Kinetic Study of Oxygen Adsorption over Nanosized Au/γ-Al2O3 Supported Catalysts under Selective CO Oxidation Conditions

Abstract
:1. Introduction
2. Results and Discussion
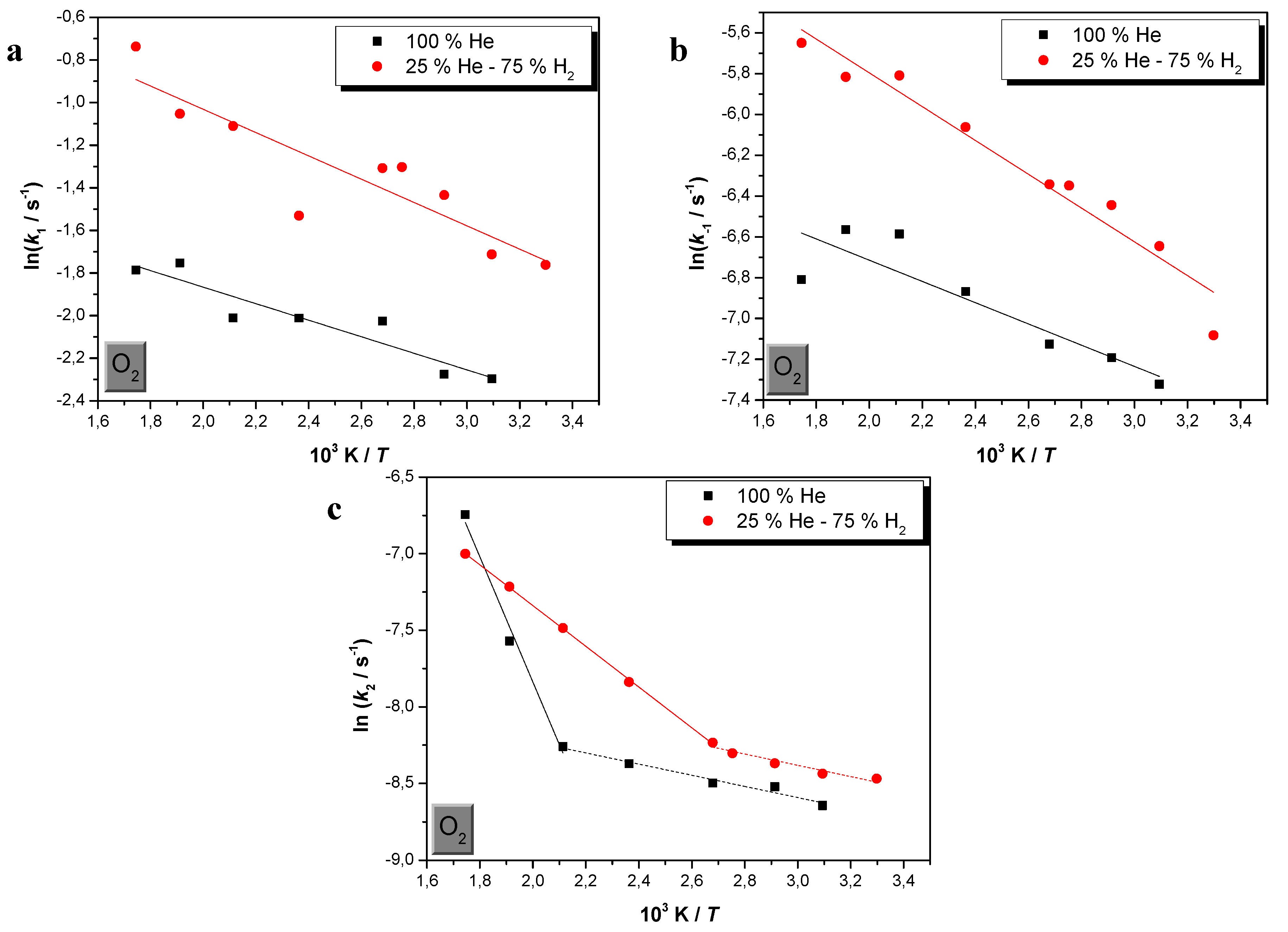
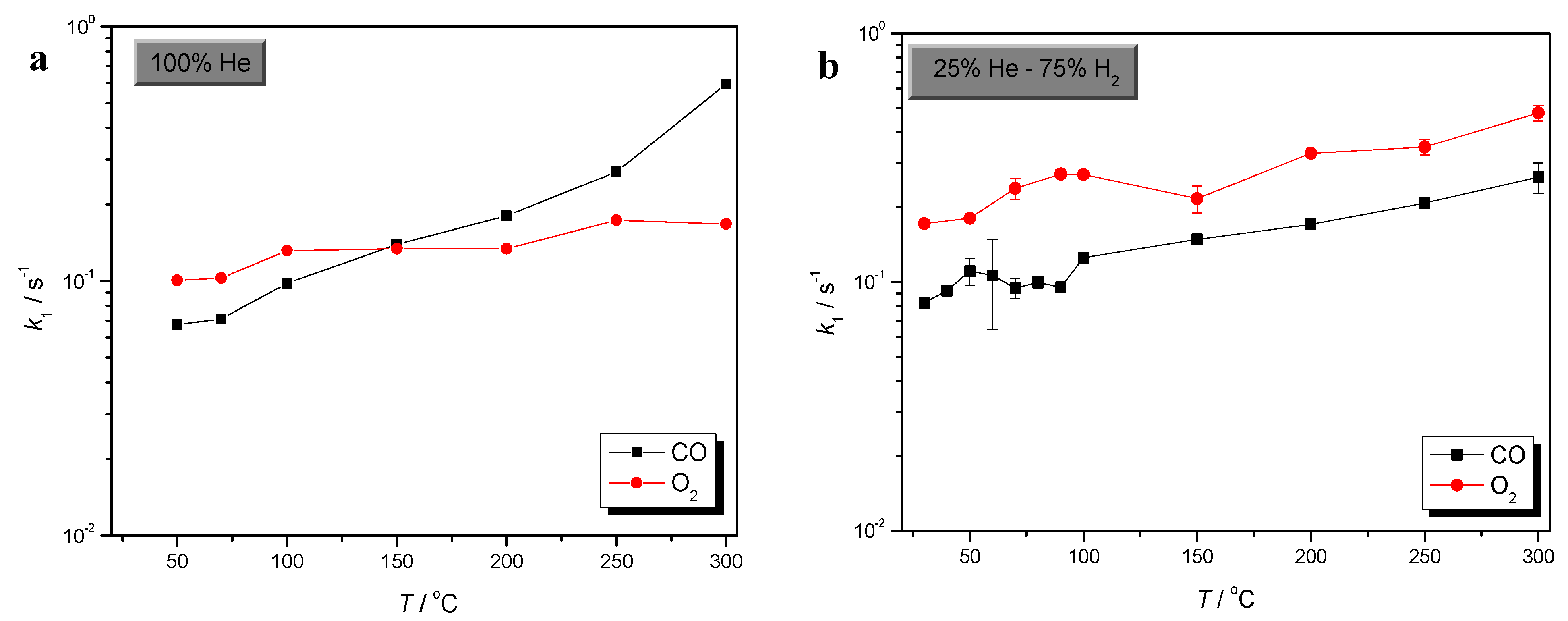
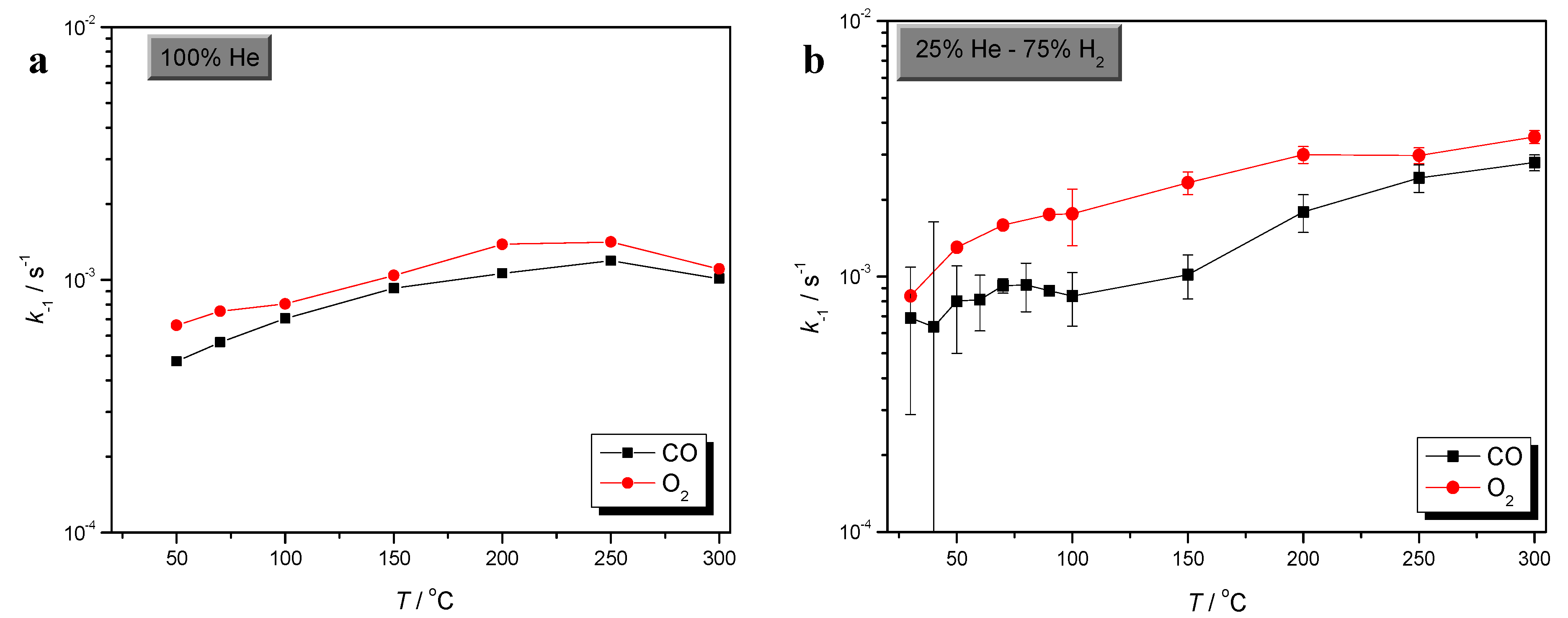
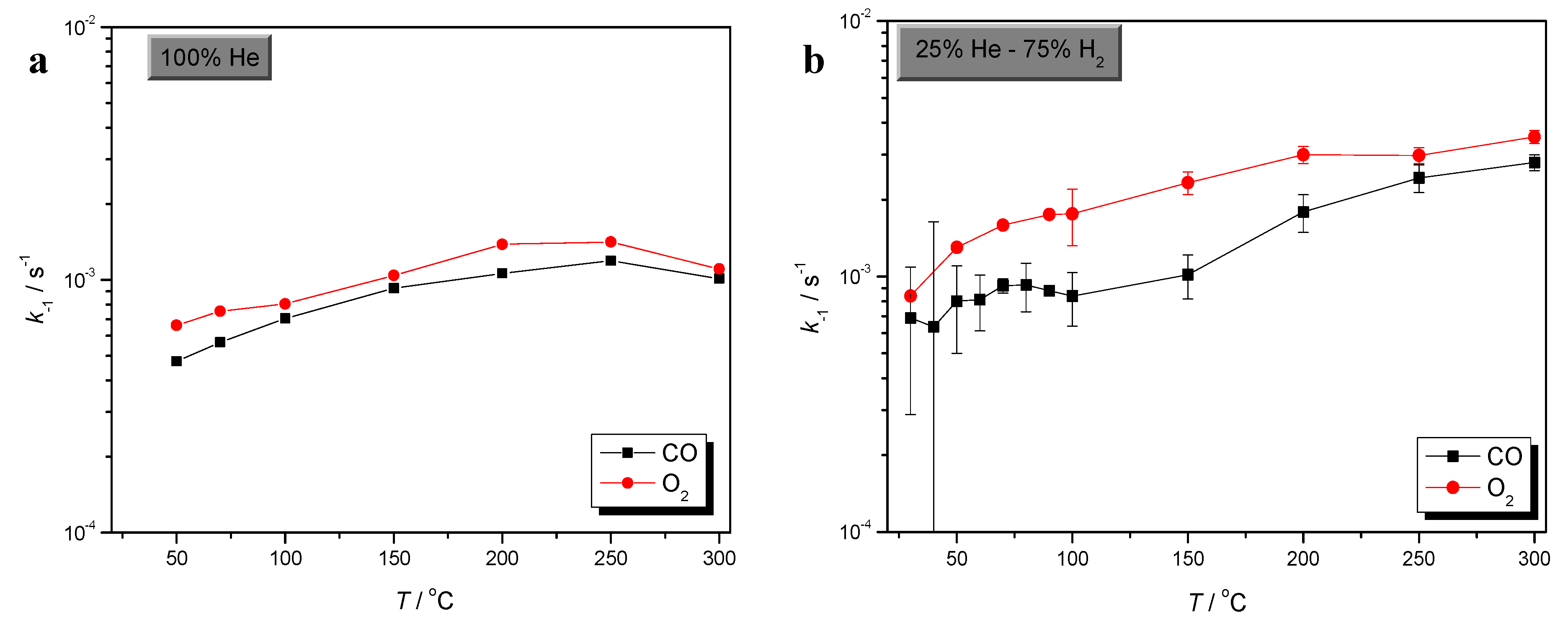
2.1. Effects of Temperature and H2 in O2 Sorption
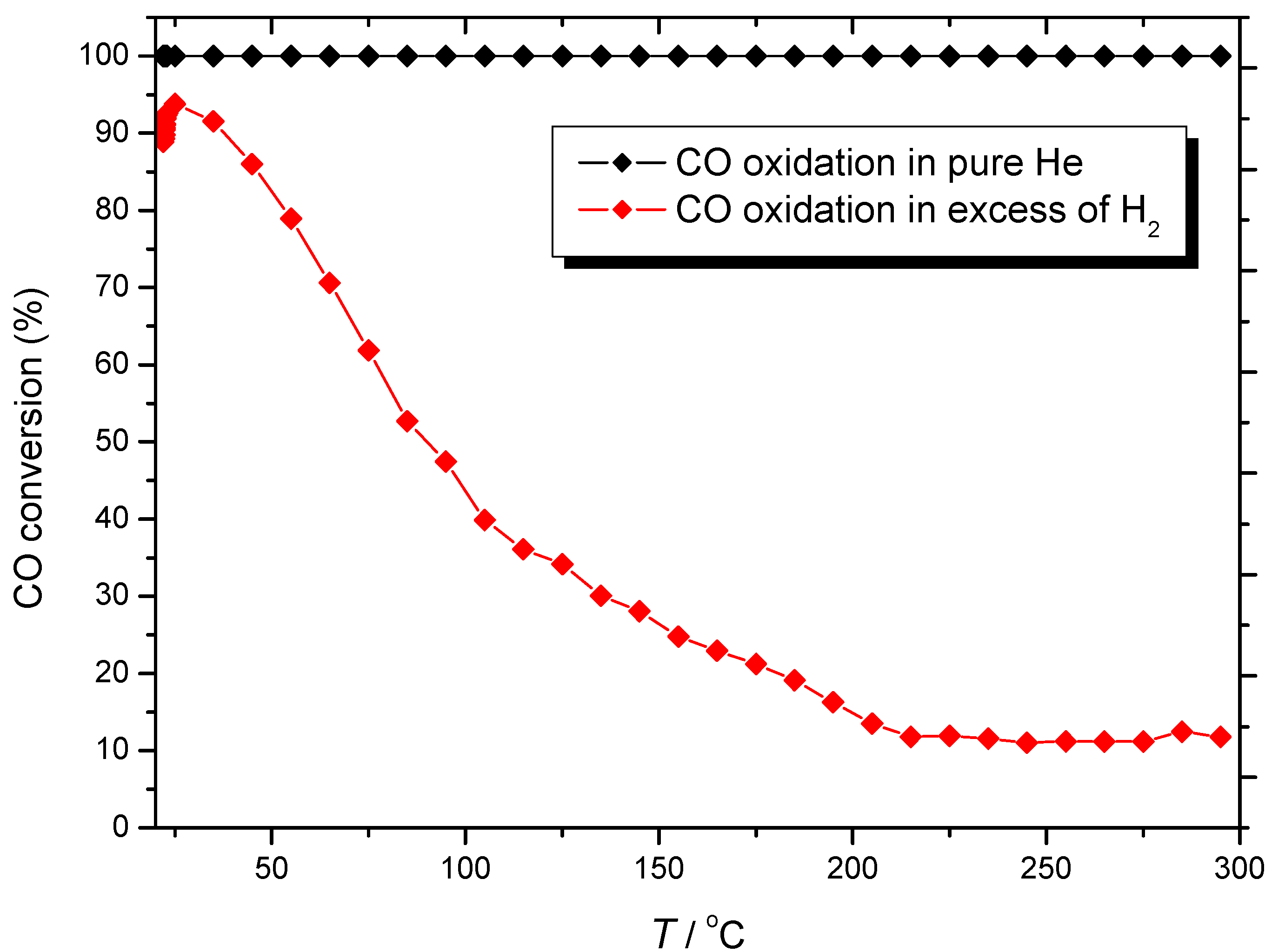
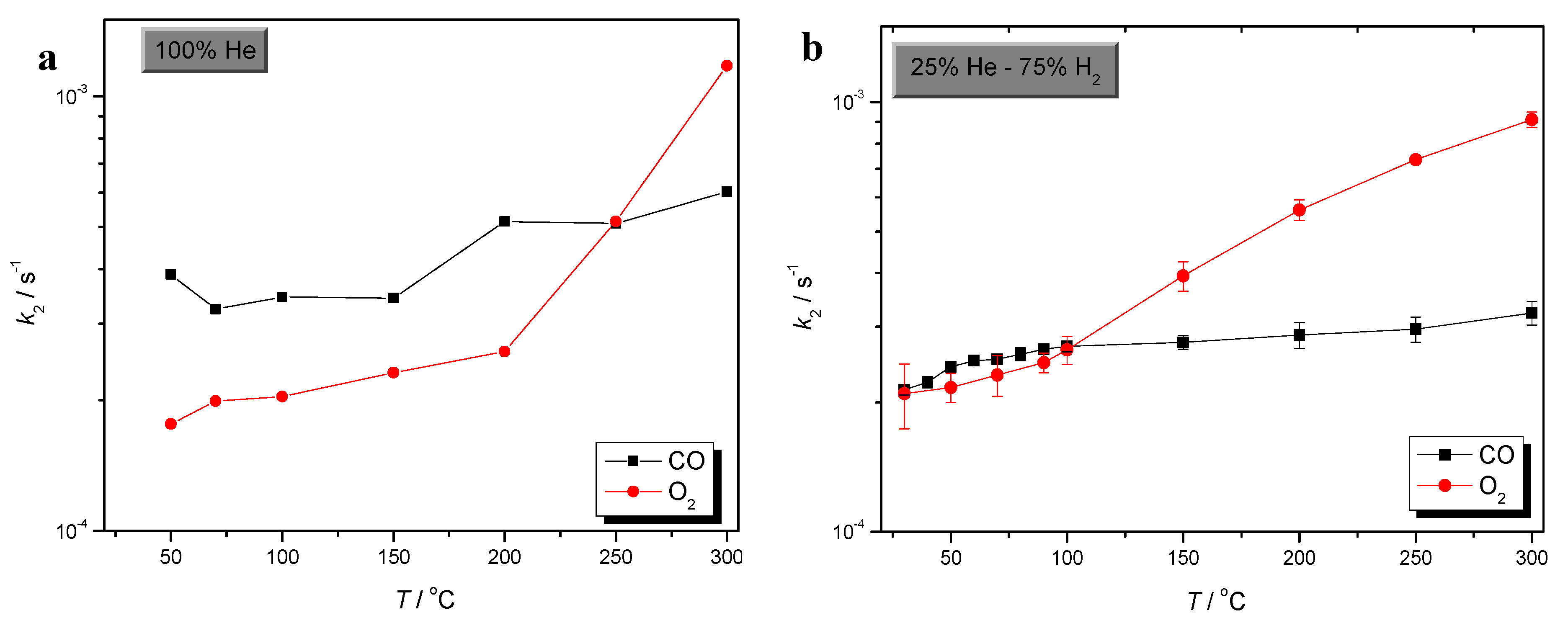
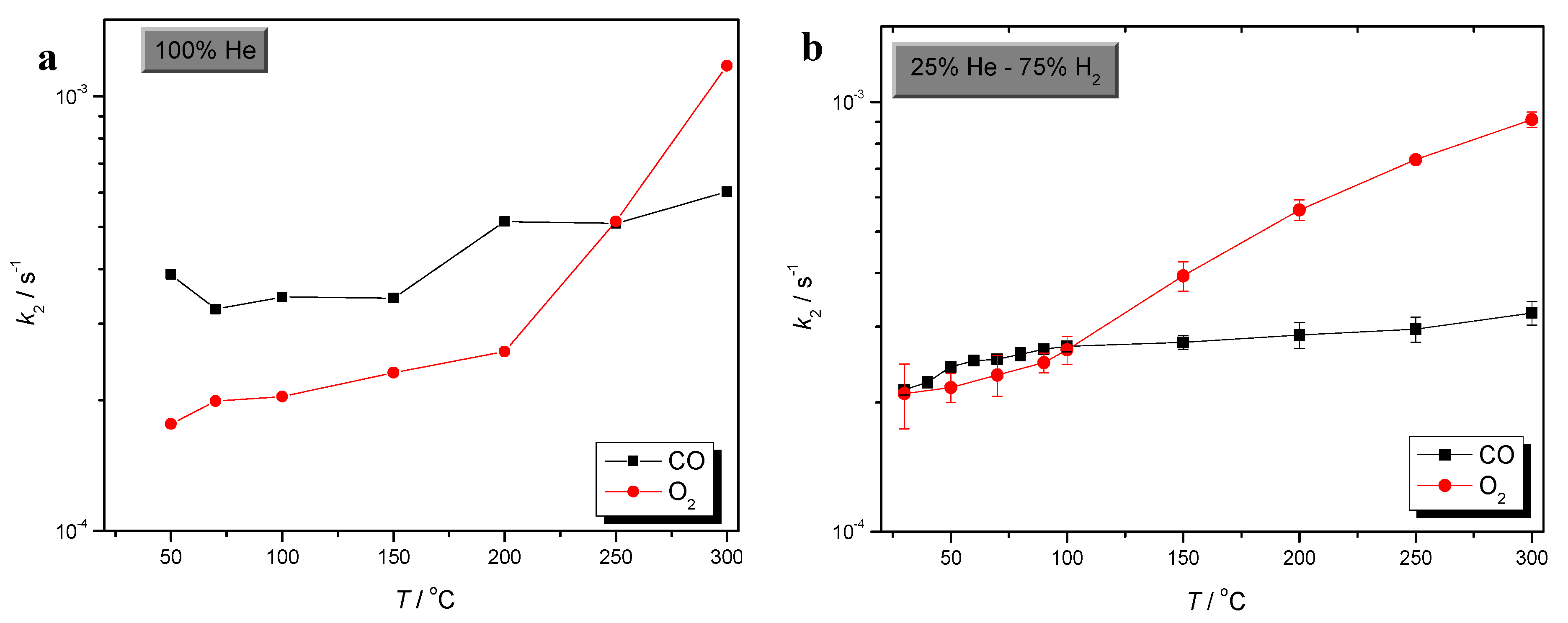
 ) CO/O2 = 1:1 in a H2-rich environment.
) CO/O2 = 1:1 in a H2-rich environment.
) CO/O2 = 1:1 in a H2-rich environment.
) CO/O2 = 1:1 in a H2-rich environment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % H2 | T/°C | k1/s−1 | % error * | 103k−1/s−1 | % error * | 103k2/s−1 | % error * |
|---|---|---|---|---|---|---|---|
| 50 | 0.09 | 0.69 | 0.17 | ||||
| 70 | 0.11 | 0.68 | 0.19 | ||||
| 100 | 0.16 | 0.73 | 0.20 | ||||
| 0 | 150 | 0.12 | 1.09 | 0.23 | |||
| 200 | 0.10 | 1.44 | 0.26 | ||||
| 250 | 0.19 | 1.53 | 0.51 | ||||
| 300 | 0.18 | 1.10 | 1.18 | ||||
| 30 | 0.17 ± 0.006 | 3.5 | 0.84 ± 0.022 | 2.6 | 0.21 ± 0.036 | 17 | |
| 50 | 0.18 ± 0.003 | 1.7 | 1.30 ± 0.017 | 1.3 | 0.22 ± 0.017 | 7.7 | |
| 70 | 0.24 ± 0.023 | 9.6 | 1.59 ± 0.024 | 1.5 | 0.23 ± 0.025 | 11 | |
| 90 | 0.27 ± 0.012 | 4.4 | 1.75 ± 0.036 | 2.1 | 0.25 ± 0.013 | 5.2 | |
| 75 | 100 | 0.27 ± 0.008 | 3.0 | 1.76 ± 0.044 | 2.5 | 0.27 ± 0.020 | 7.4 |
| 150 | 0.22 ± 0.027 | 12 | 2.33 ± 0.236 | 10 | 0.39 ± 0.031 | 7.9 | |
| 200 | 0.33 ± 0.010 | 3.0 | 3.00 ± 0.231 | 7.7 | 0.56 ± 0.031 | 5.5 | |
| 250 | 0.35 ± 0.025 | 7.1 | 2.98 ± 0.215 | 7.2 | 0.74 ± 0.009 | 1.2 | |
| 300 | 0.49 ± 0.035 | 7.7 | 3.52 ± 0.206 | 5.9 | 0.91 ± 0.038 | 4.2 | |
| Average uncertainty % | 5.8 | 4.5 | 7.5 | ||||


| % H2 | Εa1/kJ·mol−1 | Εa−1/kJ·mol−1 | Εa2/kJ·mol−1 | |
|---|---|---|---|---|
| 0 | 3.2 ± 0.5 (0.864) | 4.3 ± 1.0 (0.765) | 3.0 ± 0.3 * (0.961) | 34.0 ±3.6 * (0.977) |
| 75 | 4.6 ± 0.8 (0.781) | 6.9 ± 0.6 (0.942) | 3.1 ± 0.5 ** (0.917) | 11.1 ± 0.1 ** (0.999) |
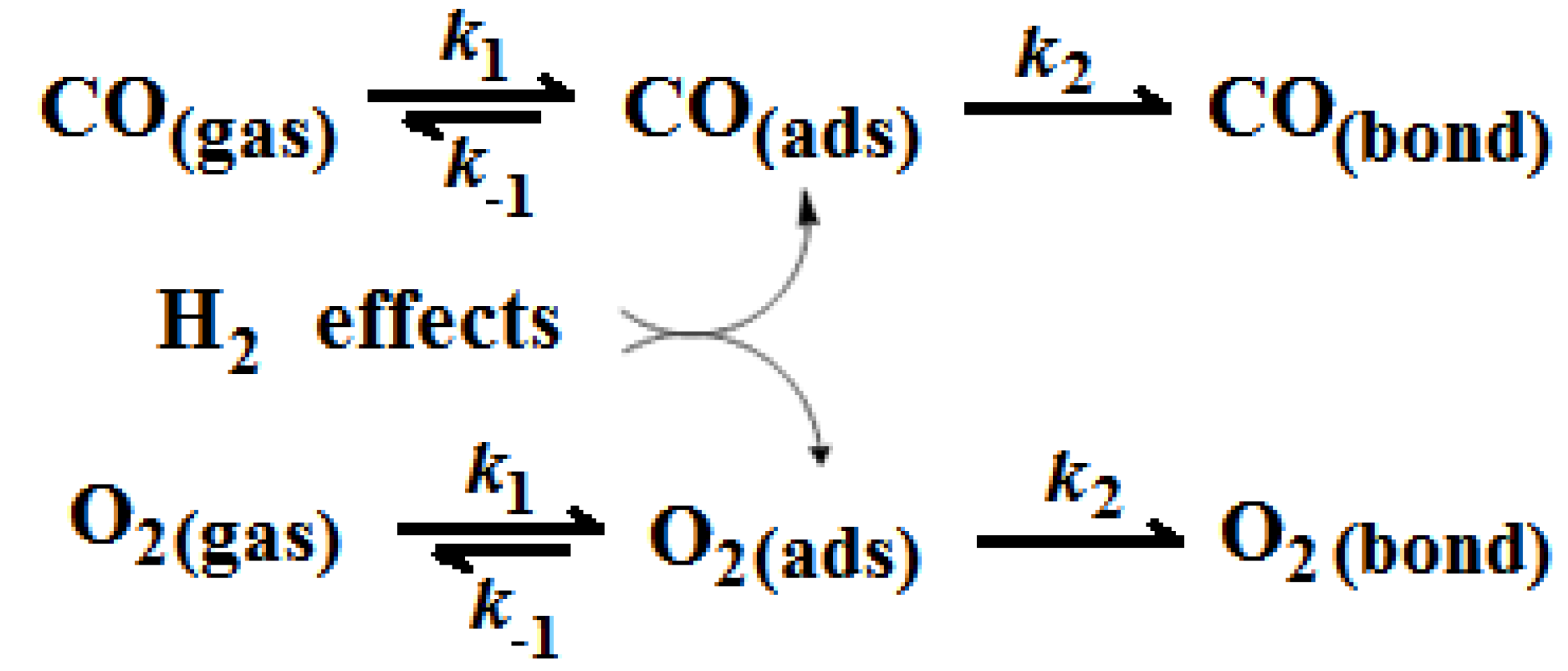
2.2. Comparison of CO and O2 Sorption

3. Experimental
3.1. Preparation of Nanometer Size Au Catalysts
3.2. Materials
