H2TPP Organocatalysis in Mild and Highly Regioselective Ring Opening of Epoxides to Halo Alcohols by Means of Halogen Elements

Abstract
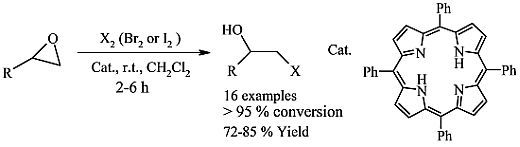
:
1. Introduction
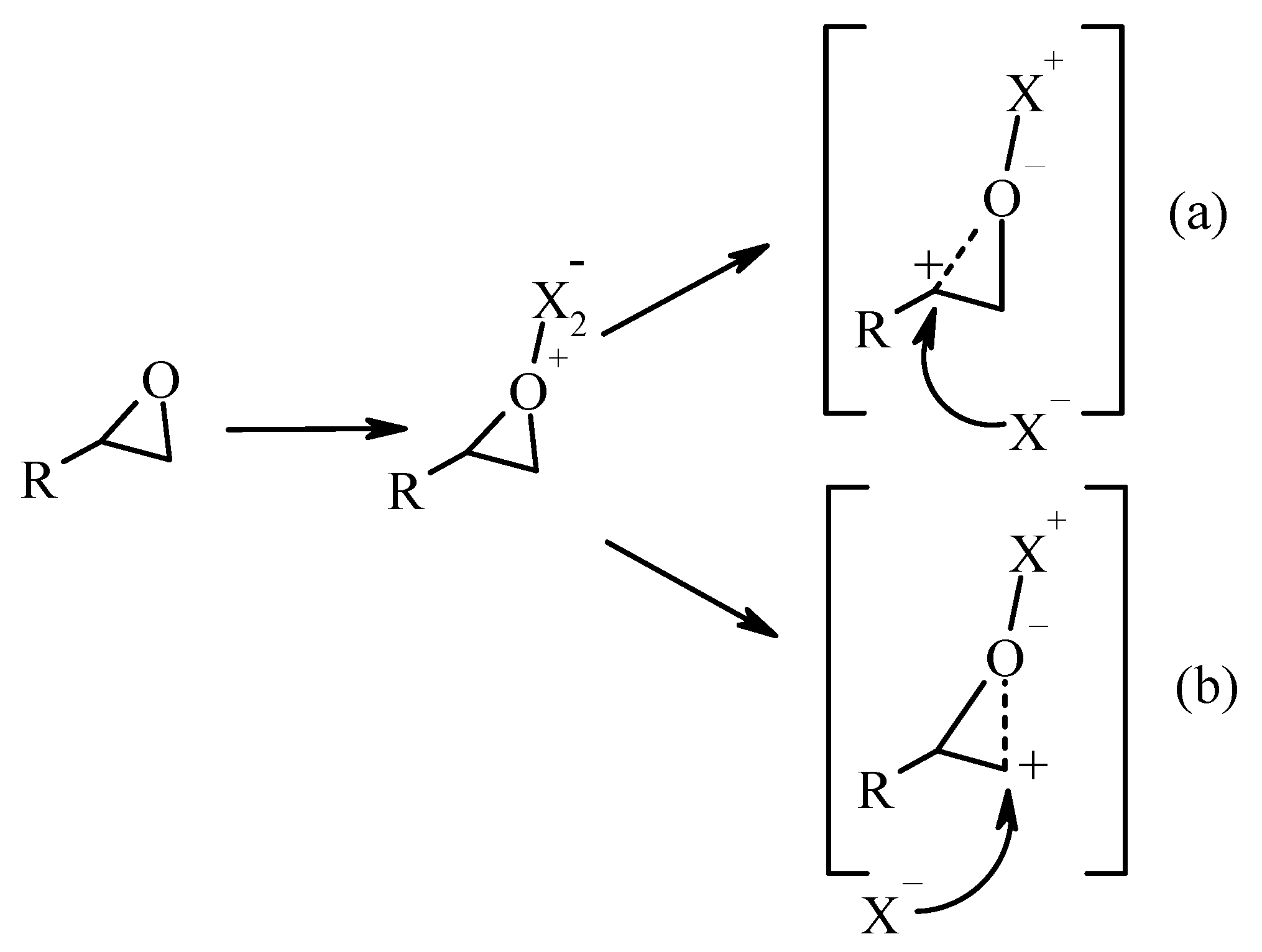
2. Results and Discussion
3. Experimental
3.1. Materials
3.2. Apparatus
3.3. General Procedure for Conversion of Epoxides to β-Halohydrins
4. Conclusions
Acknowledgments
References and Notes
- March, J. Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 2nd ed.; McGraw-Hill: New York, NY, USA, 1977; p. 1328. [Google Scholar]
- Shimizu, M.; Yoshida, A.; Fujisawa, T. Regioselective conversion of epoxides to halohydrins by titanium-(IV) halide-lithium halide complex. Synlett 1992, 23, 204–206. [Google Scholar] [CrossRef]
- Bonini, C.; Righi, G. Regio- and chemoselective synthesis of halohydrins by cleavage of oxiranes with metal halides. Synthesis 1994, 25, 225–238. [Google Scholar] [CrossRef]
- Ready, J.M.; Jacobsen, E.N. A practical oligomeric [(salen)Co] catalyst for asymmetric epoxide ring-opening reactions. Angew. Chem. Int. Ed. Engl. 2002, 41, 1374–1377. [Google Scholar] [CrossRef]
- Kumar, R.; Wiebe, L.I.; Hall, T.W.; Knaus, E.E.; Tovell, D.R.; Tyrrell, D.L.; Allen, T.M.; Fathi-Afshar, R. Synthesis of 5-[1-hydroxy(or methoxy)-2-bromo(or chloro)ethyl]-2′-deoxyuridines and related halohydrin analogues with antiviral and cytotoxic activity. J. Med. Chem. 1989, 32, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Georg, G.I.; Ali, S.M.; Stella, V.J.; Waugh, W.N.; Himes, R.H. Halohydrin analogues of cryptophycin 1: synthesis and biological activity. Bioorg. Med. Chem. Lett. 1998, 8, 1959–1962. [Google Scholar] [CrossRef]
