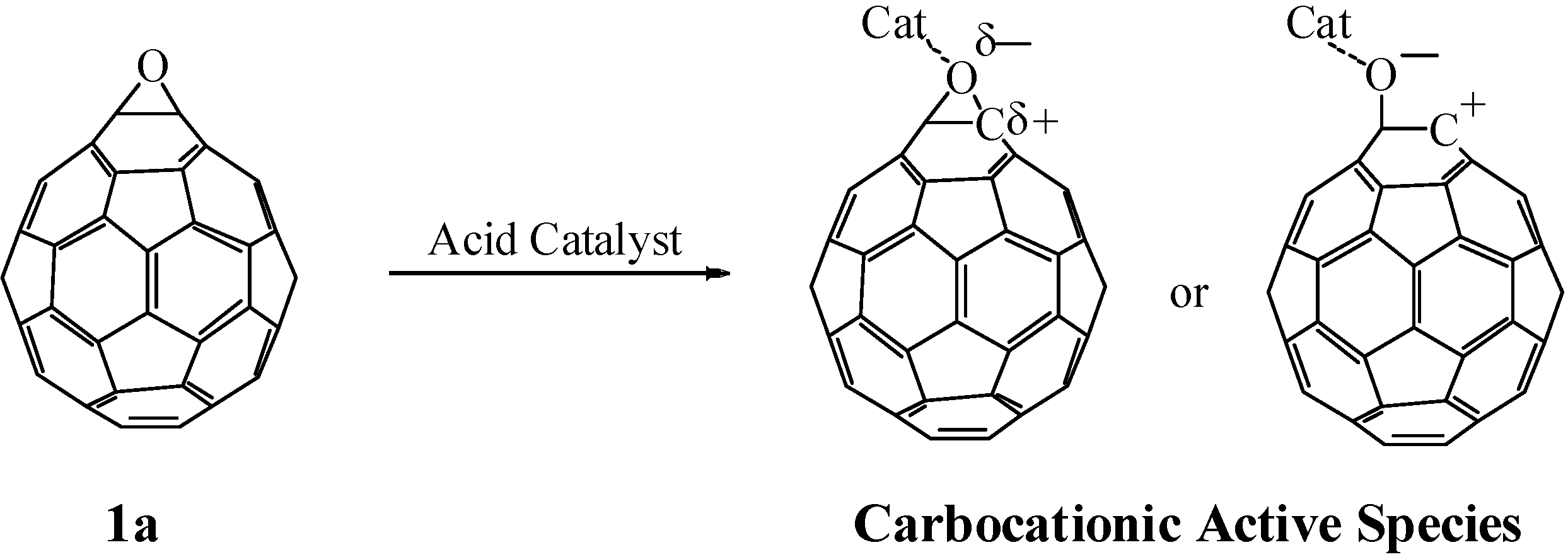
Epoxides are well-known to undergo heterolytic C–O bond cleavage in the presence of an acid catalyst to generate active carbocationic species for further reactions with nucleophiles. These active species react with various types of nucleophile such as carbonyl compounds, alcohols, and amines to afford 1,3-dioxolanes, 1,2-hydroxyethers, and 1,2-aminoalcohols, respectively. We have studied the reaction of fullerene epoxide
1a with various nucleophiles in the presence of a Lewis acid. Reaction with a carbonyl compound led to the formation of a 1,3-dioxolane derivative in high yield; interestingly, reaction with a phenol or aniline derivative afforded an O- or N-heterocycle-fused fullerene derivative. These reactions are considered to proceed via a nucleophilic reaction on the carbocationic active species generated on the carbon atom of fullerene epoxide
1a (
Scheme 1). The formation of the heterocycle-fused fullerene derivative can be accounted for by the ring closure reaction induced by intramolecular dehydration of the initially formed 1,2-substituted product. In contrast, a Lewis acid-assisted nucleophilic reaction of C
60O with an aromatic nucleophile such as toluene gave a 1,4-bisadduct. Application of these reactions to a regioisomeric fullerene polyepoxide would provide a convenient synthetic route to a variety of polyfunctionalized fullerene derivatives with a regioisomerically pure structure. These chemical transformations of fullerene epoxides are expected to play an important role in the development of functionalized fullerene cages. In this section, we review our recent progress in the work on the acid-assisted reaction of fullerene epoxides with various types of nucleophile.
