A Convenient Synthesis of Triflate Anion Ionic Liquids and Their Properties

Abstract
:1. Introduction



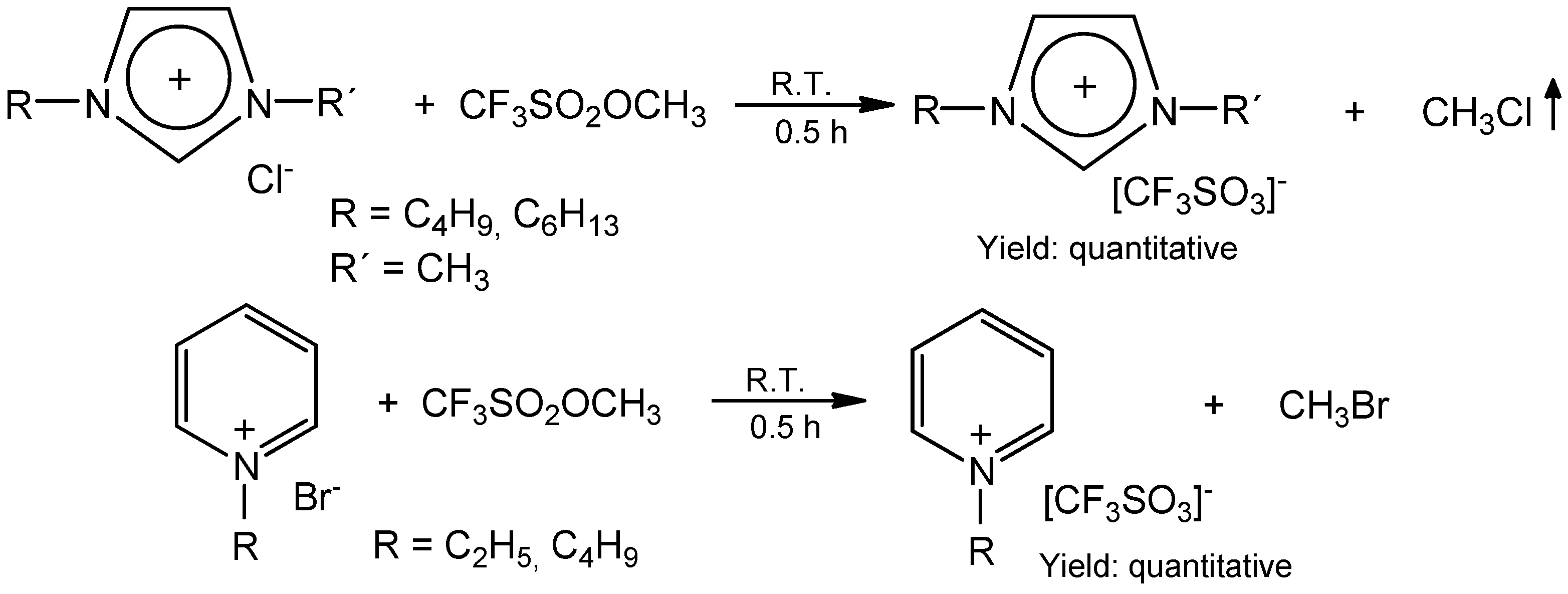
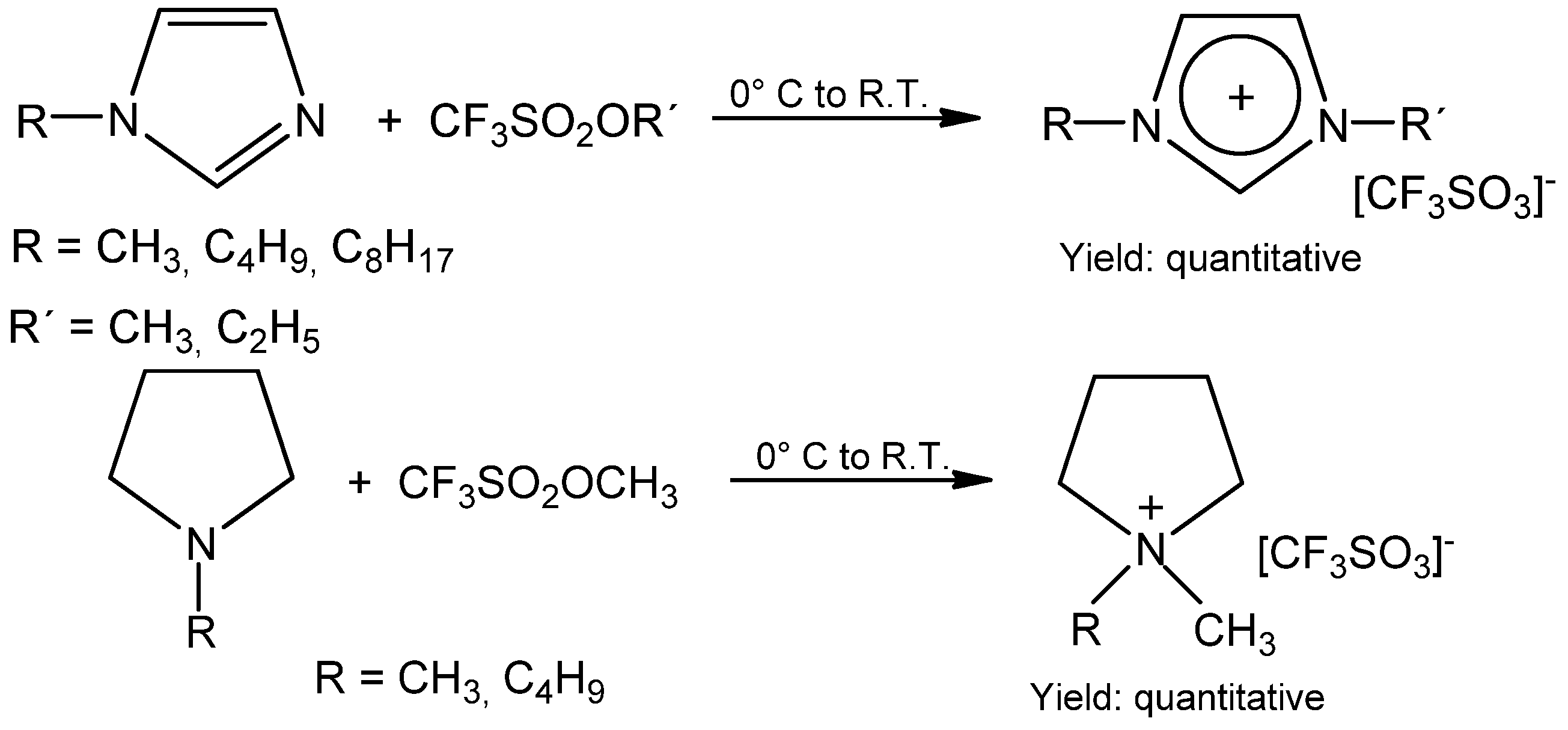
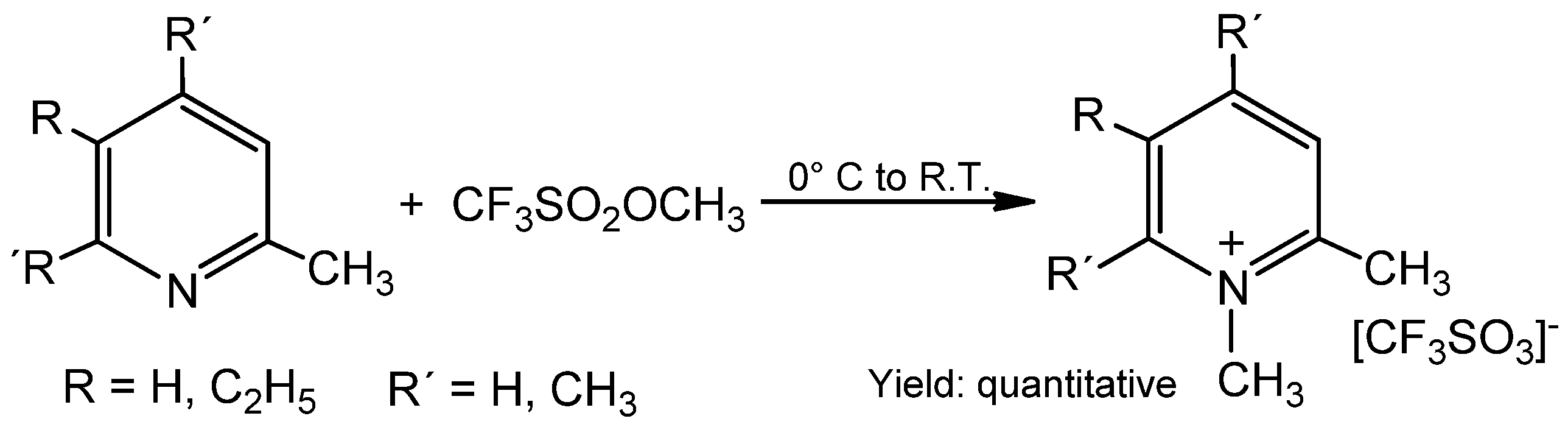
2. Results and Discussion








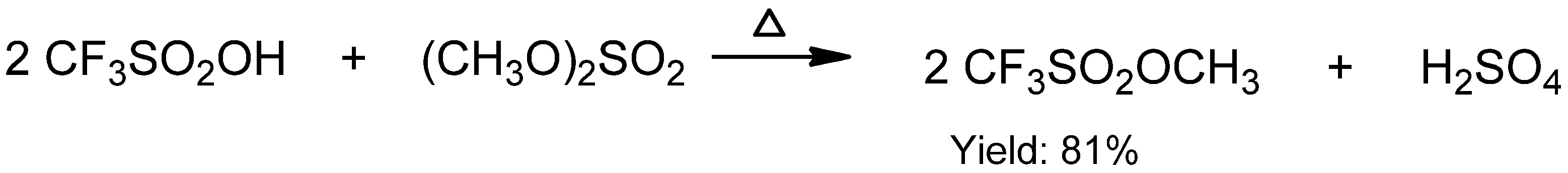
{kind=link}
{kind=link}
{kind=link}
{kind=link}
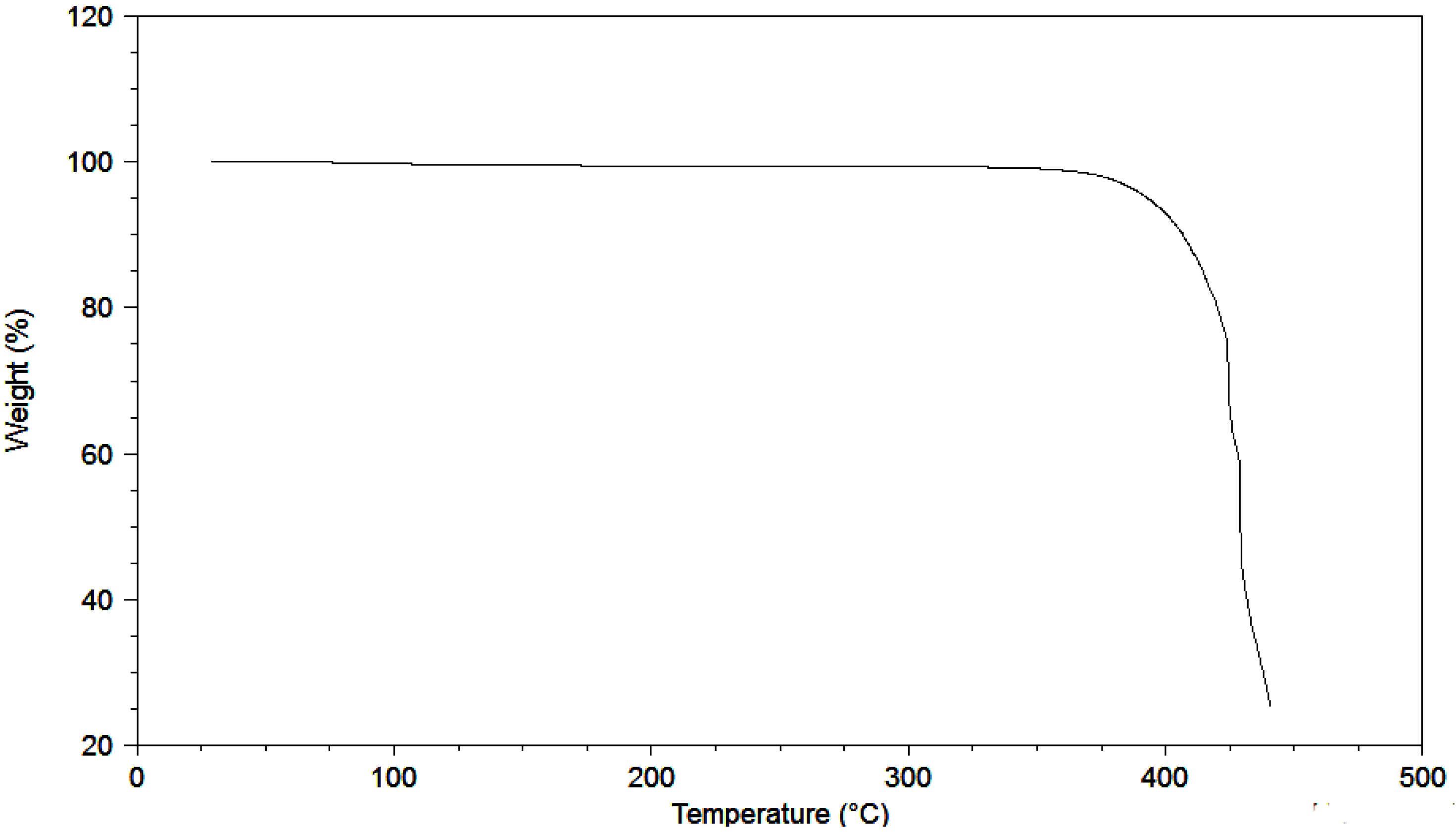
{kind=link}
{kind=link}
{kind=link}
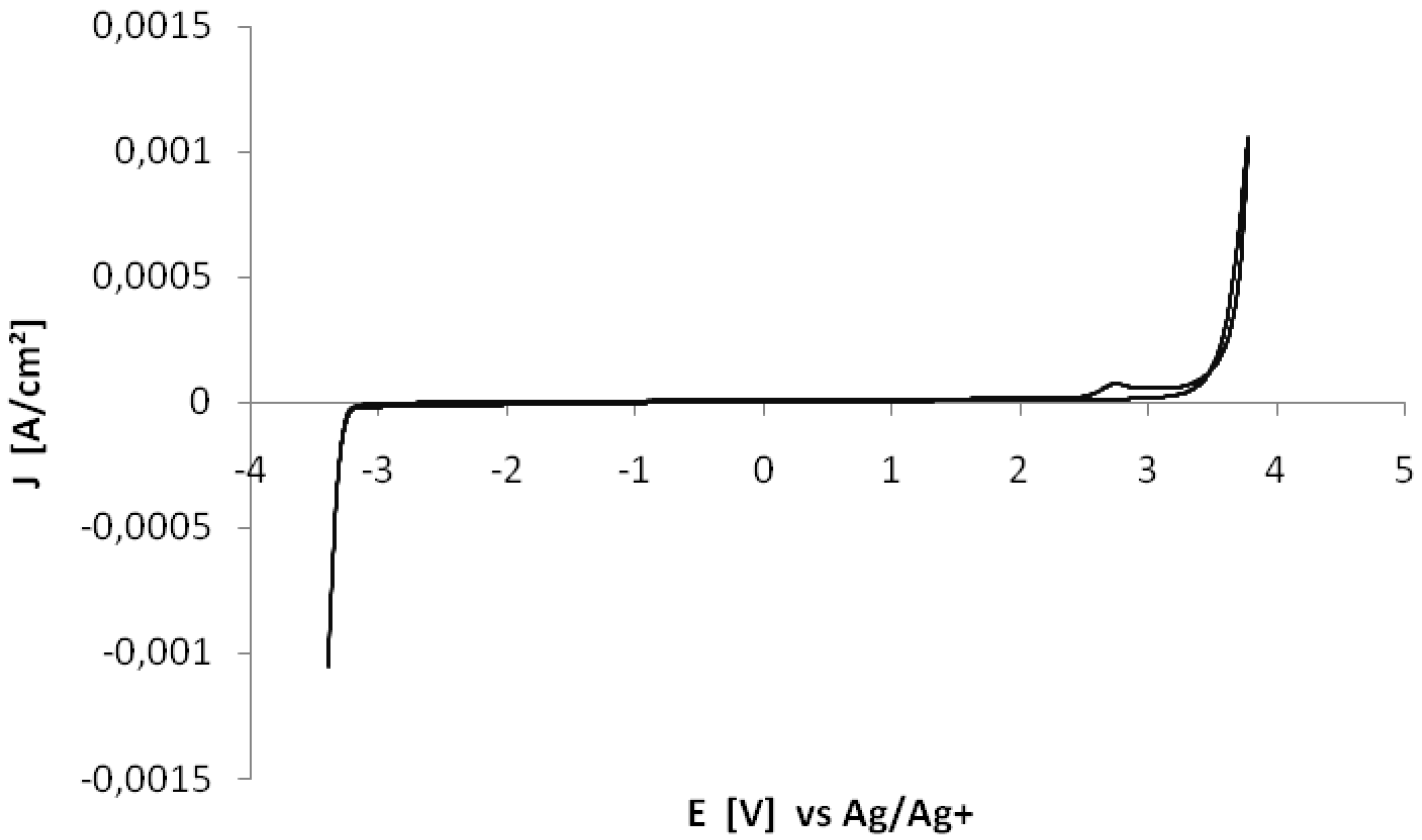
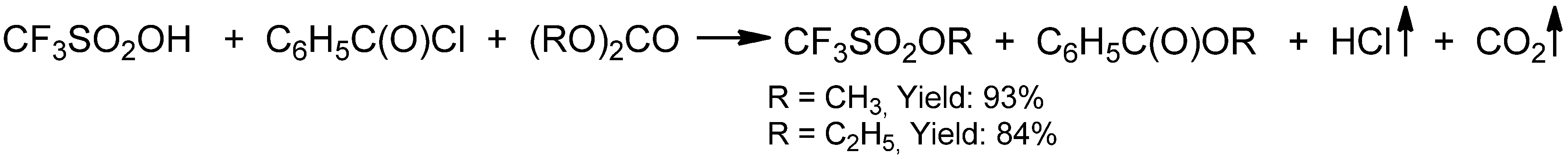{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ionic Liquid | Weight loss (% per day, 24 h), TGA isothermal mode at 200 °C | Weight loss (% per day, 24 h), TGA isothermal mode at 250 °C |
|---|---|---|
| 1-Butyl-1-methylpyrrolidinium [CF3SO3] | 0.28 | 2.8 |
| 1-Butyl-3-methylimidazolium [CF3SO3] | 0.55 | 5.9 |

| Ionic Liquid | E(ox), V | E(red), V | Window, V |
|---|---|---|---|
| Tetrabutylammonium [(C2F5)3PF3] | 3.7 | −3.3 | 7.0 |
| Methyl-trioctyl-ammonium [CF3SO3] | 3.4 | −3.4 | 6.8 |
| 1-Butyl-1-methylpyrrolidinium [CF3SO3] | 3.4 | −3.2 | 6.6 |
| 1-Butyl-3-methylimidazolium [CF3SO3] | 3.0 | −2.6 | 5.6 |
