, which was absorbed on silica and purified by column chromatography with 35–50% ethyl acetate in hexane as eluent. 150 mg of pure steviol was obtained (
1, yield = 35%). TLC (hexane: ethyl acetate, 1:1): Rf = 0.29; ESI-MS
m/z: 317.2 [M−H]
−, Calcd. 317.44 for C
20H
30O
3[M−H]
−; HPLC (
tR, 26.85 min).
Steviol (
1) - (
ii) Enzymatic Method [
3]: Stevioside (0.5 g, 0.62 mmol) was hydrolyzed at 45 °C with crude pectinase from
Aspergillus Niger (1.0 mL, Sigma-Aldrich, P2736) in 0.1 M phosphate buffer (50 mL), pH = 4.5 for 24 h. The crude steviol precipitated out during the reaction. It was filtered and then dissolved in acetonitrile. The acetonitrile layer was left overnight at room temperature when some more material precipitation occurred. It was again filtered and the acetonitrile filtrate was evaporated under reduced pressure. Crude product thus obtained was crystallized from methanol to give white needle like crystals of pure steviol (
1, yield = 25%).
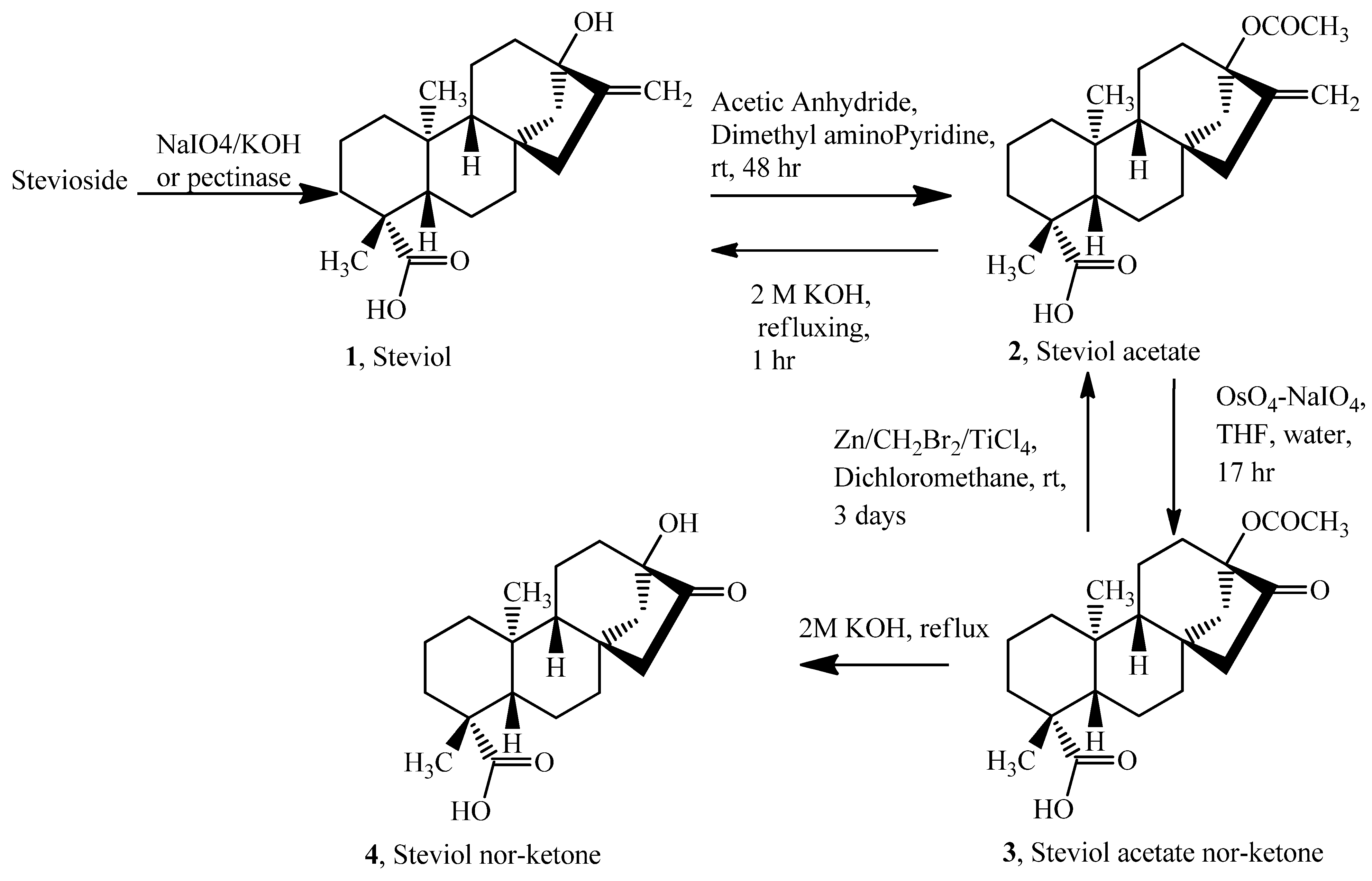
Steviol Acetate (
2). Steviol (
1, 0.954 g, 3 mmol), 4-
N,N-dimethylaminopyridine (96 mg) and acetic anhydride (1.8 mL) were dissolved in anhydrous triethylamine (5 mL) and the reaction mixture was stirred at room temperature for 48 h [
5]. To this methanol (2 mL) was added in order to remove the excess of acetic anhydride followed by evaporation on a rotavapor at 30 °C under reduced pressure. The residue was dissolved in ether (20 mL) and washed with water (3 × 8 mL), 2% HCl (3 × 8 mL) followed by washing with dilute sodium bicarbonate (5%, 3 × 8 mL) and one final wash with brine (8 mL). The ether layer was dried over sodium sulfate and evaporated on a rotavpor at 30 °C under reduced pressure to give crude steviol acetate. Recrystallization by acetone-water (5:1) gave 0.380 gm of white needle like crystals of pure steviol acetate (56%). TLC (hexane: ethyl acetate, 1:1): Rf = 0.68;
