1. Introduction
Plant phenols often occur as glycosides, such as flavonoid glycosides, and phenylethanoid glycosides. Phenol glycosides have the potential of deglycosylation and result in its aglycone metabolites
in vivo. The glycoside forms are hydrolyzed by β-glucosidases to the aglycone forms in the jejunum [
1,
2,
3], and the released aglycone forms are either absorbed intact by the intestine or further metabolized by intestinal microflora into several other products before absorption [
4,
5].
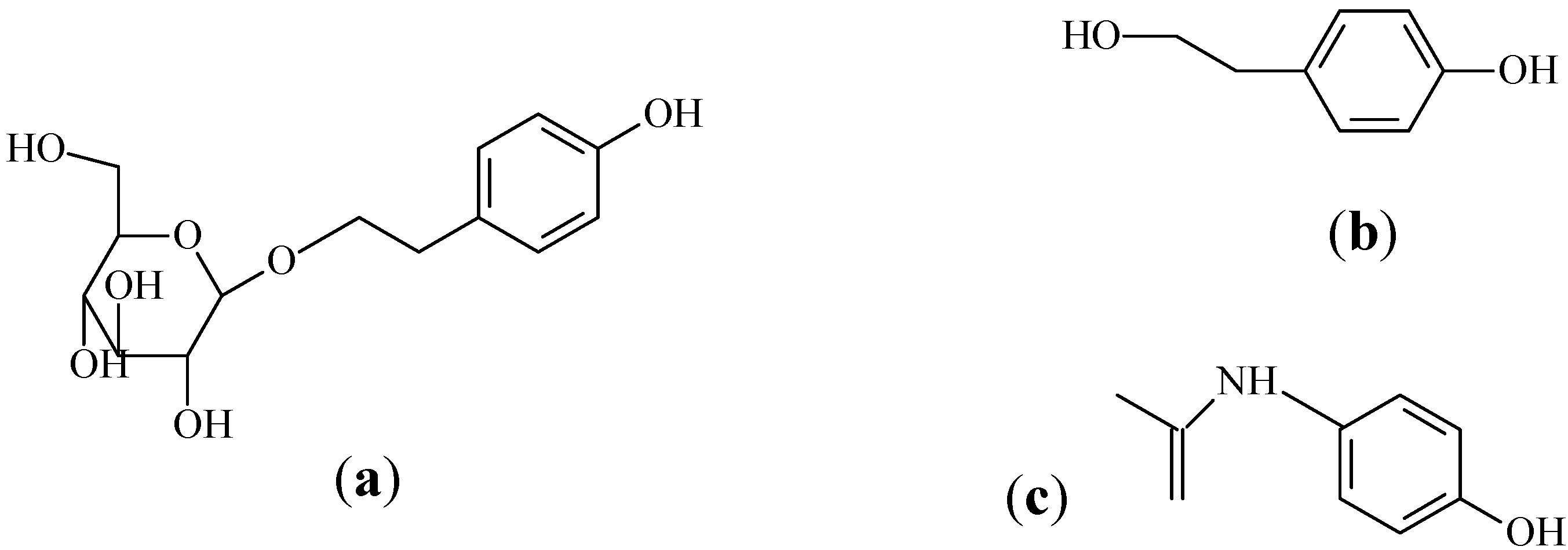
Salidroside (
p-hydroxyphenylethyl-
O-
β-D-glucopyranoside,
Figure 1a) and
p-tyrosol (the aglycone of salidroside,
Figure 1b) are two major phenols in the genus
Rhodiola. Salidroside, which possesses various pharmacological properties, is used as an adaptogen in traditional Tibetan medicines, it has also shown anti-inflammation [
6], resisting anoxia [
7], anti-aging [
8], antioxidative [
9], and anti-cancer activities [
10]
in vitro, and it also has hepatoprotective [
11] and cardioprotective effects [
12] in rats. Furthermore, the content of salidroside is one of the criteria to evaluate the medicinal quality of
Rhodiola [
13], as well as being one of active standard components for
Rhodiola rosea extract, which is valued as a strengthening tonic to increase physical and mental stamina and sold under different brand names on major websites (amazon.com, buy.com, and drugstore
etc.) and in drug stores (Walgreens and GNC) in the United States.
p-Tyrosol is also the most abundant biophenol in extra virgin olive oil [
14]. It has been proven to fully protect Caco-2 cells against the cytotoxic/apoptotic effects of ox LDL [
15], and to inhibit the activity of leukocyte 5-lipoxygenase in rat peritoneal mixed leukocytes [
16]. Moreover,
p-tyrosol could penetrate and accumulate in macrophages, and improve the intracellular antioxidant defense systems, even counteracting cardiovascular diseases [
14].