| 1-Ethyl-3-methylimidazolium [CF3SO3] | 2.8 | −2.5 | 5.3 |
| 1-Ethyl-3-methylimidazolium [BF4] | 2.6 | −2.6 | 5.2 |
| Cation | Anion | Dynamic Viscosity, mPa·s | |||
|---|---|---|---|---|---|
| 20 °C | 40 °C | 60 °C | 80 °C | ||
 | [CH3OSO3] | 84 | 37 | 20 | 12 |
| [CF3SO3] | 52 | 26 | 15 | 10 | |
| [C2H5OSO3] | 117 | 47 | 24 | 14 | |
| [C4H9OSO3] | 261 | 90 | 40 | 22 | |
| Ionic Liquid | Kinematic Viscosity (20 °C), mm2/s | Density (20 °C), g/cm3 | Dynamic Viscosity (20 °C), mPa·s |
|---|---|---|---|
| 1-Hexyl-3-methylimidazolium Chloride | 7453 | 1.05 | 7826 |
| 1-Hexyl-3-methylimidazolium [PF6] | 548 | 1.30 | 712 |
| 1-Hexyl-3-methylimidazolium [BF4] | 195 | 1.15 | 224 |
| 1-Hexyl-3-methylimidazolium [CF3SO3] | 160 | 1.24 | 198 |
| 1-Hexyl-3-methylimidazolium [(C2F5)3PF3] | 74 | 1.56 | 115 |
| 1-Hexyl-3-methylimidazolium [(CF3SO2)2N] | 65 | 1.38 | 90 |
3. Experimental
3.1. Chemicals
3.2. Analytical Procedures
3.2.1. NMR Spectroscopy
3.2.2. Elemental Analysis
3.2.3. Thermal Analyses
3.2.4. Viscosity
3.2.5. Cyclic Voltammetry
3.3. Syntheses
Methyl Trifluoromethanesulfonate (CF3SO2OCH3)
Ethyl Trifluoromethanesulfonate (CF3SO2OC2H5)