3.3. Kinetic Measurements

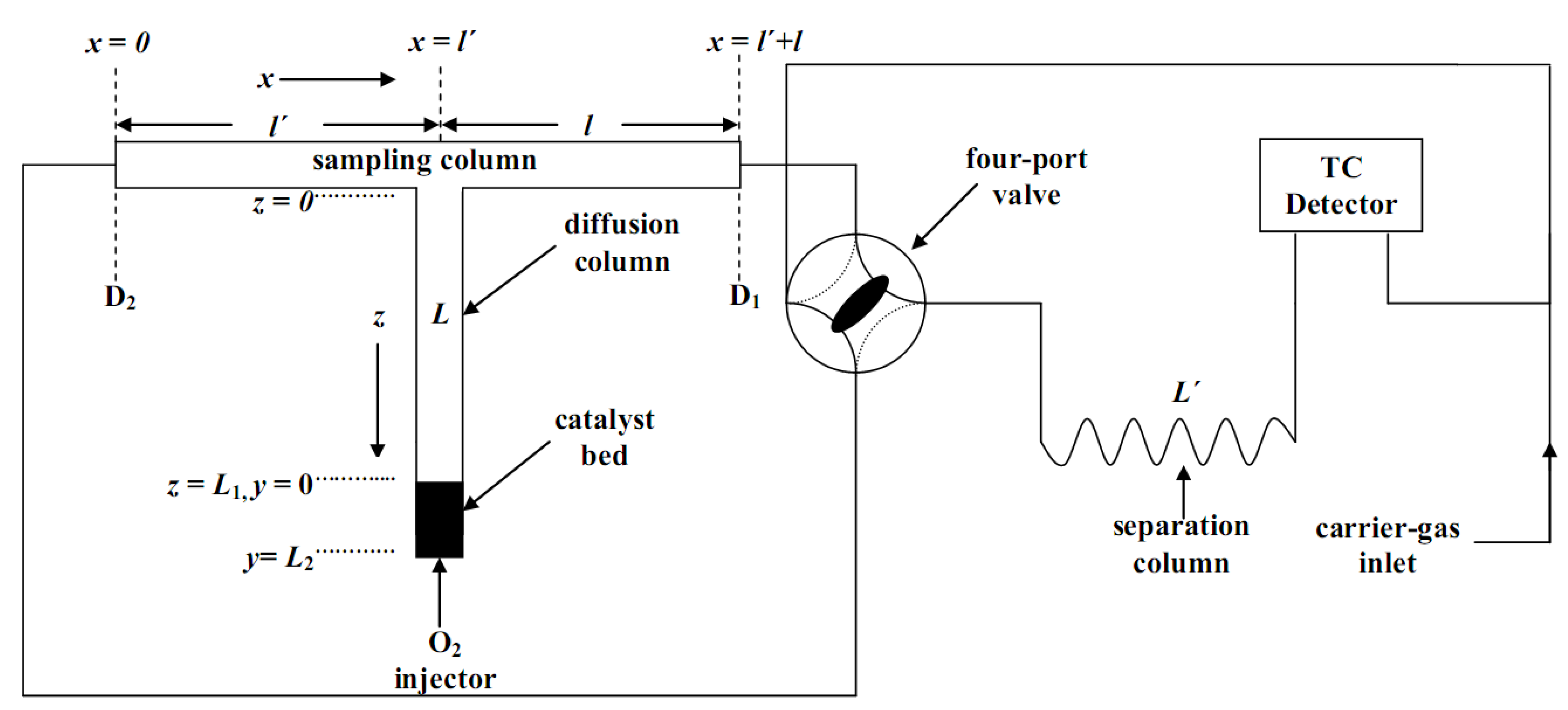
3.3.1. Apparatus and Procedure
 , Ɩ and L of the stainless-steel “sampling cell” were incorporated into a commercial gas chromatograph, Shimadzu GC-8A (Tokyo, Japan), equipped with a thermal conductivity detector as shown in Figure 6. The lengths and Ɩ of the sampling column were 36.5 cm each (4 mm ID), while the length L of the diffusion column was 70 cm (4 mm ID). The catalytic bed (0.09 g) was put in a 1 cm length at the closed end of the diffusion column L. The separation column,
, Ɩ and L of the stainless-steel “sampling cell” were incorporated into a commercial gas chromatograph, Shimadzu GC-8A (Tokyo, Japan), equipped with a thermal conductivity detector as shown in Figure 6. The lengths and Ɩ of the sampling column were 36.5 cm each (4 mm ID), while the length L of the diffusion column was 70 cm (4 mm ID). The catalytic bed (0.09 g) was put in a 1 cm length at the closed end of the diffusion column L. The separation column,  (4 mm ID), was filled with 7.6 g of silica gel (80–100 mesh)., Ɩ and . When the gas flow was restored to its original direction sample peaks, similar to those of Figure 2 of Ref. [67], were recorded. Repeating the above reversal procedure many times at each temperature, a whole series of sample peaks were recorded, corresponding to a different time from adsorbate injection. The working temperature for the chromatographic material was kept constant at 85 °C. The variation in the temperature along the catalytic bed was measured by a Fluke 2190A digital thermometer and was smaller than 1 K. The volumetric carrier gas flow rate, at ambient temperature was 1.136 cm3s−1. The pressure drop along the whole system was 0.33 atm. Plots of the height of the sample peaks against the corresponding time from adsorbate’s injection produce the so-called diffusion band”, like those shown in Figure 3 of ref. [67].