- McGowan, M.A.; Stevenson, C.P.; Schiffler, M.A.; Jacobsen, E.N. An enantioselective total synthesis of (+)-pelorusideA. Angew. Chem. Weinheim Bergstr. Ger. 2010, 122, 6283–6286. [Google Scholar]
- Agatsuma, T.; Ogawa, H.; Akasaka, K.; Asai, A.; Yamashita, Y.; Mizukami, T.; Akinaga, S.; Saitoh, Y. Halohydrin and oxime derivatives of radicicol: Synthesis and antitumor activities. Bioorg. Med. Chem. 2002, 10, 3445–3454. [Google Scholar] [CrossRef]
- Tang, L.; Lutje Spelberg, J.H.; Fraaije, M.W.; Janssen, D.B. Kinetic mechanism and enantioselectivity of halohydrin dehalogenase from Agrobacterium radiobacter. Biochemistry 2003, 42, 5378–5386. [Google Scholar] [CrossRef] [PubMed]
- Dawe, R.D.; Molinski, T.F.; Turner, J.V. Dilithium tetrabromonickelate (II) as a source of soft nucleophilic bromide: reaction with epoxides. Tetrahedron Lett. 1984, 25, 2061–2064. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, B.V.S.; Ch, S.R.; Rajasekhar, K. Green protocol for the synthesis of vicinal-halohydrins from oxiranes using the [Bmim]PF6/LiX reagent system. Chem. Lett. 2004, 33, 476–477. [Google Scholar] [CrossRef]
- Guindon, Y.; Therien, M.; Girard, Y.; Yoakim, C. Regiocontrolled opening of cyclic ethers using dimethylboron bromide. J. Org. Chem. 1987, 52, 1680–1686. [Google Scholar] [CrossRef]
- Palumbo, G.; Ferreri, C.; Caputo, R. A new general synthesis of halohydrins. Tetrahedron Lett. 1983, 24, 1307–1310. [Google Scholar] [CrossRef]
- Kwon, D.W.; Park, H.S.; Kim, Y.H. Efficient synthesis of iodohydrins by selective cleavage of epoxides with samarium iodide complex. Bull. Korean Chem. Soc. 2002, 23, 1185–1186. [Google Scholar] [CrossRef]
- Alvarez, E.; Nunez, M.T.; Martin, V.S. Mild and stereocontrolled synthesis of iodo- and bromohydrins by X2-Ti(O-i-Pr)4 opening of epoxy alcohols. J. Org. Chem. 1990, 55, 3429–3431. [Google Scholar] [CrossRef]