1,3-Dimethylimidazolium Trifluoromethanesulfonate [C5H9N2][CF3SO3]
1-Ethyl-3-methylimidazolium Trifluoromethanesulfonate [C6H11N2][CF3SO3]
1-Butyl-3-methylimidazolium Trifluoromethanesulfonate [C8H15N2][CF3SO3]
1-Hexyl-3-methylimidazolium-trifluoromethanesulfonate [C10H19N2][CF3SO3]
3-Octyl-1-methylimidazolium-trifluoromethanesulfonate [C12H23N2][CF3SO3]
1-Butyl-3-ethylimidazolium-trifluoromethanesulfonate [C9H17N2][CF3SO3]
N,N-Dimethylpyrrolidinium Trifluoromethanesulfonate [C7H14N][CF3SO3]
N-Butyl-N-methylpyrrolidinium Trifluoromethanesulfonate [C9H20N][CF3SO3]
N,N-Dimethylpiperidinium Trifluoromethanesulfonate [C8H16N][CF3SO3]
N-Methyl-3-butylpyridinium Trifluoromethanesulfonate [C9H20N][CF3SO3]
N-Ethylpyridinium Trifluoromethanesulfonate [C7H10N][CF3SO3]
N-Butylpyridinium Trifluoromethanesulfonate [C9H14N][CF3SO3]
1,2-Dimethyl-5-ethylpyridinium-trifluoromethanesulfonate
1,2,4,6-Tetramethylpyridinium Trifluoromethanesulfonate [C9H14N][CF3SO3]
1-Methyl-2,4,6,-tris(t-butyl)pyridnium Trifluoromethanesulfonate [C18H32N][CF3SO3]
Methyl trioctylammonium Trifluoromethanesulfonate [(C8H17)3NCH3][CF3SO3]
Methyl triethylphosphonium Trifluoromethanesulfonate [(C2H5)3PCH3][CF3SO3]