1H-NMR (CDCl
3) δ = 4.905 (s, 1H, H-17), 4.880 (s, 1H, H-17), 2.034 (s, 3H, OCOCH
3), 1.261 (s, 3H, H-19) 1.01(s, 3H, H-20);
13C-NMR: δ = 152.679 (
C=CH
2, C-16), 103.640 (C=
CH
2, C-17); ESI-MS
m/z: 383.4 [M+Na]
+, Calcd. 383.48 for C
22H
32O
4[M+Na]
+; HPLC (
tR, 34.8 min).
Steviol Acetate Ketone (
3) [
3]. Steviol acetate (
2, 320 mg, 0.88 mmol) was dissolved in a mixture of THF (3.2 ml) and water (2.8 mL). To this was added freshly made 4% aqueous solution of OsO
4 (0.4 mL). After 10 min the reaction mixture had become deep brown and to this NaIO
4 (672 mg) was added slowly over a period of 2–3 min and the mixture was left for stirring at room temperature for 18 h. It had become a thick white suspension by now and was extracted with ethyl acetate (3 × 30 mL). The organic layer was dried over sodium sulfate and evaporated on a rotavapor at 30 °C under reduced pressure to get crude steviol acetate ketone, which was flash chromatographed on silica gel 60 (70–230 mesh) with a stepwise gradient from 10% ethyl acetate in hexane to 40% ethyl acetate in hexane. Pure steviol acetate ketone came out in fractions collected at 35% ethyl acetate in hexane, yield = 230 mg (71%). TLC (hexane: ethyl acetate, 1:1): Rf = 0.41;
1H-NMR (CDCl
3) δ = 2.069 (s, 3H, OCOCH
3), 1.273 (s, 3H, H-19) 1.038(s, 3H, H-20);
13C-NMR: δ = 214.106 (C=O, C-16), peaks at 152.679 and 103.640 present in
2 disappear here; ESI-MS
m/z: 385.4 [M+Na]
+, Calcd. 385.45 for C
21H
30O
5 [M+Na]
+, HPLC(
tR, 25 min).