- Andrews, G.C.; Crawford, T.C.; Contillo, J.L.G. Nucleophilic catalysis in the insertion of silicon halides into oxiranes: A synthesis of o-protected vicinal halohydrins. Tetrahedron Lett. 1981, 22, 3803–3806. [Google Scholar] [CrossRef]
- Bajwa, J.S.; Anderson, R.C. A highly regioselective conversion of epoxides to halohydrins by lithium halides. Tetrahedron Lett. 1991, 32, 3021–3024. [Google Scholar] [CrossRef]
- Young-Ger, S.U.H.; Bon-Am, K.O.O.; Jung-Ae, K.O.; Youn-Sang, C.H.O. A facile and highly regioselective conversion of epoxides to bromohydrins using tetrabutylammonium bromide and magnesium nitrate. Chem. Lett. 1993, 22, 1907–1910. [Google Scholar]
- Mandal, A.K.; Soni, N.R.; Ratnam, K.R. Boron trifluoride etherate/iodide ion as a mild, convenient and regioselective ether cleaving reagent. Synthesis 1985, 1985, 274–275. [Google Scholar] [CrossRef]
- Einhorn, C.; Luche, J.L. An easy and efficient epoxide opening to give halohydrins using tin(II) halides. J. Chem. Soc. Chem. Commun. 1986, 1368–1369. [Google Scholar] [CrossRef]
- Bell, T.W.; Ciaccio, J.A. Conversion of epoxides to bromohydrins by B-bromobis(dimethylamino)borane. Tetrahedron Lett. 1986, 27, 827–830. [Google Scholar] [CrossRef]
- Righi, G.; Bonini, C.; Pandalai, S.G. Metal halides opening of oxirane and aziridine ring: Recent methodologies and applications to the synthesis of natural products. Recent Res. Dev. Org. Chem. 1999, 3, 343–356. [Google Scholar] [CrossRef]
- Sharghi, H.; Naeimi, H. Schiff-Base complexes of metal (II) as new catalysts in the high-regioselective conversion of epoxides to halo alcohols by means of elemental halogen. Bull. Chem. Soc. Jpn. 1999, 72, 1525–1531. [Google Scholar] [CrossRef]