Trihexyl tetradecylphosphonium Trifluoromethanesulfonate [(C6H13)3PC14H29][CF3SO3]
N"-Ethyl-N,N,N',N'-tetramethylguanidinium Trifluoromethanesulfonate [(CH3)2NC(NHC2H5)N(CH3)2] [CF3SO3]
N,N,N',N'-Tetramethyl-O-methylisouronium Trifluoromethanesulfonate [(CH3)2NC(OCH3)N(CH3)2] [CF3SO3]
4. Conclusions
Acknowledgements
References and Notes
- Wasserscheid, P.; Welton, T. Ionic Liquids in Synthesis, 2nd ed; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008. [Google Scholar]
- Ohno, H. Electrochemical Aspects of Ionic Liquids; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Dyson, P.J.; Geldbach, T.J. Metal Catalysed Reactions in Ionic Liquids; Springer: Dordrecht, The Netherlands, 2005. [Google Scholar]
- Holbrey, J.D. Industrial applications of ionic liquids. Chem. Today 2004, June, 35–37. [Google Scholar]
- Zulfigar, F.; Kitazume, T. Lewis acid-catalysed sequential reaction in ionic liquids. Green Chem. 2000, 2, 296–298. [Google Scholar] [CrossRef]
- Serguchev, Y.S.; Lourie, L.F.; Ponomarenko, M.V.; Rusanov, E.B.; Ignat’ev, N.V. Fluorolactonization of unsaturated carboxylic acids with F-TEDA-BF4 in ionic liquids. Tetrahedron Lett. 2011, 52, 5166–5169. [Google Scholar] [CrossRef]