The oral or intravenous (i.v.) administration of salidroside and
Rhodiola extracts to rat or beagle dog and the pharmacokinetic parameters of salidroside have been reported [
13,
17,
18,
19]. The pharmacokinetic studies of
p-tyrosol via i.v. injection have also been elucidated [
20,
21]. The analysis methods of salidroside have been developed and applied to plant materials and biological matrices, including liquid chromatography with ultraviolet detection (LC-UV) [
18], liquid chromatography with mass spectrometry (LC-MS) [
17,
19], and liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) [
13,
22], and the simultaneous determination of salidroside and
p-tyrosol in
Rhodiola were established by HPLC [
23,
24]. However, in all these studies the metabolic characteristics of salidroside
in vivo has not been mentioned so far.
To explore whether the deglycosylation of salidroside into
p-tyrosol occurs
in vivo, and to elucidate the contribution of
p-tyrosol to the bioavailability of salidroside, in the present study intragastric gavage (i.g.) and i.v. administration of salidroside to rats were performed. The identification of salidroside and
p-tyrosol in plasma samples was conducted by HPLC with photodiode array (PDA) detector and LC-MS/MS, respectively. The PDA detector records a full UV spectrum of the contents of the detector flow cell in real-time and provides the possibility for identification. LC-MS/MS using Electrospray Ionization (ESI) followed by two stages of mass selection: a first stage (MS1) selecting the mass of the intact analyte (parent ion) and, after fragmentation of the parent by collision with gas atoms, a second stage (MS2) selecting a specific fragment of the parent, collectively generating a multiple reaction monitoring (MRM) assay. The mass filters produce a very specific and sensitive response for the selected analyte that can be used to detect and integrate a peak in a simple chromatographic separation of the sample [
25]. LC-MS/MS with MRM mode was also used for the following quantitative assay of salidroside and
p-tyrosol. Then, a specific and rapid LC-MS/MS method was developed and validated to simultaneously determine salidroside and
p-tyrosol with paracetamol (
Figure 1c) as the internal standard (IS). The pharmacokinetic studies of salidroside in rats after i.g. and i.v. administration were carried out afterwards.
Figure 1.
Chemical structures of (a) salidroside; (b) p-tyrosol; and (c) paracetamol (IS).
Figure 1.
Chemical structures of (a) salidroside; (b) p-tyrosol; and (c) paracetamol (IS).
3. Experimental
3.1. Chemicals and Reagents
Salidroside was purchased from the National Institutes for Food and Drug Control (Beijing, China). Paracetamol, the IS (purity ≥ 98%) was purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). p-Tyrosol and HPLC-grade acetonitrile was obtained from Sigma (St. Louis, MO, USA). Ultra pure water was produced by a Millipore Milli-Q system (Billerica, MA, USA). All other reagents or solvents used were commercially available and of reagent grade. Blank rat plasma was collected from healthy Male Wistar rats weighting 200 ± 20 g (Laboratory Animal Center of Jilin University, Changchun, Jilin Province, China).