- Sharghi, H.; Naeimi, H. Efficient, mild and highly regioselective cleavage of epoxides with elemental halogen catalyzed by 2-phenyl-2-(2-pyridyl) imidazolidine (PPI). Synlett 1998, 1998, 1343–1344. [Google Scholar] [CrossRef]
- Sharghi, H.; Eskandari, M.M. Conversion of epoxides into halohydrins with elemental halogen catalyzed by thiourea. Tetrahedron 2003, 59, 8509–8514. [Google Scholar] [CrossRef]
- Sharghi, H.; Paziraee, Z.; Niknam, K. Halogenated cleavage of epoxides into halohydrins in the presence of a series of diamine podands as catalyst with elemental idoine and bromine. Bull. Korean Chem. Soc. 2002, 23, 1611–1615. [Google Scholar]
- Venkatasubbaiah, K.; Zhu, X.; Kays, E.; Hardcastle, K.I.; Jones, C.W. Co(III)-Porphyrin-mediated highly regioselective ring-opening of terminal epoxides with alcohols and phenols. ACS Catal. 2011, 1, 489–492. [Google Scholar] [CrossRef]
- 2Zakavi, S.; Karimipour, G.R.; Gharab, N.G. Meso-tetraarylporphyrin catalyzed highly regioselective ring opening of epoxides with acetic acid. Catal. Commun. 2009, 10, 388–390. [Google Scholar] [CrossRef]
- Konaklieva, M.I.; Dahl, M.L.; Turos, E. Halogenation reactions of epoxides. Tetrahedron Lett. 1992, 33, 7093–7096. [Google Scholar] [CrossRef]
- Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Shaibani, R. Rapid and efficient ring opening of epoxides catalyzed by a new electron deficient tin(IV) porphyrin. Tetrahedron 2004, 60, 6105–6111. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Kampas, F.; Kim, J. On the preparation of metalloporphyrins. J. Inorg. Nucl. Chem. 1970, 32, 2443–2445. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Shergalis, W. Mechanistic investigations of porphyrin syntheses. I. Preliminary studies on ms-tetraphenylporphin. J. Am. Chem. Soc. 1964, 86, 3145–3149. [Google Scholar] [CrossRef]