- Epishina, M.A.; Kulikov, A.S.; Ignat’ev, N.V.; Schulte, M.; Makhova, N.N. The first example of the Schmidt reaction in ionic liquids. Mendeleev Commun. 2010, 20, 335–336. [Google Scholar] [CrossRef]
- Vygodskii, Y.S.; Mel’nik, O.A.; Lozinskaya, E.I.; Shaplov, A.S.; Malyshkina, I.A.; Gavrilova, N.D.; Lyssenko, K.A.; Antipin, M.Y.; Golovanov, D.G.; Korlyukov, A.A.; et al. The influence of ionic liquid’s nature on free radical polymerization of vinyl monomers and ionic conductivity of the obtained polymeric materials. Polym. Adv. Technol. 2007, 18, 50–63. [Google Scholar] [CrossRef]
- Choi, D.S.; Choi, D.W.; Park, E.J.; Chang, S.K.; Byun, I.S.; Kim, W.J. A Purification Method of Ionic Liquids to Obtain Their High Purity. WO2004/080974, 2004. [Google Scholar]
- Cammarata, L.; Kazarian, S.; Salter, P.; Welton, T. Molecular states of water in room temperature ionic liquids. Phys. Chem. Chem. Phys. 2001, 3, 5192–5200. [Google Scholar]
- Wasserscheid, P.; Hilgers, C. Halogen-Free Preparation of Ionic Fluids. EP118 EP1182196, 2002. [Google Scholar]