(4 mm ID), was filled with 7.6 g of silica gel (80–100 mesh)., Ɩ and . When the gas flow was restored to its original direction sample peaks, similar to those of Figure 2 of Ref. [67], were recorded. Repeating the above reversal procedure many times at each temperature, a whole series of sample peaks were recorded, corresponding to a different time from adsorbate injection. The working temperature for the chromatographic material was kept constant at 85 °C. The variation in the temperature along the catalytic bed was measured by a Fluke 2190A digital thermometer and was smaller than 1 K. The volumetric carrier gas flow rate, at ambient temperature was 1.136 cm3s−1. The pressure drop along the whole system was 0.33 atm. Plots of the height of the sample peaks against the corresponding time from adsorbate’s injection produce the so-called diffusion band”, like those shown in Figure 3 of ref. [67].3.3.2. Theoretical
 , of the sampling cell, at time, t:
, of the sampling cell, at time, t:

 and
and  , are calculated by a non-linear regression analysis program. The auxiliary parameters X, Y and Z are functions of the rate constants k1, k−1 and k2 as well as of the geometric characteristics of the diffusion column and the diffusivity of the adsorbate into the carrier gas. By adding the two exponential coefficients of time B1 and B2, the value of X is found, while subtracting B1 and B2, the value of Y is obtained. The value of Z is found from the ratio of the two pre-exponential factors A1 and A2 (
, are calculated by a non-linear regression analysis program. The auxiliary parameters X, Y and Z are functions of the rate constants k1, k−1 and k2 as well as of the geometric characteristics of the diffusion column and the diffusivity of the adsorbate into the carrier gas. By adding the two exponential coefficients of time B1 and B2, the value of X is found, while subtracting B1 and B2, the value of Y is obtained. The value of Z is found from the ratio of the two pre-exponential factors A1 and A2 (  ):
):





 are the gaseous volumes of empty sections L1 and L2, respectively, a1, a2 functions of the diffusion coefficient, D, of the adsorbate at the experimental temperature and pressure and ε is the external porosity of catalyst bed.
are the gaseous volumes of empty sections L1 and L2, respectively, a1, a2 functions of the diffusion coefficient, D, of the adsorbate at the experimental temperature and pressure and ε is the external porosity of catalyst bed.3.3.3. Precision Analysis of Rate Constants Determination by RF-IGC