Methylenation of Steviol Acetate Ketone [
4]. Steviol acetate ketone (
3, 100 mg, 0.27 mmol) was taken up in anhydrous dichloromethane (4 mL) and to this solution was slowly added Zn/CH
2Br
2/TiCl
4 reagent (5 mL, 7.1 equivalents, cold reagent). The reaction mixture was stirred for five days at room temperature and then quenched by pouring into a biphasic 2:1 mixture of sodium bicarbonate-water (10 mL) and ether (10 mL). The aqueous layer was extracted two more times with ether (10 mL ea.). The combined ether layer was dried over sodium sulfate and evaporated on a rotavapor at 30 °C under reduced pressure to afford a crude product that was chromatographed on silica gel 60 (70–230 mesh) with a stepwise gradient from 5% ethyl acetate in hexane to 20% ethyl acetate in hexane. First an impurity eluted at 15% ethyl acetate/hexane and after this at 20% ethyl acetate /hexane came a product similar to steviol acetate (
2) on TLC, yield = 60 mg (60%). TLC (hexane-ethyl acetate, 1:1): Rf = 0.68;
1H-NMR (CDCl
3) δ = 4.916 (s, 1H, H-17), 4.876 (s, 1H, H-17), 2.060 (s, 3H, OCOCH
3), 1.292 (s, 3H, H-19), 1.001(s, 3H, H-20);
13C-NMR: δ = 152.604 (
C=CH
2, C-16), 103.867 (C=
CH
2, C-17);. ESI-MS
m/z: 383.4 [M+Na]
+, Calcd. 383.48 for C
22H
32O
4[M+Na]
+, HPLC (
tR, 34.8 min).
Steviol nor-ketone (4). Steviol acetate nor-ketone (40 mg, 0.11 mmol) was refluxed in 2 M KOH (20 mL) for one hour and then cooled. The cooled reaction mixture was then acidified with 30% HCl to pH = 2.5 whereupon the solution becomes milky and a white precipitate appears. It was extracted with ethyl acetate (3 × 15 mL), dried over sodium sulfate and then evaporated on a rotavapor at 30 °C under reduced pressure, yield = 30 mg, 85%. TLC (hexane-ethyl acetate, 1:1): Rf = 0.28; 1H-NMR (CDCl3) δ = 1.205 (s, 3H, H-19), 0.940 (s, 3H, H-20); 13C-NMR: δ = 214.201 (C=O, C-16); ESI-MS m/z: 319.4 [M−H]−, Calcd. 319.42 for C19H28O4[M−H]−, HPLC (tR, 27 min). A similar procedure was followed for de-acetylation of 2 to 1.
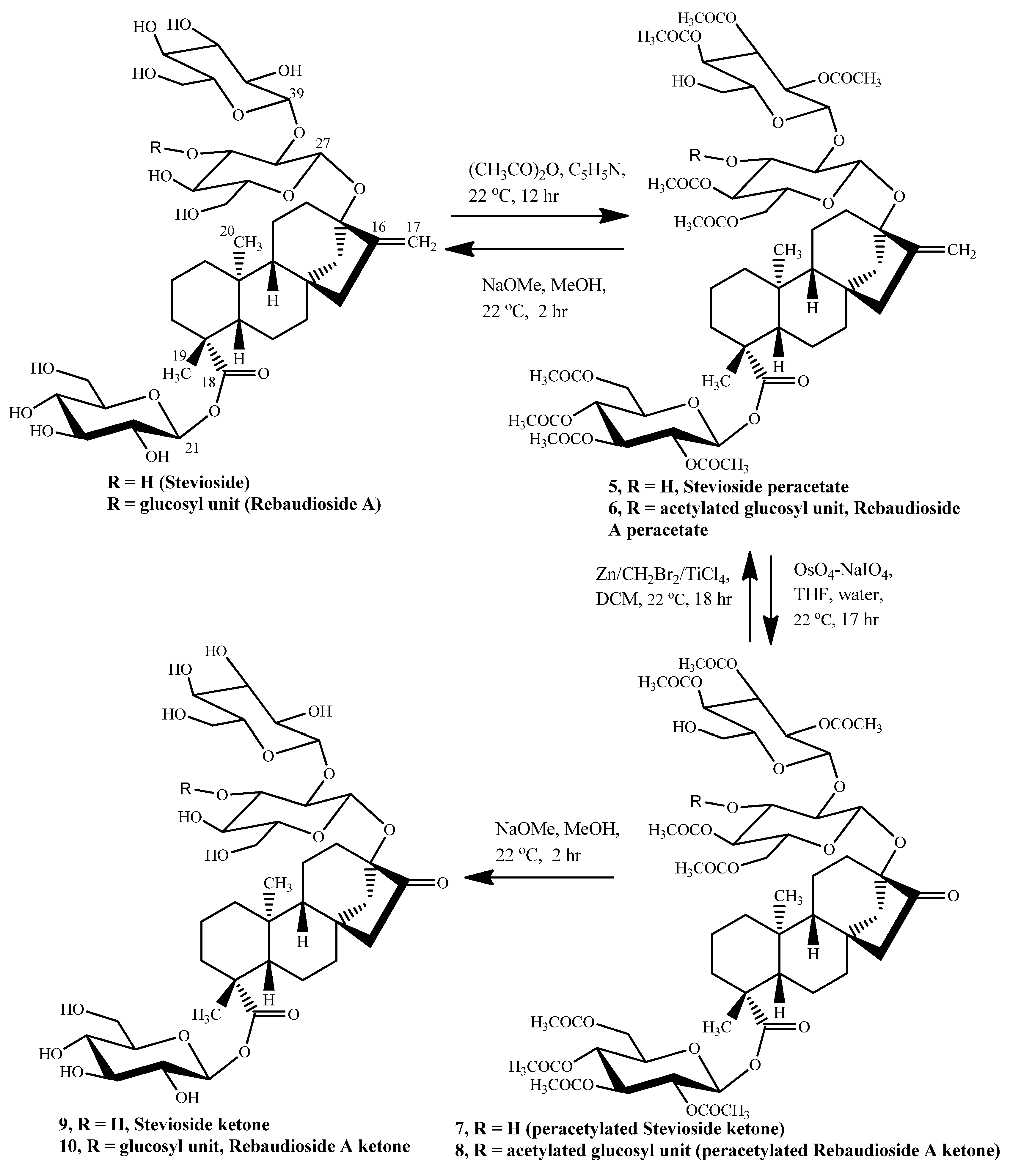