Scheme 1.
Generation of active carbocationic species from 1a in the presence of an acid catalyst.
3.1. Acetalization of Fullerene Epoxides
Several methods have been reported for the preparation of fullerene-fused 1,3-dioxolane derivatives, such as the reactions of C
60 fullerene with dioxiranes [
3], acyl hypohalogenites [
19], and diacyl peroxides [
20], or the reaction of C
60 fullerene with a sodium alkoxide in the presence of air [
21]. However, all of these reactions afforded fullerene-fused 1,3-dioxolane derivatives in only low to moderate yields, (
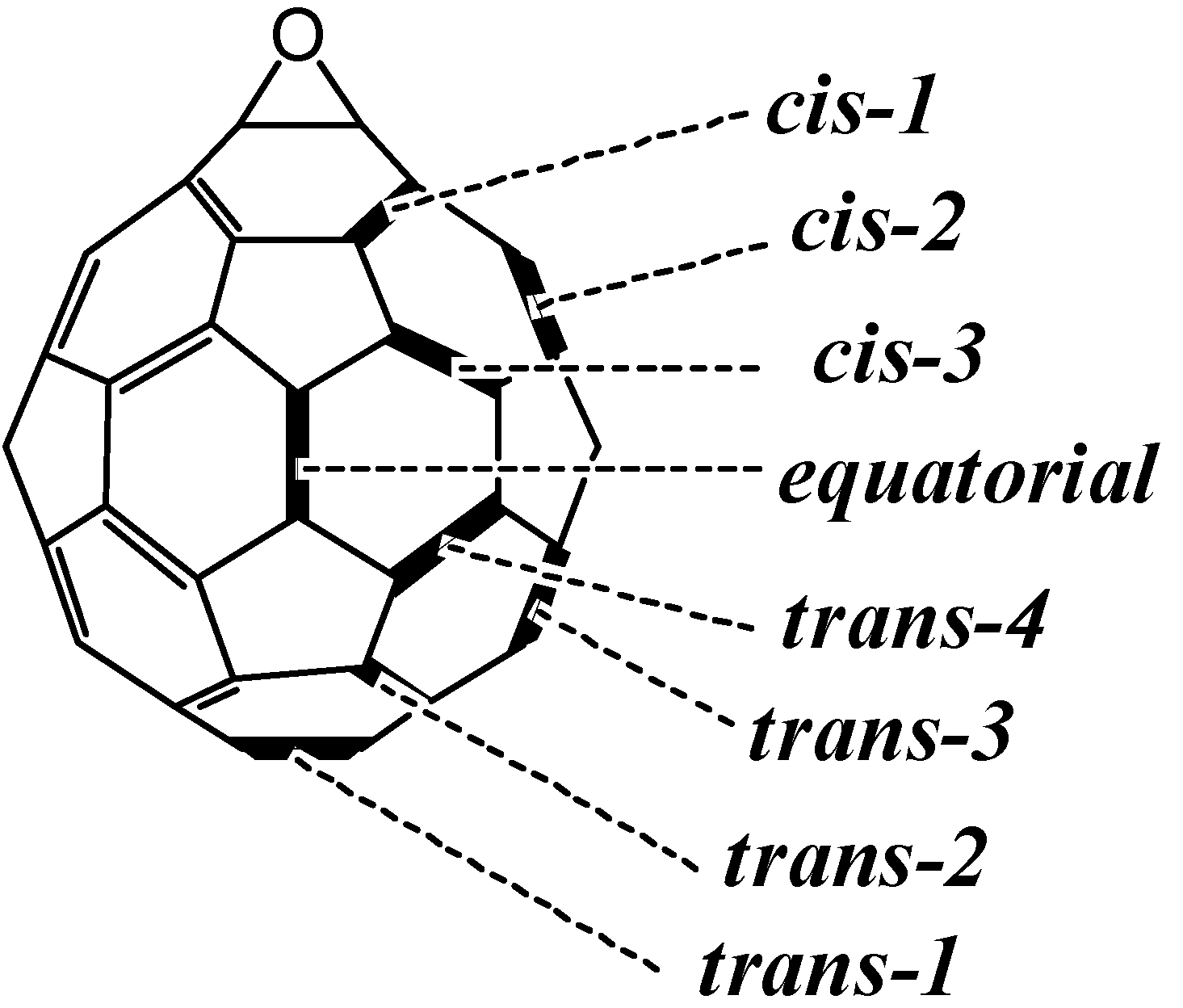
i.e., <50%). As described in the previous section, we recently developed a preparative HPLC isolation method for some regioisomeric fullerene epoxides (
Figure 3) [
7]. Isolation of these epoxides prompted us to employ them as a starting material for efficient synthesis of fullerene-fused 1,3-dioxolane derivatives. We have found that the acetalization reactions of fullerene epoxide
1a with various carbonyl compounds occur in the presence of a Lewis acid catalyst, an ion-exchange resin such as Amberlite, and a clay mineral such as montmorillonite to afford the corresponding 1,3-dioxolane derivatives in very high yields (
Table 2) [
9,
22].