4. Conclusions
4.1. Effects of Temperature and H2 in O2 Sorption
4.2. Comparison of CO and O2 Sorption
Acknowledgements
Conflict of Interest
- Sample Availability: Samples of the catalysts are available from the authors.
References and Notes
- Haruta, M. Preparation and environmental applications of supported gold catalysts. Now Future 1992, 7, 13–20. [Google Scholar]
- Hutchings, G.J. Catalysis: A golden future. Gold Bull. 1996, 29, 123–130. [Google Scholar] [CrossRef]
- Haruta, M. Size and support dependency in the catalysis by gold. Catal. Today 1997, 36, 153–166. [Google Scholar]
- Thompson, D.T. New advances in gold catalysis. Gold Bull. 1998, 31, 111–118, 1999, 32, 12-19.. [Google Scholar]
- Bond, G.C.; Thompson, D.T. Catalysis by gold. Cat. Rev. Sci. Eng. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Gold catalysed oxidation of carbon monoxide. Gold Bull. 2000, 33, 41–50. [Google Scholar] [CrossRef]
- Haruta, M.; Date, M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A Gen. 2001, 222, 427–437. [Google Scholar] [CrossRef]
- Thompson, D.T. A golden future for catalysis. Chem. Br. 2001, 37, 43–44. [Google Scholar]
- Haruta, M. Catalysis of gold nanoparticles deposited on metal oxides. Cattech 2002, 6, 102–115. [Google Scholar] [CrossRef]
- Hutchings, G.J. Gold catalysis in chemical processing. Catal. Today 2002, 72, 11–17. [Google Scholar] [CrossRef]
- Cortie, M.B.; van der Lingen, E. Catalytic gold nanoparticles. Mater. Forum 2002, 26, 1–14. [Google Scholar]
- Haruta, M. When gold is not noble: Catalysis by nanoparticles. Chem. Record 2003, 3, 75–87. [Google Scholar] [CrossRef]
- Thompson, D.T. Perspective on industrial and scientific aspects of gold catalysis. Appl. Catal. A Gen. 2003, 243, 201–205. [Google Scholar] [CrossRef]
- Cameron, D.; Holliday, R.; Thompson, D. Gold’s future role in fuel cell systems. J. Power Sources 2003, 118, 298–303. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Homogeneous catalysis by gold. Gold Bull. 2004, 37, 51–65. [Google Scholar] [CrossRef]
- Hutchings, G.J. Catalysts: Golden opportunities. Chem. Eng. 2004, 34–36. [Google Scholar]