Stevioside peracetate (5). In a 500 mL round bottom flask, stevioside (55% stevioside by HPLC analysis, 15 g, nearly 10.25 mmol) was dried overnight in a vacuum oven at 75 °C. To this was added dry pyridine (200 mL) followed by cooling to 0 °C and then addition of acetic anhydride (350 mL). It was allowed to warm up to room temperature and then stirred at room temperature for 48 h. The reaction was then poured on a beaker containing ice and shaken vigorously. It was then extracted with diethyl ether (3 × 50 mL) followed by washing the organic layer with cold water (3 × 30 mL), 5% hydrochloric acid (3 × 30 mL), cold saturated sodium bicarbonate till no further evolution of CO2 was seen (3 × 40 mL) and then finally by brine (30 mL). The organic layer was dried over sodium sulfate and then evaporated to get the crude per-acetylated stevioside which was purified by silica gel flash chromatography (230–400 mesh) using a stepwise gradient of dichloromethane-methanol from pure dichloromethane up to 10% methanol in dichloromethane, yield = 10.00 g (77%). TLC (dichloromethane-methanol, 10:0.5): Rf = 0.52; 1H-NMR (DMSO) δ = 6.016 (d, 1H, H-21, J = 8.4 Hz); 13C NMR δ = 153.016 (C=CH2, C-16), 103.792 (C=CH2, C-17); ESI-MS m/z: 1289.8 [M+Na]+, Calcd. 1290.27 for C60H82O29 [M+Na]+.
Stevioside per acetate Ketone (7). Stevioside per-acetate (5, 866 mg, 1.1 mmol) was taken up in a mixture of THF (4.0 ml) and water (3.5 mL). To this was added freshly made 4% aqueous solution of OsO4 (0.5 mL). After 10 min the reaction mixture had become deep brown and to this NaIO4 (840 mg) was added slowly over a period of 2–3 min and the mixture was left for stirring at room temperature for 17 h. It had become a thick white suspension by now and was extracted with ethyl acetate (2 × 40 mL). The organic layer was dried over sodium sulfate and evaporated on arotavapor at 30 °C under reduced pressure to give the crude stevioside per-acetate ketone, which was chromatographed on silica gel 60 (70–230 mesh) with a stepwise gradient from pure dichloromethane to 2.5% methanol in dichloromethane. Pure ketone started coming out in fractions collected at 2% dichloromethane in methanol, yield = 378 mg (27%). TLC (DCM–MeOH, 10:0.5): Rf = 0.42; 1H-NMR (DMSO) δ = 6.024 (d, 1H, H-21, J = 8.4 Hz); 13C-NMR δ = 191.228 (C=O, C-16), peaks at δ 153.016 & 103.792 present in 5 disappear here; ESI-MS m/z: 1292.0 [M+Na]+, Calcd. 1292.24 for C59H80O30 [M+Na] +.