Table 2.
Acetalization of fullerene epoxide 1a with carbonyl compound in the presence of an acid catalyst.
The reaction of a toluene solution of fullerene epoxide
1a with excess amounts (200 equiv.) of a benzaldehyde derivative at 75 °C in the presence of a Lewis acid catalyst led to the formation of 1,3-dioxolane
3 in very high yield.
Table 2 also shows that a similar acetalization of
1a with other ketone compounds such as acetophenone, methyl ethyl ketone or cyclohexanone took place to give the corresponding 1,3-dioxolanes in moderate yields. Furthermore, a similar reaction of
1a with γ-butyrolactone in benzene afforded an ortho-ester type 1,3-dioxolane derivative in high yield, although in toluene, it resulted in the formation of a 1,4-addition product of toluene to
1a. This fact suggests that the nucleophilicity of a carbonyl group of an ester compound is not as strong as that of an aldehyde or a ketone, such that toluene instead acts preferentially as a nucleophile to the carbocationic species to yield a 1,4-addition product (1,4-bis(p-tolyl)-1,4-dihydro[60]fullerene). The Lewis acid catalyzed 1,4-addition reaction of aromatic nucleophiles to
1a will be discussed in
Section 3.2. Recently Gan and Zhang
et al. reported that a Lewis acid such as BF
3·Et
2O catalyzes the acetalization reaction of the fullerene epoxide bearing
t-butylperoxo groups to yield the corresponding 1,3-dioxolane derivatives in moderate yields [
23].
An epoxy ring is known to undergo a Lewis acid catalyzed acetalization to afford 1,3-dioxolane [
24,
25,