- Pattrick, G.; van der Lingen, E.; Corti, C.W.; Holliday, R.J.; Thompson, D.T. The potential for use of gold in automotive pollution control technologies: A short review. Topics Catal. 2004, 30–31, 273–279. [Google Scholar]
- Thompson, D.T. Catalysis by gold/platinum group metals. Plat. Metals Rev. 2004, 48, 169–172. [Google Scholar] [CrossRef]
- Corti, C.W.; Holliday, R.J.; Thompson, D.T. Commercial aspects of gold catalysis. Appl. Catal. A Gen. 2005, 291, 253–261. [Google Scholar] [CrossRef]
- Hutchings, G.J. Catalysis by gold. Catal. Today 2005, 100, 55–61. [Google Scholar]
- Hashmi, A.S.K. The catalysis gold rush: New claims. Angew Chem. Int. Ed. 2005, 44, 6990–6993. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Status of catalysis by gold following an AURICAT workshop. Appl. Catal. A Gen. 2006, 302, 1–4. [Google Scholar] [CrossRef]
- Thompson, D.T. An overview of gold-catalysed oxidation processes. Topics Catal. 2006, 38, 231–240. [Google Scholar]
- Carabineiro, S.A.C.; Thompson, D.T. Catalytic Applications for Gold Nanotechnology. In Nanocatalysis; Heiz, U., Landman, U., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2007; pp. 377–489. [Google Scholar]
- Hashmi, A.S.K.; Hutchings, G.J. Gold catalysis. Angew Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef]
- Della Pina, C.; Falletta, E.; Rossi, M. Update on selective oxidation using gold. Chem. Soc. Rev. 2012, 41, 350–369. [Google Scholar]
- Liu, Z.-P.; Hu, P.; Alavi, A. Catalytic role of gold in gold-based catalysts: A density functional theory study on the CO oxidation on gold. J. Am. Chem. Soc. 2002, 124, 14770–14779. [Google Scholar]
- Hussain, A.; Gracia, J.; Niemantsverdriet, J.W.; Nieuwenhuys, B.E. The beneficial effect of hydrogen on CO oxidation over Au catalysts. A computationalstudy. Molecules 2011, 16, 9582–9599. [Google Scholar] [CrossRef]
- Kandoi, S.; Gokhale, A.A.; Grabow, L.C.; Dumesic, J.A.; Mavrikakis, M. Why Au and Cu are more selective than Pt for preferential oxidation of CO at low temperature. Catal. Lett. 2004, 93, 93–100. [Google Scholar] [CrossRef]
- Piccolo, L.; Daly, H.; Valcarcel, A.; Meunier, F.C. Promotional effect of H2 on CO oxidation over Au/TiO2 studied by operando infrared spectroscopy. Appl. Catal. B-Environ. 2009, 86, 190–195. [Google Scholar]