Methylenation of stevioside acetate ketone and re-generation of stevioside. Stevioside peracetate ketone (500 mg, 0.393 mmol) was taken up in anhydrous dichloromethane (10.5 mL) and to this solution was slowly added, under nitrogen, Zn/CH2Br2/TiCl4 reagent (10 mL, 8.3 equivalents, cold reagent). The reaction mixture was stirred for 18 h. at room temperature when the TLC in DCM–MeOH (20:1) showed disappearance of ketone. The reaction was further monitored by HPLC on an amino column. On the basis of HPLC and LC-MS of t = 0 and t = 18 h. samples, the reaction appeared complete. It was then quenched by pouring into a biphasic mixture of saturated sodium bicarbonate (20 mL) and ethyl acetate (10 mL). The aqueous layer was extracted two more times with ethyl acetate (10 mL ea.). The combined ethyl acetate layer was dried over sodium sulfate and evaporated on a rotavapor at 30 °C under reduced pressure to afford a crude product that was taken up in anhydrous methanol (10 mL). To this was added anhydrous sodium methoxide (200 mg). It was stirred at room temperature for 2 h and then neutralized by 0.5% HCl to pH ~ 6.5–7.0. The methanol was removed on a rotavapor under reduced pressure. A 1.0 mL size short column of reverse phase silica, C18, was packed in 50% acetonitrile-water and then washed with 50–100 mL double distilled water. The de-acetylated product was dissolved in minimum amount of water and loaded on this C18 column followed by washing with water (100 mL). Elution with 90% acetonitrile-water (50 mL) resulted in pure stevioside, as shown by HPLC (tR, 3.51, same as standard stevioside). The acetonitrile solution was evaporated and dried to get pure stevioside, yield = 208 mg (68%).
Stevioside ketone (9). Stevioside peracetate ketone (500 mg, 0.393 mmol) was taken up in anhydrous methanol (10 mL). To this was added anhydrous sodium methoxide (200 mg). It was stirred at room temperature for 2 h. and then neutralized by 0.5% HCl to pH ~ 6.5–7.0. The methanol was removed on a rotavapor under reduced pressure followed by desalting on a pre-packed C-18 cartridge (500 mg) from Varian. The de-acetylated product was further purified via preparative HPLC on a C-18 column to get pure stevioside ketone, yield = 158 mg (55%). 1H-NMR (DMSO) δ = 1.212 (s, 3H, H-19), 0.983 (s, 3H, H-20); 13C-NMR: δ = 191.230 (C=O, C-16); ESI-MS m/z: 809.1 [M+H]+, Calcd. 807.85 for C37H58O19 [M+H] +.
Per-acetylated reb A (6). In a 500 mL round bottom flask, rebaudioside A (+99% by crystallization, 5 g, 5.17 mmol) was dried overnight in a vacuum oven at 75 οC. To this was added dry pyridine (150 mL) followed by cooling it to 0 οC and then addition of acetic anhydride (250 mL). It was allowed to warm up to room temperature and then stirred for 48 h. The reaction was then poured on a beaker containing ice and shaken vigorously. It was then extracted with diethyl ether (3 × 50 mL) followed by washing the organic layer with cold water (3 × 30 mL), 5% hydrochloric acid (3 × 30 mL), cold saturated sodium bicarbonate till no further evolution of CO2 was seen (3 × 40 mL) and then finally by brine (30 mL). The organic layer was dried over sodium sulfate and then evaporated to get the per-acetylated rebaudioside A as a single spot on TLC, yield = 6.5 g (80%). TLC (DCM–MeOH, 10:0.5): Rf = 0.36; 1H-NMR (DMSO) δ = 5.982 (d, 1H, H-21, J = 7.8 Hz); 13C-NMR δ = 152.890 (C=CH2, C-16), 104.292 (C=CH2, C-17); ESI-MS m/z: 1578.5 [M+Na]+, Calcd. 1578.52 for C72H98O37[M+Na]+.