- Kim, H.G.; Lee, S.D.; Ahn, B.S.; Lee, J.S.; Kim, U.S.; Cho, B.W.; Kim, H.S. The Lithium Salts of Pyrrolidinium Type Zwitterion and the Preparation Method of the Same. WO2006/104305, 2006. [Google Scholar]
- Cama, L.D.; Wilkening, R.R.; Ratcliffe, R.W.; Wildonger, K.J.; Sun, W. Carbapenem Antibacterial Compounds, Compositions Containing such Compounds and Method of Treatment. US5994345, 1999. [Google Scholar]
- Fuller, J.; Carlin, R.T. Facile Preparation of Tetrafluoroborate and Trifluoromethanesulfonate Room-Temperature Ionic Liquids. In Proceedings of the 11th International Symposium on Molten Salts XI; Truvol, P.C., De Long, H.C., Stafford, C.R., Deki, S., Eds.; The Electrochemical Society, Inc.: Pennington, NJ, USA, 1998; 98–11, pp. 227–230. [Google Scholar]
- Tetsuo, N.; Yasutaka, T.; Megumi, T.; Masashi, Y.; Kazutaka, H.; Akihiro, N.; Hiroaki, T.; Kenji, S.; Takashi, H. Quaternary Ammonium Salt, Electrolyte, and Electrochemical Device. EP1642894 2006. EP1837333, 2007.. [Google Scholar]
- Ignatiev, N.; Welz-Biermann, U. New Technologies for Ionic Liquids Production. In Presented at the First Congress on Ionic Liquids, Salzburg, Austria, 19-23 June 2005.