3.2. Chromatographic and Mass Spectrometric Conditions
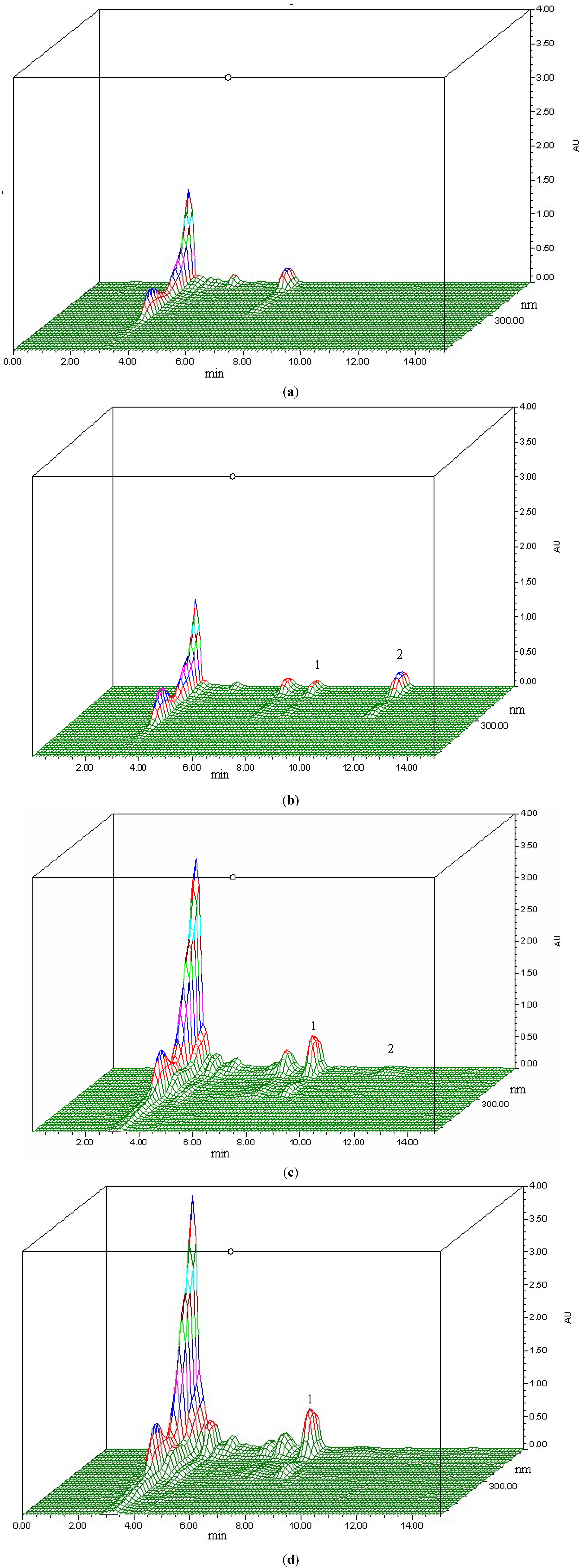
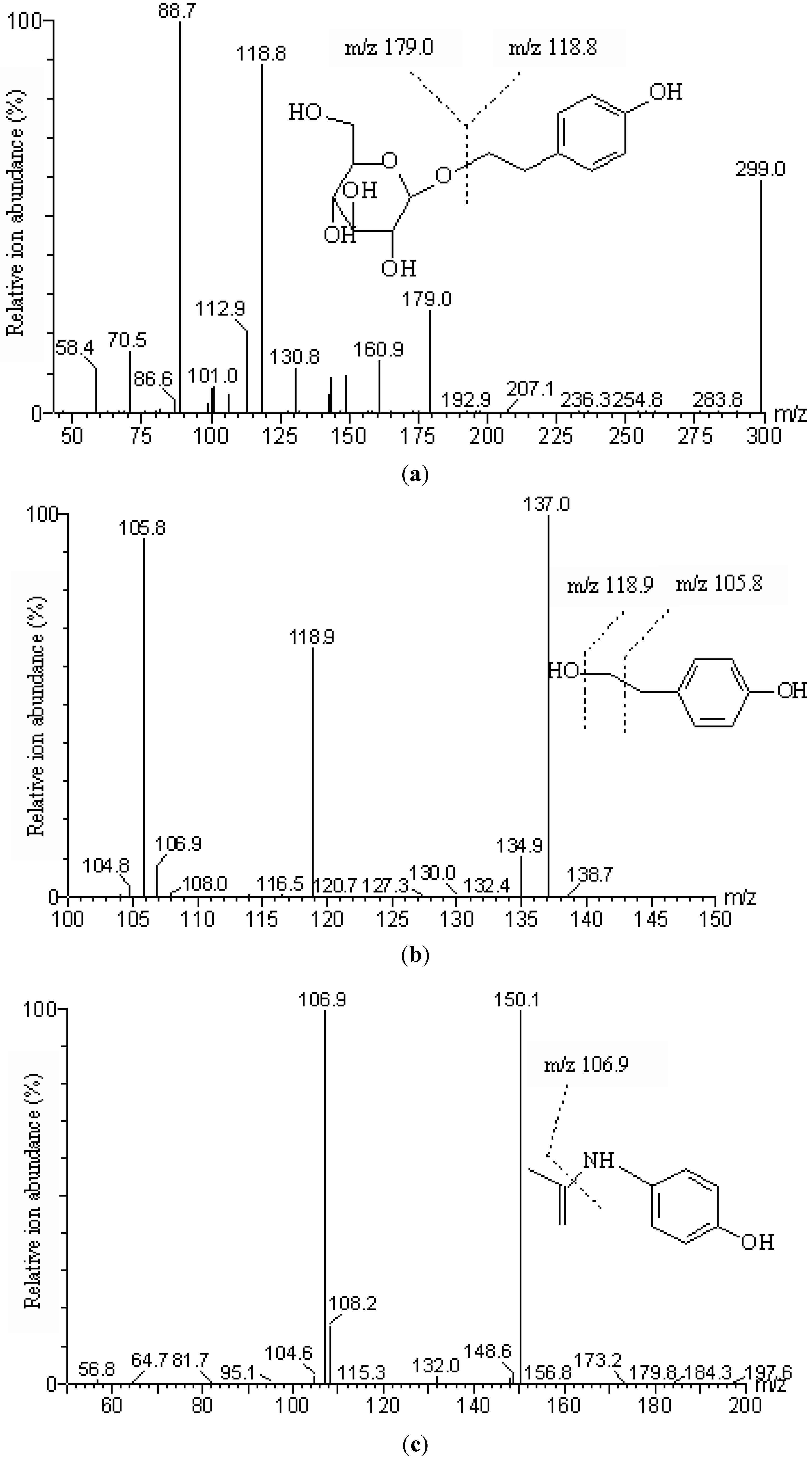
The identification of salidroside and p-tyrosol in plasma samples was conducted by HPLC with a PDA detector and LC-MS/MS. The quantitative analysis of salidroside and p-tyrosol was performed on a LC-MS/MS.
The HPLC system consisted of a 1525 HPLC Pump, a 717 plus autosampler, and a 2996 PDA detector (Waters Co., Milford, MA, USA). The column used for separation was a Thermo Hypersil Gold C18 column (4.6 mm × 250 mm, 5 μm, USA). The mobile phase was a mixture of acetonitrile and water (1/9, v/v).
The LC-MS/MS system was comprised of an Alliance 2695 HPLC and a Quattro-Micro
TM mass spectrometer (Waters Co.). An xTerra C18 mass column (3.5 µm, 50 × 3.0 mm, Waters Co., Milford, MA, USA) was used for separation, which was equilibrated and eluted with an isocratic mixture of acetonitrile-water (1:9, v/v) at a flow rate of 0.3 mL/min. The injection volume was 20 µL. The ESI-MS/MS detection was performed under negative ion mode under the following conditions: Capillary voltage 3.00 kV, extractor 2.00 V, RF Lens 0.1 V, desolvation gas 500 L/Hr, cone gas 50 L/Hr, source temperature 120 °C, desolvation temperature 350 °C, entrance voltage −2 V, exit voltage 1 V. ESI-MS/MS parameters were shown in