- Joshi, N.N.; Srebnik, M.; Brown, H.C. Enantioselective ring cleavage of meso-epoxides with b-halodiisopinocampheylboranes. J. Am. Chem. Soc. 1988, 110, 6246–6248. [Google Scholar] [CrossRef] [PubMed]
- Chini, M.; Crotti, P.; Gardelli, C.; Macchia, F. Regio-and stereoselective synthesis of β-halohydrins from 1,2-epoxides with ammonium halides in the presence of metal salts. Tetrahedron 1992, 48, 3805–3812. [Google Scholar] [CrossRef]
- Hopkins, H.P., Jr.; Jahagirdar, D.V.; Windler, I.F.J. Molecular complexes in solutions containing macrocyclic polyethers and iodine. J. Phys. Chem. 1978, 82, 1254–1257. [Google Scholar] [CrossRef]
- Lang, R.P. Molecular complexes of iodine with trioctylphosphine oxide and triethoxyphosphine sulfide. J. Phys. Chem. 1974, 78, 1657–1662. [Google Scholar] [CrossRef]
Sample Availability: Samples of the H2TPP compounds 2a−e are available from the author. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
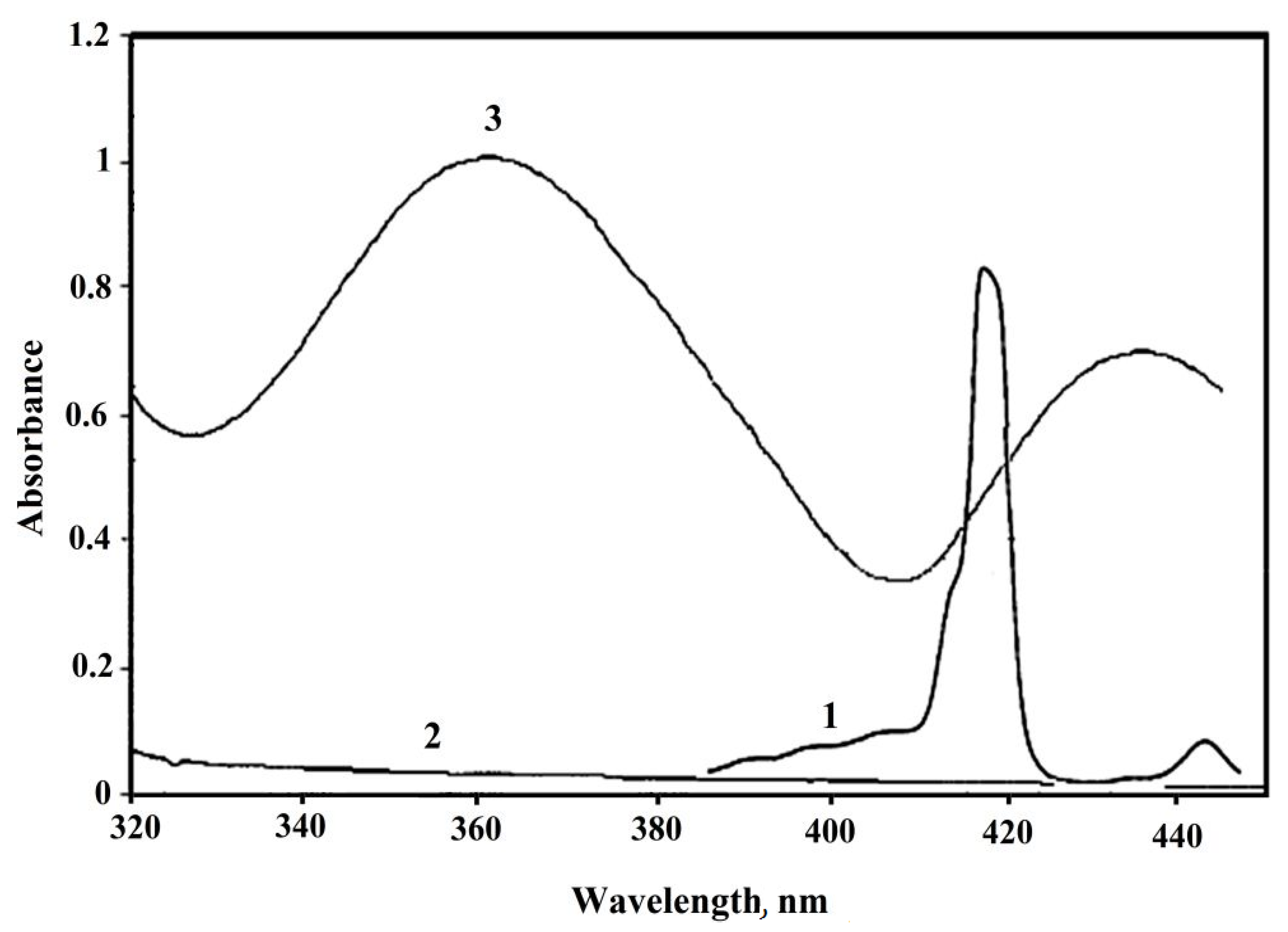
| Entry | Catalyst | Iodination | Bromination | ||
|---|---|---|---|---|---|
| Time /h | Yield a /% | Time /h | Yield a /% | ||
| 1 | 2a | 2.1 | >95 | 1.7 | >95 |
| 2 | 2b | 2.1 | 90 | 1.7 | >95 |
| 3 | 2c | 2.2 | 87 | 1.8 | 90 |
| 4 | 2d | 2.3 | 88 | 2.1 | 82 |
| 5 | 2e | 2.5 | 76 | 2.0 | 75 |
| 6 b | - | Several days | 0 | 1 | 31 c |
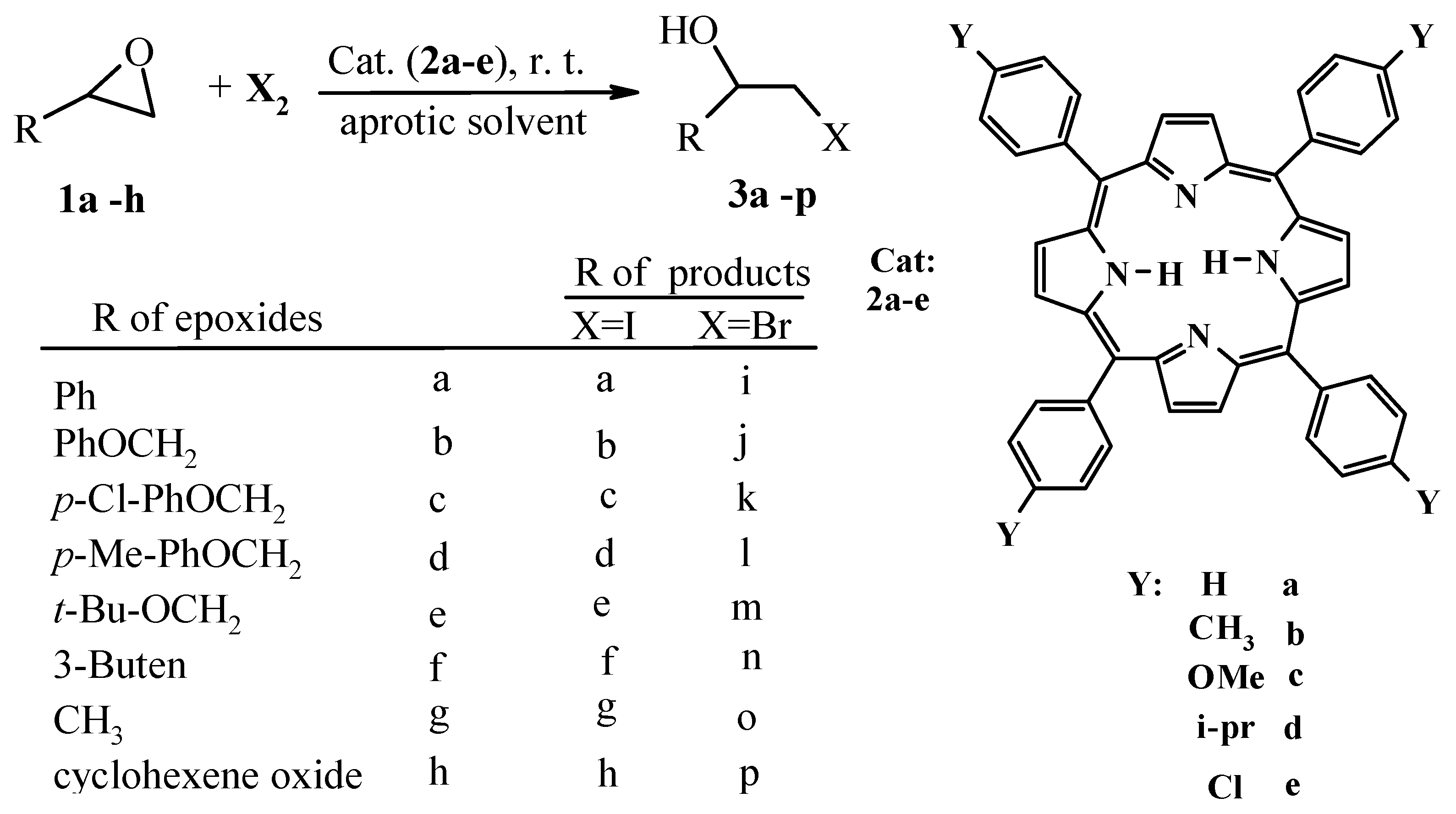
| Entry | Epoxide (1a–h) | Conditions | Time/h | Yield a /% | Product (s) (3a–p) |
|---|---|---|---|---|---|
| 1 |  | I2, 2a, r.t., CH2Cl2 | 2.1 | 81 |  |
| 2 |  | ״ | 3.9 | 80 |  |
| 3 |  | ״ | 4.3 | 81 |  |
| 4 |  | ״ | 4.5 | 82 |  |
| 5 |  | ״ | 6.5 | 78 |  |
| 6 |  | ״ | 4.6 | 82 |  |
| 7 |  | ״ | 5.8 | 77 |  |
| 9 |  | ״ | 3.5 | 72 |  |
| 10 b |  | I2, r.t., acetone | 2 | 83 |  |
| 11 c | ״ | [n-Bu4N]Br/Mg(NO3)2, CHCl3 | 5 | 78 (5:1) |  |