26]. Generally the acetalization is assumed to follow a concerted S
N2-like mechanism involving backside attack of a carbonyl oxygen on an epoxide carbon followed by rotation of the C–C bond of the epoxide and then the second C–O bond formation to produce a 1,3-dioxolane [
26]. However, such a backside attack on a fullerene epoxide is not expected to occur owing to occupation of the entire side opposite the epoxy ring by the fullerene cage. According to this view, an acetalization of a fullerene epoxide is not expected to occur unless an alternative mechanism is involved in a front side attack of a carbonyl compound on the epoxy ring of the fullerene epoxide. The acetalization reaction of fullerene epoxide
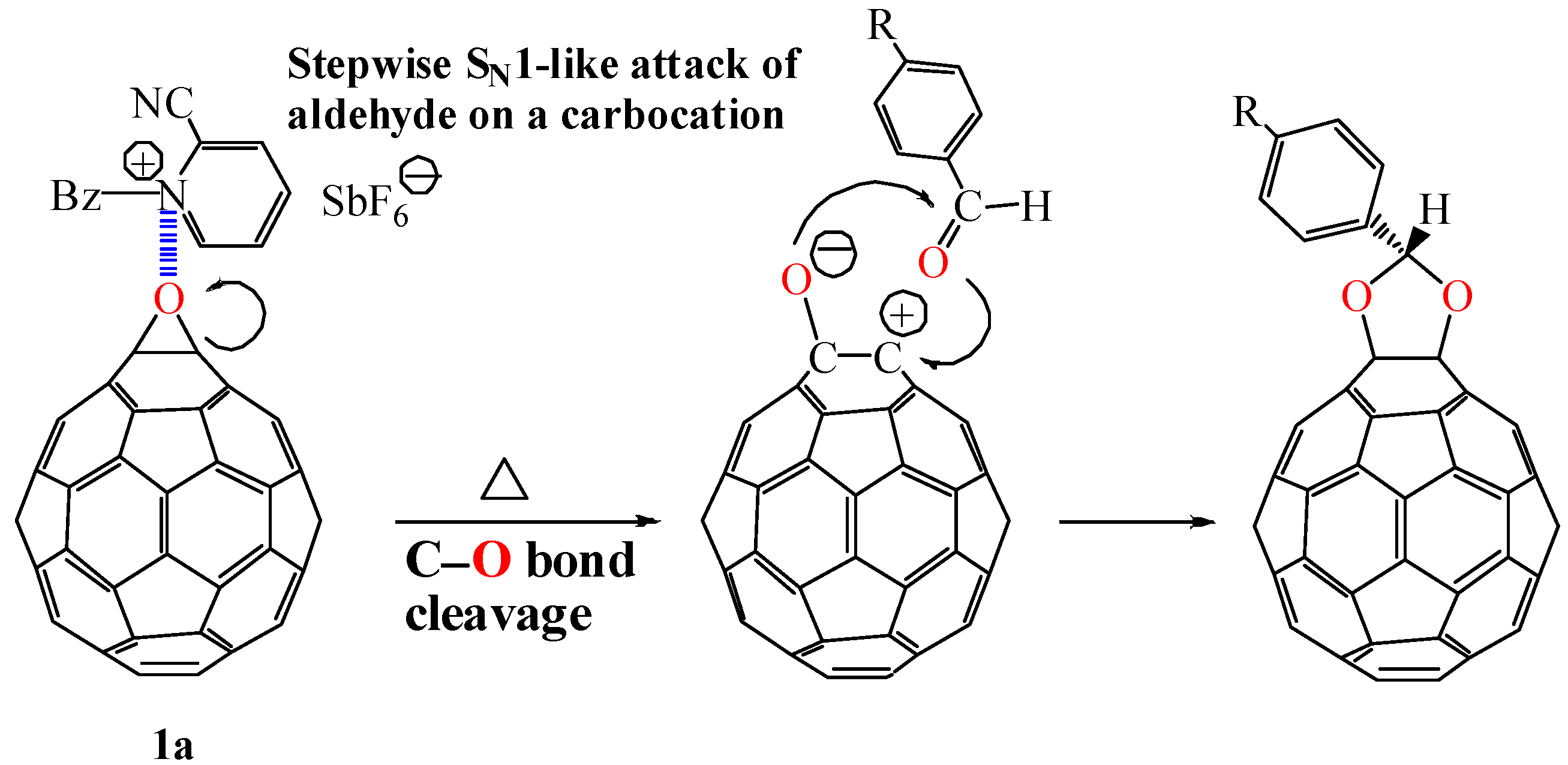
1a with benzaldehyde at 75 °C for 1 d did not proceed at all in the absence of a Lewis acid and resulted in the complete recovery of
1a. This result suggests that the Lewis acid catalyzes the acetalization reaction to proceed by a stepwise S
N1-like mechanism (
Scheme 2). A Lewis acid induces a catalytic C–O bond cleavage of the epoxy ring to generate a carbocation on the carbon atom of the fullerene moiety. This is followed by nucleophilic attack of a carbonyl compound on the carbocation. An energetically unfavorable S
N1-like attack of a carbonyl compound on the carbocation from the fullerene surface would be forced to occur with the requirement of relatively high activation energy for the C–O bond cleavage of the epoxy ring.
Scheme 2.
Stepwise SN1-like acetalization of fullerene epoxide 1a with benzaldehyde derivative.