- Koichi, M. Green Reaction Media in Organic Synthesis; Blackwell Publishing Ltd.: Oxford, UK, 2005; p. 50. [Google Scholar]
- Cravotto, G.; Gaudino, E.C.; Boffa, L.; Lévêque, J.-M.; Estager, J.; Bonrath, W. Preparation of second generation ionic liquids by efficient solvent-free alkylation of N-heterocycles with chloroalkanes. Molecules 2008, 13, 149–156. [Google Scholar] [CrossRef]
- Estager, J.; Lévêque, J.-M.; Cravotto, G.; Boffa, L.; Bonrath, W.; Draye, M. One-pot and solventless synthesis of ionic liquids under ultrasonic irradiation. Synlett 2007, 13, 2065–2068. [Google Scholar]
- Deetlefs, M.; Seddon, K.J. Assessing the greenness of some typical laboratory ionic liquid preparations. Green Chem. 2010, 12, 17–30. [Google Scholar] [CrossRef]
- Olah, G.A.; Prakash, G.K.S.; Molnar, A.; Sommer, J. Superacid Chemistry, 2nd ed; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; p. 38. [Google Scholar]
- Rakita, P.H. Triflic acid and its derivatives. A family of useful reagents for synthesis. Chem. Today 2004, 48–50, March/April. [Google Scholar]
- Gramstadt, T.; Haszeldine, R.N. Perfluoroalkyl derivatives of sulphur. Part VII. Alkyl trifluoromethunesulphonates as alkylating agents, trifluoromethanesulphonic anhydride as a promoter for esterification, and some reactions of trifluoromethanesulphonic acid. J. Chem. Soc. 1956, 173–180. [Google Scholar]
- Booth, B.L.; Haszeldine, R.N.; Laali, K. Alkyltrifluoromethanesulphonates as alkylating reagents for aromatic compounds. J. Chem. Soc. Perkin Trans. I 1980, 2887–2894. [Google Scholar]
- Burdon, J.; McLoughlin, V.C.R. Trifluoromfthanesulphonate esters and their alkylating properties. Tetrahedron 1965, 21, 1–4. [Google Scholar] [CrossRef]
- Dobbs, A.P.; Jones, K.; Veal, K.T. The generation and cyclisation of pyridinium radicals as a potential route to indolizidine alkaloids. Tetrahedron Lett. 1997, 38, 5383–5386. [Google Scholar] [CrossRef]
- Beard, C.D.; Baum, K.; Grakauskas, V. Synthesis of some novel trifluoromethanesulfonates and their reactions with alcohols. J. Org. Chem. 1973, 38, 3673–3677. [Google Scholar] [CrossRef]
- Ignatyev, N.; Schmidt, M.; Heider, U.; Sartori, P.; Kucheryna, A. Method for Producing Perfluoroalkanesulfonic Acid Esters. Merck Patent GmbH WO 2002/098844, EP 1 399 417 B1, 2002. [Google Scholar]
- Ignatyev, N.; Schmidt, M.; Heider, U.; Sartori, P.; Kucheryna, A. Method for Producing Perfluoroalkane Sulfonic Acid Esters and the Salts Thereof. Merck Patent GmbH WO 2003/053918, EP 1 472 217 B1, 2003. [Google Scholar]
- Bonhöte, P.; Dias, A.; Papageorgiou, N.; Kalyanasundaram, K.; Grätzel, M. Hydrophobic, highly conductive ambient-temperature molten salts. Inorg. Chem. 1996, 35, 1168–1178. [Google Scholar] [CrossRef]
- Denmark, S.E.; Forbes, D.C.; Hays, D.S.; DePue, J.S.; Wilde, R.G. Catalytic epoxidation of alkenes with oxone. J. Org. Chem. 1995, 60, 1391–1407. [Google Scholar] [CrossRef]
- Beer, L.; Britten, J.F.; Brusso, J.L.; Cordes, A.W.; Haddon, R.C.; Itkis, M.E.; MacGregor, D.S.; Oakley, R.T.; Reed, R.W.; Robertson, C.M. Prototypal dithiazolodithiazolyl radicals: Synthesis, structures, and transport properties. J. Am. Chem. Soc. 2003, 125, 14394–14403. [Google Scholar]
- Canac, Y.; Duhayon, C.; Chauvin, R. A diaminocarbene-phosphonium ylide: Direct access to C,C chelating ligands. Angew. Chem. 2007, 119, 6429–6431. [Google Scholar] [CrossRef]
- Ferreira, L.M.; Marques, M.M.B.; Glória, P.M.C.; Chaves, H.T.; Franco, J.-P.P.; Mourato, I.; Antunes, J.-R.T.; Rzepa, H.S.; Lobo, A.M.; Prabhakar, S. Reaction of aromatic nitroso compounds with chemical models of ‘thiamine active aldehyde’. Tetrahedron 2008, 64, 7759–7770. [Google Scholar]
