Diterpenoid Alkaloids from the Chinese Traditional Herbal “Fuzi” and Their Cytotoxic Activity

Abstract
:1. Introduction
2. Results and Discussion
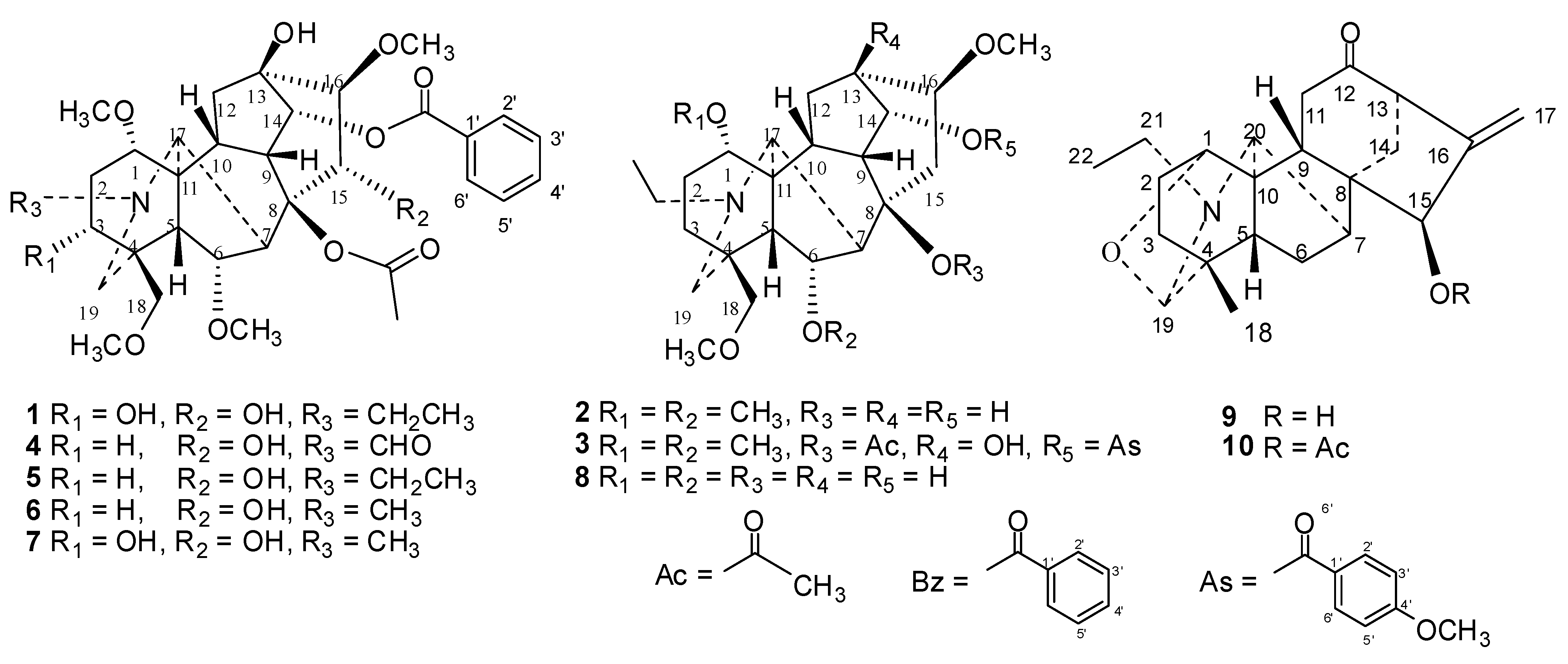

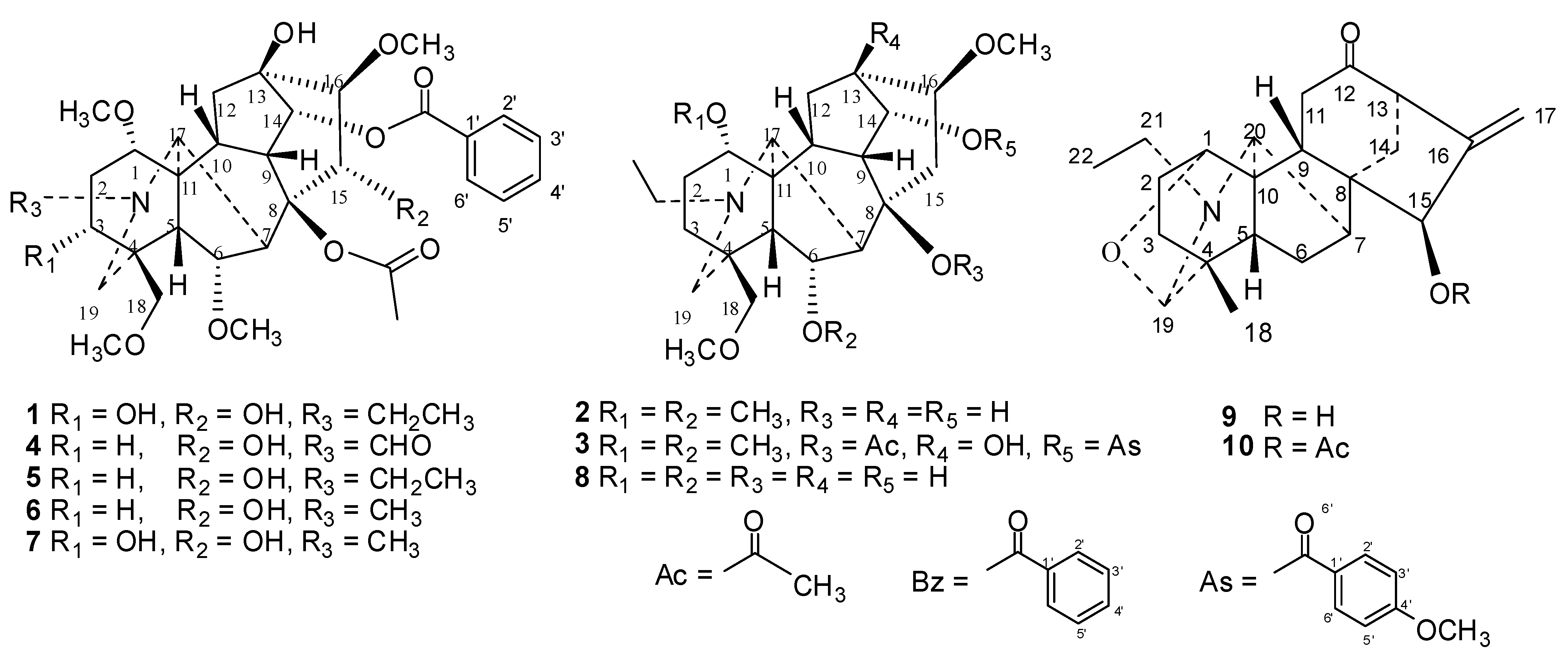{kind=link}
| Compds | IC50 (×10−8 M) | Compds | IC50 (×10−8 M) | ||||
|---|---|---|---|---|---|---|---|
| HCT8 | MCF7 | HePG2 | HCT8 | MCF7 | HePG2 | ||
| Taxol | 4.49 ± 0.83 | 0.71 ± 0.25 | 0.12 ± 0.47 | Taxol | 4.49 ± 0.83 | 0.71 ± 0.25 | 0.12 ± 0.47 |
| 1 | 8.12 ± 0.23 | 2.45 ± 1.02 | 0.85 ± 0.06 | 6 | 12.05 ± 0.76 | 6.46 ± 1.26 | 0.92 ± 0.06 |
| 2 | >1,000 | >1,000 | >1,000 | 7 | 13.16 ± 1.58 | 4.57 ± 0.29 | 1.45 ± 0.01 |
| 3 | 16.45 ± 1.55 | 12.86 ± 3.52 | 2.36 ± 0.42 | 8 | >1,000 | >1,000 | >1,000 |
| 4 | 29.48 ± 3.12 | 3.13 ± 0.14 | 8.61 ± 1.31 | 9 | >1,000 | >1,000 | >1,000 |
| 5 | 5.14 ± 0.68 | 10.35 ± 1.24 | 9.21 ± 2.33 | 10 | >1,000 | >1,000 | 58.55 ± 9.17 |
3. Experimental
3.1. General
3.2. Plant Material Collection and Identification
3.3. Extraction and Isolation
3.4. NMR Spectroscopic Data
3.5. Determination of Cell Viability by MTT Assay
4. Conclusions
Acknowledgments
- Samples Availability: Samples of the compounds 1–3, 5–7, and 9 are available from the corresponding author.