Table 1. The total run time was 3.5 min. The retention time of salidroside,
p-tyrosol and IS were 1.94 min, 2.73 min and 1.80 min respectively. Waters MassLynx 4.0 software was used for system control and data acquisition.
3.3. Preparation of Stock Solutions, Calibration Standard (CS) and Quality Control (QC) Samples
Stock solutions of salidroside, p-tyrosol and IS (paracetamol) were prepared at 200 μg/mL in acetonitrile-water (1:9, v/v), respectively, and further diluted with acetonitrile-water (1:9, v/v) to give a series of working solutions. All solutions were stored at −20 °C until use.
Calibration curves were prepared by spiking 20 μL of appropriate working solution with 100 μL of blank rat plasma. The effective concentrations were 50, 100, 200, 500, 1,000, 1,500 and 2,000 ng/mL for salidroside, and 20, 40, 80, 100, 140, 180, 200 ng/mL for p-tyrosol, respectively. QC samples were prepared in pool as a single batch for each concentration at concentrations of 50, 500 and 2,000 ng/mL for salidroside and of 20, 100, 200 ng/mL for p-tyrosol, and then divided into aliquots and stored in the freezer at −20 °C until use. The IS working solution of 250 ng/mL was diluted from stock solution as needed. The spiked rat plasma (CSs and QCs) were treated following the sample processing procedure as for the unknown samples.