- Chen, M.S.; Goodman, D.W. The structure of catalytically active Au OnTitania. Science 2004, 306, 252–255. [Google Scholar]
- Carretin, S.; Concepcion, P.; Corma, A.; Lopez-Nieto, J.M.; Puntes, V.F. Nanocrystalline CeO2 increases the activity of Au for CO oxidation by two orders of magnitude. Angew. Chem. Int. Ed. 2004, 43, 2538–2540. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.V.J.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Nørskov, J.K. On the origin of the catalytic activity of nanometer gold particles for low temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar]
- Schumacher, B.; Denkwitz, Y.; Plzak, V.; Kinne, M.; Behm, R.J. Kinetics, mechanism and the influence of H2 on the CO oxidation reaction on a Au/TiO2 catalyst. J. Catal. 2004, 224, 449–462. [Google Scholar]
- Rossignol, C.; Arrii, S.; Morfin, F.; Piccolo, L.; Caps, V.; Rousset, J.L. Selective oxidation of CO over model gold-based catalysts in the presence of H2. J. Catal. 2005, 230, 476–483. [Google Scholar]
- Guzman, J.; Carrettin, S.; Corma, A. Spectroscopic evidence for the supply of reactive oxygen during CO oxidation catalyzed by gold supported on nanocrystalline CeO2. J. Am. Chem. Soc. 2005, 127, 3286–3287. [Google Scholar] [CrossRef]
- Remediakis, I.N.; Lopez, N.; Nørskov, J.K. CO oxidation on rutile-supported Au nanoparticles. Angew. Chem. Int. Ed. 2005, 44, 1824–1826. [Google Scholar] [CrossRef]
- Hernandez, N.C.; Sanz, J.F.; Rodriguez, J.A. Unravelling the origin of the high-catalytic activity of supported Au: A density-functional theory-based interpretation. J. Am. Chem. Soc. 2006, 128, 15600–15601. [Google Scholar]
- Weiher, N.; Beesley, A.M.; Tsapatsaris, N.; Delannoy, L.; Louis, C.; van Bokhoven, J.A.; Schroeder, S.L.M. Activation of oxygen by metallic gold in Au/TiO2 catalysts. J. Am. Chem. Soc. 2007, 129, 2240–2241. [Google Scholar]
- Janssens, T.V.W.; Clausen, B.S.; Hvolbaek, B.; Falsig, H.; Christensen, C.H.; Bligaard, T.; Nørskov, J.K. Insights into the reactivity of supported Au nanoparticles: Combining theory and experiments. Top. Catal. 2007, 44, 15–26. [Google Scholar]