References and Notes
- Xiao, P.G.; Wang, F.P.; Gao, F.; Yan, L.P.; Chen, D.L.; Liu, Y. A pharmacophylogenetic study of Aconitum L. (Ranunculaceae) from China. Acta Phytotaxon. Sin. 2006, 44, 1–46. [Google Scholar]
- Wang, F.P.; Chen, Q.H.; Liu, X.Y. Diterpenoid alkaloids. Nat. Prod. Rep. 2010, 27, 529–570. [Google Scholar] [CrossRef]
- Wada, K.; Hazawa, M.; Takahashi, K.; Mori, T.; Kawahara, N.; Kashiwakura, I. Inhibitory effects of diterpenoid alkaloids on the growth of A172 human malignant cells. J. Nat. Prod. 2007, 70, 1854–1858. [Google Scholar] [CrossRef]
- Wada, K.; Hazawa, M.; Takahashi, K.; Mori, T.; Kawahara, N.; Kashiwakura, I. Structure-activity relationships and the cytotoxic effects of novel diterpenoid alkaloid derivatives against A549 human lung carcinoma cells. J. Nat. Med. 2011, 65, 43–49. [Google Scholar] [CrossRef]
- Shen, Y.; Zuo, A.X.; Zhang, X.M.; Wang, H.L.; Chen, J.J. Two new C20-diterpenoid alkaloids from Aconitum carmichaelii. Helv.Chim. Acta 2011, 94, 122–126. [Google Scholar] [CrossRef]
- Borcsa, B.; Widowitz, U.; Cspupor, D.; Forgo, P.; Bauer, R.; Hohmann, J. Semisynthesis and pharmacologicalinvestigation of lipo-alkaloidsprepared from aconitine. Fitoterapia 2011, 82, 365–368. [Google Scholar] [CrossRef]
- Yang, J.; Liu, W.; Yang, X.; Zhao, J.; Liu, F.; Li, L. Diterpenoid alkaloids from Aconitum handelianum. Zhongguo Zhong Yao Za Zhi 2009, 34, 1927–1929. [Google Scholar]
- Wang, F.P.; Fang, Q.C. Alkaloids from roots of Aconitum crassicaule. Planta Med. 1981, 42, 375–379. [Google Scholar] [CrossRef]
- Yunusov, M.S. Diterpenoid alkaloids. Nat. Prod. Rep. 1991, 8, 499–526. [Google Scholar] [CrossRef]
- Yunusov, M.S. Diterpenoid alkaloids. Nat. Prod. Rep. 1993, 10, 471–486. [Google Scholar] [CrossRef]
- Zhapova, T.; Modonava, L.D.; Semenov, A.A. Mesaconitine and hypaconitine from Aconitum czekanovskyi. Chem. Nat. Prod. 1985, 21, 678–679. [Google Scholar]
- Konno, C.; Shirasaka, M.; Hikino, H. Structure of senbusine A, B and C, diterpenie alkaloids of Aconitum carmichaeli roots from China. J. Nat. Prod. 1982, 45, 128–133. [Google Scholar] [CrossRef]
- Chen, H.C.; Wang, X.K.; Zhao, T.F.; Liao, X. Chemical constituents from Aconitum carmichaeli. Nat. Prod. Res. Dev. 2003, 15, 324–325. [Google Scholar]
- Fan, Z.C.; Zhang, Z.Q. Molecular and crystal structure of 15-acetylsongorine and songoramine isolated from Aconitum szechenyianum Gray. Struct.Chem. 2008, 19, 413–419. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gao, F.; Li, Y.-Y.; Wang, D.; Huang, X.; Liu, Q. Diterpenoid Alkaloids from the Chinese Traditional Herbal “Fuzi” and Their Cytotoxic Activity. Molecules 2012, 17, 5187-5194. https://doi.org/10.3390/molecules17055187
Gao F, Li Y-Y, Wang D, Huang X, Liu Q. Diterpenoid Alkaloids from the Chinese Traditional Herbal “Fuzi” and Their Cytotoxic Activity. Molecules. 2012; 17(5):5187-5194. https://doi.org/10.3390/molecules17055187
Chicago/Turabian StyleGao, Feng, Yuan-Yuan Li, Dan Wang, Xing Huang, and Qian Liu. 2012. "Diterpenoid Alkaloids from the Chinese Traditional Herbal “Fuzi” and Their Cytotoxic Activity" Molecules 17, no. 5: 5187-5194. https://doi.org/10.3390/molecules17055187