3.4. Sample Processing
To identify salidroside and the potential metabolite p-tyrosol in plasma sample, an aliquot (500 µL) of plasma sample and methanol (1,500 µL) were mixed by vortex-mixing for 1 min followed by ultrasonic incubation for 10 min. After centrifuging at 45,000 g for 5 min, the clear supernatant was transferred into a new polypropylene tube and evaporated to dryness under a nitrogen stream. The residue was reconstituted in 200 µL of mobile phase. After filtering through a membrane (0.22 µm pore size), each 20 µL aliquot was injected into the HPLC-PDA or LC-MS/MS system.
The above sample preparation method was employed with minor modifications for pharmacokinetic studies. One hundred micro liters of plasma sample (blank plasma, spiked plasma or pharmacokinetics study plasma sample) and IS working solution (20 µL) was pipetted out into a 1.5 mL polypropylene tube, then methanol (280 µL) was added followed by vortex mixing for 1 min and 10 min of ultrasonic incubation. After centrifuging at 45,000 g for 5 min, the clear supernatant was transferred to a new tube and evaporated to dryness under a nitrogen stream. The residue was reconstituted in 200 µL of mobile phase. After centrifuging at 45,000 g for 10 min, an aliquot of 20 µL was injected into the LC-MS/MS system. When the concentration of salidroside in rat plasma was over the range of calibration curve, appropriate dilutions were applied to the plasma sample with blank rat plasma before sample processing.
3.5. Method Validation
The method for simultaneous determination of salidroside and p-tyrosol in rat plasma by LC-MS/MS was validated to meet the acceptance criteria as the guidance from Food Drug Administration (FDA) and per guidelines of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH).
The selectivity was assessed by comparing the chromatograms of six individual blank rat plasma samples with the corresponding spiked plasma samples. Each blank plasma sample was processed through the proposed extraction procedure and tested to ensure no interference of the analyte from the rat plasma.
Lower limit of quantification (LLOQ) was defined as 10 times the signal-to-noise (S/N) ratio, and limit of detection (LOD) was done as three times the S/N ratio. The lowest concentration on the calibration curve was to be accepted as the LLOQ, if the analyte response was at least 10 times more than that of blank plasma sample.
Linearity was assessed by assaying calibration curves ranging from 50 to 2,000 ng/mL for salidroside and 20–200 ng/mL for p-tyrosol in duplicate on six consecutive days. Plasma samples were quantified using the peak area ratio of salidroside or p-tyrosol to that of IS standard curves are in the form of y = A + Bx, where y represents the plasma concentration of analyte and x represents the ratio of analyte peak area to that of IS The acceptance criterion for a calibration curve was a correlation coefficient (r) of 0.99 or better, and each back-calculated standard concentration must be within 100 ± 15% except at LLOQ, for which the maximum acceptable deviation was at 20%.
Accuracy and precision were evaluated at three QC levels of 50, 500 and 2,000 ng/mL for salidroside and of 20, 100, 200 ng/mL for p-tyrosol. The assays of intra- or inter-day accuracy were preformed in six separate runs on the same day or on six consecutive days, and expressed as (observed concentration/spiked concentration) ×100%. Intra- and inter-day precisions were obtained by one-way analysis of variance (ANOVA) test, and were expressed as relative standard deviation (RSD). The accuracy was required to be within 100 ± 15% and the precision should not exceed ±15%.
The extraction recoveries of salidroside and p-tyrosol were determined at three QC levels, respectively. Recoveries were calculated by comparing the analyte/IS peak area ratios of each analyte in spiked plasma samples with those of analytes in the matrices by spiking extracted analyte-free plasma samples prior to chromatography.
The matrix effects from endogenous substances present in extracted rat plasma may cause ion suppression or enhancement of the signal. The matrix effects were investigated by post-extraction spike method in the present study. Peak area (A) of standard analyte in spiked blank plasma was compared with the corresponding peak area (B) obtained by directly injecting the standard analyte in the mobile phase at concentrations of 50, 500 and 2,000 ng/mL for salidroside and 20, 100 and 200 ng/mL for
p-tyrosol in triplicates. The peak area ratio of A/B (as a percentage) was used as a quantitative measure of the matrix effects [
33].