| 12 d | ״ | (Me2N)2BBr/CH2Cl2,N2 atm. | 12 | 75 (1:4.5) | ״ |
| 13e | ״ | SmI2 (2 eq.), THF, −78 °C | >5 min | 93 |  |
| 14 f | ״ | NH4+X−/M+., CH3CN | 1.3 | 87 (1:2) |  |
| 15 | ״ | Br2, 2a, r.t., CH2Cl2 | 1.7 | 91 |  |
| 16 |  | ״ | 2.0 | 84 |  |
| 17 |  | ״ | 2.4 | 82 |  |
| 18 |  | ״ | 2.8 | 83 |  |
| 19 |  | ״ | 2.7 | 80 |  |
| 20 |  | ״ | 2.5 | 76 |  |
| 21 |  | ״ | 3.5 | 78 |  |
| 22 |  | ״ | 2.2 | 73 |  |
| Entry | Solvent | Iodination | Bromination | ||
|---|---|---|---|---|---|
| Time /h | Yield a /% | Time /h | Yield /% | ||
| 1 | CH2Cl2 | 2.1 | >95 | 1.7 | >95 |
| 2 | CHCl3 | 2.2 | 93 | 2.0 | 95 |
| 3 | CH3CN | 2.8 | 90 | 2.5 | 91 |
| 4 | DMSO | 3.2 | 85 | 3.0 | 88 |
| 5 | THF | 3.5 | 83 | 3.2 | 85 |
| 6 | Diethyl ether | 3.5 | 83 | 3.2 | 85 |
| Entry | Run | Time (h) | Yield (%) b |
|---|---|---|---|
| 1 | 0 | 2.1 | >95 |
| 2 | 1 | 2.1 | 91 |
| 3 | 2 | 2.1 | 88 |
| 4 | 3 | 2.1 | 82 |
| 5 | 4 | 2.1 | 77 |
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Torabi, P.; Azizian, J.; Zomorodbakhsh, S. H2TPP Organocatalysis in Mild and Highly Regioselective Ring Opening of Epoxides to Halo Alcohols by Means of Halogen Elements. Molecules 2012, 17, 5508-5519. https://doi.org/10.3390/molecules17055508
Torabi P, Azizian J, Zomorodbakhsh S. H2TPP Organocatalysis in Mild and Highly Regioselective Ring Opening of Epoxides to Halo Alcohols by Means of Halogen Elements. Molecules. 2012; 17(5):5508-5519. https://doi.org/10.3390/molecules17055508
Chicago/Turabian StyleTorabi, Parviz, Javad Azizian, and Shahab Zomorodbakhsh. 2012. "H2TPP Organocatalysis in Mild and Highly Regioselective Ring Opening of Epoxides to Halo Alcohols by Means of Halogen Elements" Molecules 17, no. 5: 5508-5519. https://doi.org/10.3390/molecules17055508