- Fu, Q.; Kudriavtseva, S.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Gold-ceria catalysts for low-temperature water-gas shift reaction. Chem. Eng. J. 2003, 93, 41–53. [Google Scholar] [CrossRef]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active non-metallic Au and Pt species on ceria-based water-gas shift catalysts. Science 2003, 301, 935–938. [Google Scholar]
- Gonzalez-Arellano, C.; Corma, A.; Iglesias, M.; Sanchez, F. Enantioselective hydrogenation of alkenes and imines by a gold catalyst. Chem. Commun. 2005, 27, 3451–3453. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Gold-catalyzed synthesis of N,O-heterocycles. Pure Appl. Chem. 2010, 82, 657–668. [Google Scholar] [CrossRef]
- Milone, C.; Tropeano, M.L.; Guline, G.; Neri, G.; Ingoglia, R.; Galvagno, S. Selective liquid phase hydrogenation of citral on Au/Fe2O3 catalysts. Chem. Commun. 2002, 868–869. [Google Scholar]
- Tabakova, T.; Idakiev, V.; Tenchev, K.; Boccuzzi, F.; Manzoli, M.; Chiorino, A. Pure hydrogen production on a new gold-thoria catalyst for fuel cell applications. Appl. Catal. B 2006, 63, 94–103. [Google Scholar] [CrossRef]
- Bowker, M.; Millard, L.; Greaves, J.; James, D.; Soares, J. Photocatalysis by Au nanoparticles: Reforming of methanol. Gold Bull. 2004, 37, 170–173. [Google Scholar] [CrossRef]
- Grisel, R.J.H.; Nieuwenhuys, B.E. Selective oxidation of CO, over supported Au catalysts. J. Catal. 2001, 199, 48–59. [Google Scholar]
- Molina, L.M.; Hammer, B. Active role of oxide support during CO oxidation at Au/MgO. Phys. Rev. Lett. 2003, 90, 206102–206105. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Stoltze, P.; Norskov, J.K. Making gold less noble. Catal. Lett. 2000, 64, 101–106. [Google Scholar] [CrossRef]
- Oh, S.H.; Sinkevitch, S. Carbon monoxide removal from hydrogen-rich fuel cell feedstreams by selective catalytic oxidation. J. Catal. 1993, 142, 254–262. [Google Scholar]
- Kahlich, M.J.; Gasteiger, H.A.; Behm, R.J. Kinetics of the selective CO oxidation in H2-Rich gas on Pt/Al2O3. J. Catal. 1997, 171, 93–105. [Google Scholar]
- Grisel, R.J.H. Supported gold catalysts for environmental. Applications. Leiden University, Leiden, The Netherlands, 2002.