- Riegel, S.D.; Burford, N.; Lumsdenb, M.D.; Decken, A. Synthesis and characterization of elusive cyclo-di-and-tri-phosphino-1,3-diphosphonium salts: Fundamental frameworks in catenaorganophosphorus chemistry. Chem. Commun. 2007, 4668–4670. [Google Scholar]
- Weigand, J.J.; Burford, N.; Decken, A.; Schulz, A. Preparation and characterization of a ligand-stabilized trimethylphosphane dication. Eur. J. Inorg. Chem. 2007, 4868–4872. [Google Scholar]
- Levin, V.V.; Kozlov, M.A.; Song, Y.-H.; Dilman, A.D.; Belyakov, P.A.; Struchkova, M.I.; Tartakovsky, V.A. Nucleophilic fluoroalkylation of iminium salts. Tetrahedron Lett. 2008, 49, 3108–3111. [Google Scholar]
- Maruoka, K.; Sato, J.; Yamamoto, H. Methylaluminum bis(4-bromo-2,6-di-tert-butylphenoxide) as a key reagent for effecting primary α-alkylation of carbonyl compounds. J. Am. Chem. Soc. 1992, 114, 4422–4423. [Google Scholar] [CrossRef]
- Zoller, U. The cheletropic fragmentation of hypervalent three-membered thiaheterocyclic intermediates. Tetrahedron 1988, 44, 7413–7426. [Google Scholar] [CrossRef]
- Bonhöte, P.; Dias, A.; Papageorgiou, N.; Kalyanasundaram, K.; Grätzel, M. Hydrophobic, highly conductive ambient-temperature molten salts. Inorg. Chem. 1996, 35, 1168–1178. [Google Scholar] [CrossRef]
- Kevill, D.N.; Lin, G.M.L. A comparison of leaving-group abilities in reactions of powerful methylating agents. Tetrahedron Lett. 1978, 11, 949–952. [Google Scholar] [CrossRef]
- Ignat’ev, N.V.; Welz-Biermann, U.; Kucheryna, A.I.; Willner, H. Close the Gap: Synthesis of Highly Unsymmetrical Ionic Liquids. In 231st ACS National Meeting, Atlanta, GA, USA, March 26-30, 2006. Abstract I&EC 126..
- Ignatyev, N.; Welz-Biermann, U.; Kucheryna, A.; Willner, H. Method for Producing Onium Salts Comprising Alkyl- or Aryl-Sulfonate Anions or Alkyl-Aryl-Carboxylate Anions Having a Low Halide Content. Merck Patent GmbH WO 2006/063656 A1, 2006. [Google Scholar]
- Ignat’ev, N.V.; Kucheryna, A.; Bissky, G.; Willner, H. How to Make Ionic Liquids More Liquid. In Ionic Liquids: Not Just Solvents Anymore; Brennecke, J.F., Rogers, R.D., Seddon, K.R., Eds.; American Chemical Society: Washington, DC, USA, 2007; ACS Symposium Series 975, Chapter 22. [Google Scholar]
- Ignat’ev, N.V.; Welz-Biermann, U.; Kucheryna, A.; Bissky, G.; Willner, H. New ionic liquids with tris(perfluoroalkyl)trifluorophosphate (FAP) anions. J. Fluorine Chem. 2005, 126, 1150–1159. [Google Scholar] [CrossRef]
- Ignatyev, N.; Welz-Biermann, U.; Koppe, K.; Barthen, P.; Frohn, H.-J. Dehydration of Alcohols to Form Alkenes or Ethers. Merck Patent GmbH WO 2007/014613, 2007. [Google Scholar]
- Ignatyev, N.; Koppe, K.; Frohn, H.-J.; Barthen, P. Splitting of Dialkoxyalkanes in Ionic Liquids. Merck Patent GmbH WO 2007/104380, 2007. [Google Scholar]
- Ignatyev, N.; Koppe, K.; Barthen, P.; Frohn, H.-J. Synthesis of Chromane Derivatives. Merck Patent GmbH WO 2008/086847 A2, 2008. [Google Scholar]
- Sample Availability: Ionic Liquids are available from Merck KGaA, Darmstadt, Germany.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ignat’ev, N.V.; Barthen, P.; Kucheryna, A.; Willner, H.; Sartori, P. A Convenient Synthesis of Triflate Anion Ionic Liquids and Their Properties. Molecules 2012, 17, 5319-5338. https://doi.org/10.3390/molecules17055319
Ignat’ev NV, Barthen P, Kucheryna A, Willner H, Sartori P. A Convenient Synthesis of Triflate Anion Ionic Liquids and Their Properties. Molecules. 2012; 17(5):5319-5338. https://doi.org/10.3390/molecules17055319
Chicago/Turabian StyleIgnat’ev, Nikolai V., Peter Barthen, Andryi Kucheryna, Helge Willner, and Peter Sartori. 2012. "A Convenient Synthesis of Triflate Anion Ionic Liquids and Their Properties" Molecules 17, no. 5: 5319-5338. https://doi.org/10.3390/molecules17055319