3.6. Stability
The stability of standard solutions was tested at room temperature for 2 h and upon refrigeration (4 °C) for 30 days. The stability of analytes was examined by keeping replicates of salidroside and p-tyrosol QC samples in the autosampler tray for 24 h and in a freezer at −20 °C for 30 days; the freeze-thaw stability was obtained over three freeze-thaw cycles, by thawing at room temperature for 2–3 h and then refreezing at −20 °C for 12–24 h. For each concentration and each storage condition, 6 replicates were analyzed in one analytical batch. The concentration of analytes after each storage period was related to the initial concentration, which was determined when the samples were originally prepared and processed.
3.7. Pharmacokinetic and Bioavailability Studies
Male rats (ICR, 200 ± 20 g) were obtained from Laboratory Animal Center of Jilin University (Changchun, Jilin Province, China). Animal handling procedures were according to standard operating procedure approved by the institutional animal care and use committee. All rats were dosed following an overnight fasting (except for water).
For the identification of salidroside and p-tyrosol, six male rats were divided into two groups. Rats in one group underwent an i.v. administration through the vena caudalis with sodium chloride solution of salidroside at the dose of 50 mg of salidroside (12.5 mg/mL in saline) per kg body weight (i.v. 50 mg/kg). Five min after administration, blood samples were collected in heparinized tubes via cardiac puncture. Rats in the other group received an i.g. dose of 100 mg of salidroside (25 mg/mL in saline) per kg body weight (i.g. 100 mg/kg), and blood samples were collected at 0.5 h after administration. Plasma was separated by centrifuging at 4,000 g for 30 min at 4 °C and stored frozen at −20 °C until analysis.