- Georgaka, A.; Gavril, D.; Loukopoulos, V.; Karaiskakis, G.; Nieuwenhuys, B.E. H2 and CO2 coadsorption effects in CO adsorption over nanosized Au/γ-Al2O3 catalysts. J. Chromatogr. A 2008, 1205, 128–136. [Google Scholar]
- Gavril, D.; Georgaka, A.; Loukopoulos, V.; Karaiskakis, G.; Nieuwenhuys, B.E. On the mechanism of selective CO oxidation on nanosized Au/γ-Al2O3 catalysts. Gold Bull. 2006, 39, 192–199. [Google Scholar] [CrossRef]
- Molina, L.M.; Hammer, B. Some recent theoretical advances in the understanding of the catalytic activity of Au. Appl. Catal. A Gen. 2005, 291, 21–31. [Google Scholar] [CrossRef]
- Karaiskakis, G.; Gavril, D. Determination of diffusion coefficients by gas chromatography. J. Chromatogr. A 2004, 1037, 147–189. [Google Scholar]
- Rashid, K.A.; Gavril, D.; Karaiskakis, G. A new gas chromatographic methodology for the estimation of the composition of binary gas mixtures. J. Chrom. Sci. 2003, 41, 123–132. [Google Scholar]
- Gavril, D.; Rashid, K.A.; Karaiskakis, G. Determination of collision cross sectional parameters from experimentally measured gas diffusion coefficients. Fluid Phase Equil. 2004, 218, 177–188. [Google Scholar] [CrossRef]
- Gavril, D.; Rashid, K.A.; Karaiskakis, G. Study of the mechanism of the interaction of vinylchloride with water by reversed-flow gas chromatography. J. Chromatogr. A 2001, 919, 349–356. [Google Scholar] [CrossRef]
- Rashid, K.A.; Gavril, D.; Katsanos, N.A.; Karaiskakis, G. Flux of gases across the air-water interface studied by reversed flow gas chromatography. J. Chromatogr. A 2001, 934, 31–49. [Google Scholar] [CrossRef]
- Gavril, D.; Rashid, K.Α.; Karaiskakis, G. Study of the evaporation of pollutant liquids under the influence of surfactants by reversed-flow inverse gas chromatography. AIChE J. 2006, 52, 2381–2390. [Google Scholar]
- Gavril, D.; Gabriel, A. Investigation of the determination of diffusion coefficients of gases into liquids from reversed-flow gas chromatography. Instrum. Sci. Technol. 2006, 34, 435–454. [Google Scholar] [CrossRef]
- Gavril, D.; Loukopoulos, V.; Karaiskakis, G. Study of CO dissociative adsorption over Pt and Rh catalysts by inverse gas chromatography. Chromatographia 2004, 59, 721–729. [Google Scholar]
- Gavril, D.; Loukopoulos, V.; Georgaka, A.; Gabriel, A.; Karaiskakis, G. Inverse gas chromatographic investigation of the effect of hydrogen in carbon monoxide adsorption over silica supported Rh and Pt-Rh alloy catalysts, under hydrogen-rich conditions. J. Chromatogr. A 2005, 1087, 158–168. [Google Scholar]
- Gavril, D. Reversed flow gas chromatography: A tool for instantaneous monitoring of the concentrations of reactants and products in heterogeneous catalytic processes. J. Liq. Chrom. Rel. Technol. 2002, 25, 2079–2099. [Google Scholar] [CrossRef]
- Gavril, D.; Koliadima, A.; Karaiskakis, G. Adsorption studies of gases on Pt-Rh bimetallic catalysts by reversed-flow gas chromatography. Langmuir 1999, 15, 3798–3806. [Google Scholar] [CrossRef]
- Gavril, D. An inverse gas chromatographic tool for the measurement of local isotherms on heterogeneous surfaces. Instrum. Sci. Technol. 2002, 30, 409–425. [Google Scholar]
- Loukopoulos, V.; Gavril, D.; Karaiskakis, G. An inverse gas chromatographic instrumentation for the study of carbon monoxide’s adsorption on Rh/SiO2, under hydrogen-rich conditions. Instrum. Sci. Technol. 2003, 31, 165–181. [Google Scholar] [CrossRef]
- Gavril, D.; Neuwenhuys, B.E. Investigation of the surface heterogeneity of solids from reversed-flow inverse gas chromatography. J. Chromatogr. A 2004, 1037, 161–172. [Google Scholar]
© 2012 by MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gavril, D.; Georgaka, A.; Karaiskakis, G. Kinetic Study of Oxygen Adsorption over Nanosized Au/γ-Al2O3 Supported Catalysts under Selective CO Oxidation Conditions. Molecules 2012, 17, 4878-4895. https://doi.org/10.3390/molecules17054878
Gavril D, Georgaka A, Karaiskakis G. Kinetic Study of Oxygen Adsorption over Nanosized Au/γ-Al2O3 Supported Catalysts under Selective CO Oxidation Conditions. Molecules. 2012; 17(5):4878-4895. https://doi.org/10.3390/molecules17054878
Chicago/Turabian StyleGavril, Dimitrios, Aglaia Georgaka, and George Karaiskakis. 2012. "Kinetic Study of Oxygen Adsorption over Nanosized Au/γ-Al2O3 Supported Catalysts under Selective CO Oxidation Conditions" Molecules 17, no. 5: 4878-4895. https://doi.org/10.3390/molecules17054878