Rebaudioside A Per-Acetate Ketone (8). Rebaudioside A peracetate (800 mg, 0.51 mmol) was taken up in a mixture of THF (4.0 mL) and water (3.5 mL). To this was added freshly made 4% aqueous solution of OsO4 (0.5 mL). After 15 minthe reaction mixture had become deep brown and to this NaIO4 (840 mg) was added slowly over a period of 2–3 min and the mixture was left for stirring at room temperature for 18 h. It had become a thick white suspension by now and was extracted with ethyl acetate (2 × 40 mL). Organic layer was dried over sodium sulfate and evaporated on a rotavapor at 30 °C under reduced pressure to get crude rebaudioside A per-acetate ketone that was chromatographed on silica gel 60 (70–230 mesh) with a slow stepwise gradient from pure dichloromethane to 2.5% methanol in dichloromethane, yield = 220 mg (30%). TLC (DCM–MeOH, 10:0.5): Rf = 0.28; 1H-NMR (DMSO) δ = 5.982 (d, 1H, H-21, J = 7.8 Hz), 13C-NMR δ = 215.749 (C=O, C-16), peaks at δ152.890 and 104.292 present in 6 disappear here; ESI-MS m/z: 1580.0 [M+Na]+, Calcd. 1580.5 for C71H96O38[M+Na]+.
Methylenation of Rebaudioside A Acetate Ketone and re-generation of reb A. Rebaudioside A per- acetate ketone (8, 233 mg, 0.149 mmol) was taken up in anhydrous dichloromethane (4 Ml). To this solution was slowly added under nitrogen, Zn/CH2Br2/TiCl4 reagent (4 mL, 8.7 equivalents, cold reagent). The reaction mixture was stirred at room temperature and was monitored by HPLC on an amino column (it was hard to interpret the TLC data due to the appearance of several spots below the actual ketone spot, which were probably the de-acetylation products). A series of aliquots of the reaction mixture were taken out at t = 18, t = 41 and t = 72 h (each ~400 μL, 7 μmol) to monitor the reaction. They were first quenched with sat. sodium bicarbonate and ethyl acetate (1:1) followed by isolation, drying and evaporation of ethyl acetate layer and then de-acetylation. On the basis of HPLC and LC-MS of t = 0 and t = 18, 41, 72 h. samples the reaction seemed complete after 72 h. The reaction was then quenched by pouring into a biphasic mixture of saturated sodium bicarbonate (10 mL) and ethyl acetate (5 mL). The aqueous layer was extracted two more times with ethyl acetate (5 mL ea.). The combined ethyl acetate layer was dried over sodium sulfate and evaporated on a rotavapor at 30 οC under reduced pressure to get a crude product which was only 37 mg. The crude was taken up in anhydrous methanol (4 mL). To this was added anhydrous sodium methoxide (19 mg). It was stirred at room temperature for 2 h. and then neutralized by 0.5% HCl to pH ~ 6.5–7.0. The methanol was removed on a rotavapor under reduced pressure. A 500 mg pre-packed C-18 cartridge from Varian was first treated with 50% acetonitrile-water and then washed with 15–20 mL double distilled water. The de-acetylated product was dissolved in minimum amount of water and loaded on C-18 column followed by washing with water (15 mL). Elution with 90% acetonitrile-water (15 mL) resulted in pure reb A as shown by HPLC. The acetonitrile solution was evaporated and dried to get pure reb A, yield = 17 mg (14.5% yield).
Reb A ketone (10). Rebaudioside A peracetate ketone (8, 233 mg, 0.149 mmol) was taken up in anhydrous methanol (4 mL). To this was added anhydrous sodium methoxide (19 mg). It was stirred at room temperature for 2 h. and then neutralized by 0.5% HCl to pH ~ 6.5–7.0. The methanol was removed on a rotavapor under reduced pressure followed by desalting on a pre-packed C-18 cartridge (500 mg) from Varian. The de-acetylated product was further purified via preparative HPLC on a C-18 column to get pure reb A ketone, yield = 31 mg (22%). 1H-NMR (DMSO) δ = 1.204 (s, 3H, H-19), 0.979 (s, 3H, H-20); 13C-NMR: δ = 215.457 (C=O, C-16); ESI-MS m/z: 992.1 [M+Na]+, Calcd. 991.98 for C43H68O24[M+Na]+.



{kind=link}
{kind=link}
{kind=link}