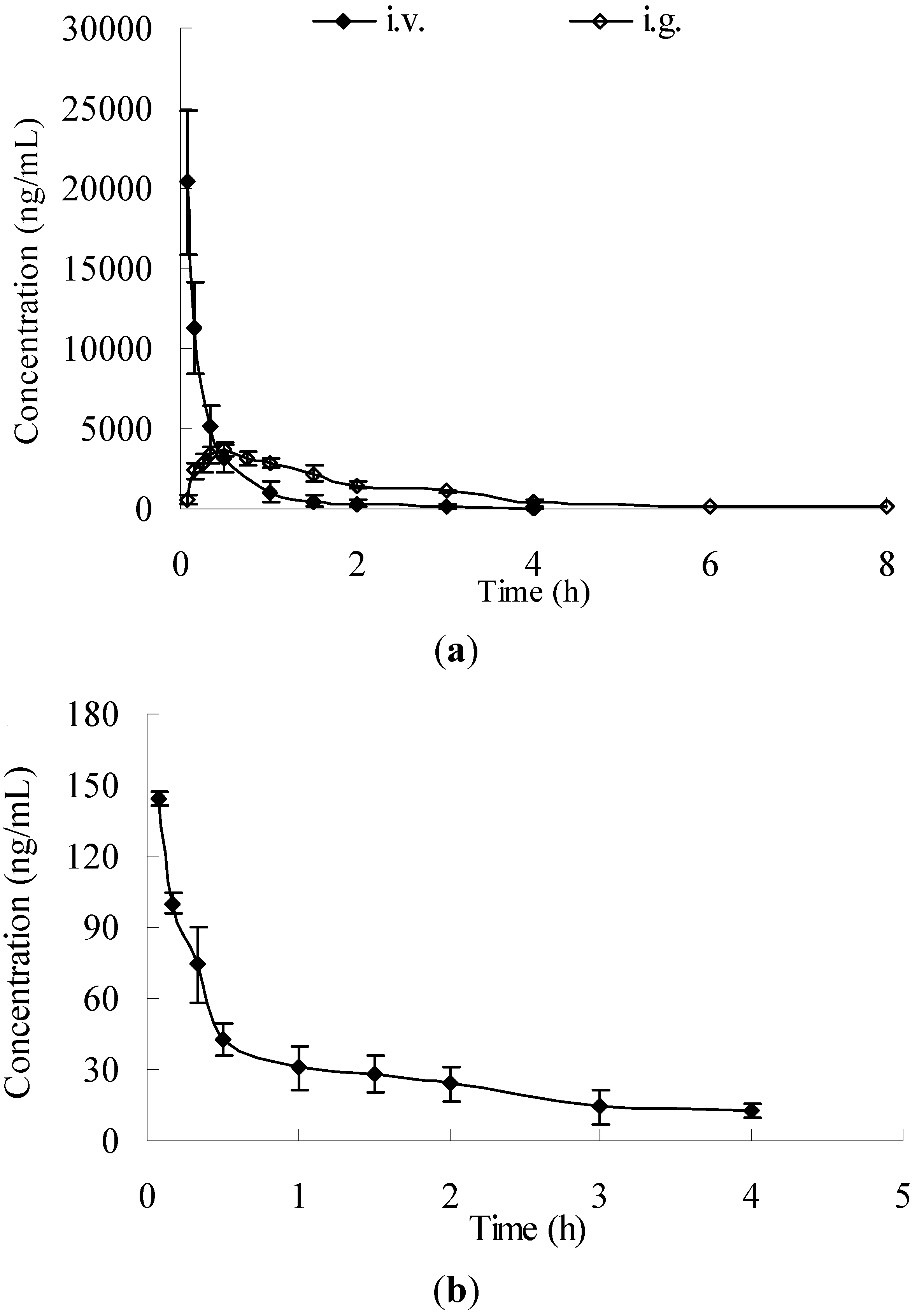
For pharmacokinetic study, 12 male rats underwent jugular vein cannulation [
34] and were randomly divided into two groups. Rats in group 1 were i.v. administered with 50 mg/kg. Serial blood samples (about 0.3 mL) were collected in heparinized tubes via the jugular vein before and at the time points of 0, 0.08, 0.17, 0.33, 0.5, 1.0, 1.5, 2.0, 3.0, 4.0 h after administration. In group 2, each rat received the administration of i.g. 100 mg/kg. Blood samples were collected before and at the time points of 0, 0.08, 0.17, 0.25, 0.33, 0.5, 0.75, 1.0, 1.5, 2.0, 3.0, 4.0, 6.0, 8.0 h. Plasma was separated and stored frozen at −20 °C until analysis.
A noncompartmental pharmacokinetic analysis using KineticaTM software package (version 5.0, Thermo Fisher Scientific Inc., Pittsburgh, PA, USA) was performed to determine the key parameters including maximum concentration (Cmax), time-to-maximum concentration (Tmax), half life time (T1/2), total body clearance (Cl), mean residence time (MRT), steady state apparent volume of distribution (Vss), area under curve from zero to the last measurable plasma concentration point (AUC0−t, t = 4.0 h for i.v. administration, t = 8.0 h for i.g. administration), and area under the plasma concentration-time curve from zero to time infinity (AUC0–∞).
The oral bioavailability (F) is defined as the fraction of unchanged drug reaching the systemic circulation following administration through the i.g. route. The absolute oral bioavailability of a drug is generally measured by comparing the respective AUCs after i.g. and i.v. administration according to the following equation [
28]:





{kind=link}
{kind=link}
{kind=link}
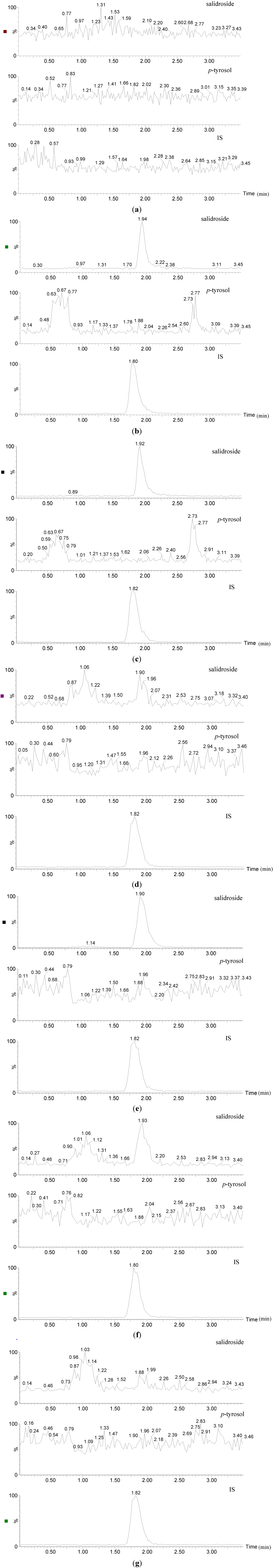{kind=link}
{kind=link}