Scheme 2.
Stepwise SN1-like acetalization of fullerene epoxide 1a with benzaldehyde derivative.
The activation energy for the reaction of
1a with benzaldehyde was determined to be 112.7 kJ·mol
−1 from the Arrhenius plots of ln k
vs. 1/T, where k is the pseudo-first order rate constant and T is the reaction temperature. Based on the activation energy determined, the rate constant at room temperature (293 K) can be calculated to be 9.5 × 10
−7 s
−1, which is more than 10
3 times smaller than that at 75 °C (k = 1.43 × 10
−3 s
−1). Therefore, that the Lewis acid catalyzed acetalization of
1a with benzaldehyde did not occur at room temperature is unsurprising, although a similar acetalization of oxiranes such as but-2-ene epoxide or styrene oxide readily proceeds at room temperature and even at 0 °C to yield 1,3-dioxolanes according to an energetically favorable S
N2-like mechanism [
24,
25,
26].
The application of the above acetalization reaction to the regioisomerically pure fullerene di-epoxide
1b (
cis-isomer) or
1c (
equatorial isomer) would lead to chemical transformation of the two epoxy rings while conserving their arrangement on the fullerene cage. Under reaction conditions similar to those for mono-epoxide
1a, the fullerene di-epoxides
1b and
1c were also subjected to acetalization with benzaldehyde derivatives to give bis-1,3-dioxolanes
5a and
5b, respectivelyinhigh yields (
Table 3) [
9,
21]. The visible absorption spectra of
5a and
5b show the bands characteristic to those of the di-epoxides
1b and
1c, respectively, that is,
5a showed a sharp band at 423 nm, which is the characteristic band for the
cis-isomer
1b [
11,
15]. This fact suggests that during the acetalization reaction, no migration of the carbocation on the fullerene moiety occurs and that the acetalization reaction of the fullerene di-epoxide proceeds while the rearrangement of the two epoxy rings on the fullerene cage is conserved.
Table 3.
Acetalization of fullerene epoxide
1b or
1c with benzaldehyde derivatives
2 in the presence of pyridinium salt
4a or
4b shown in
Table 2.
The
1H-NMR spectrum of
5a (R
3 =
n-BuO) showed the two unequivalent acetal methine protons of equal intensity. In addition, it showed two sets of four phenyl protons and nine
n-butoxy protons, also of equal intensity. Thus, the
1H-NMR spectrum of
5a clearly demonstrates that the two phenyl groups are unsymmetrically disposed with respect to the fullerene moiety. The
13C-NMR spectrum of
5a showed 51 signals for the fullerene sp
2 carbons, indicating a lack of symmetry of the fullerene cage. Based on the above NMR analysis,
bis-1,3-dioxolane
5a is confirmed to have the structure of the stereoisomer depicted in
Table 3. However, the configuration of the two phenyl groups in
5b remains undetermined because no definitive information suggesting their configuration could be obtained from the
1H-NMR spectra of
5b. We assume that
5b is likely to be a mixture of two stereoisomers.
The fullerene di-epoxide
1b (
cis-isomer) or
1c (
equatorial isomer) also reacts with a ketone compound such as cyclohexanone in the presence of a Lewis acid catalyst or Amberlyst 15
® to yield the corresponding
bis-1,3-dioxolane derivative in moderate yields (50–70%) [
27]. These acetalization reactions are presumed to proceed with conservation of their arrangement on the fullerene cage.
The present work has clearly shown that the epoxy moiety of fullerene epoxides can be readily converted to 1,3-dioxolane derivatives in high yield by treatment with a carbonyl compound in the presence of a Lewis acid, ion-exchange resin or clay mineral. The acetalization reaction proceeds by a stepwise SN1-like mechanism which involves the nucleophilic attack of a carbonyl moiety at the carbocationic active species generated on the carbon atom of fullerene epoxides. Furthermore, the efficient bis-acetalization of a regioisomerically pure fullerene di-epoxide occurs under conservation of the arrangement of the two epoxy moieties to yield bis-1,3-dioxolane derivatives in high yield. These results could lead to development of a new methodology for facile synthesis of a variety of regioisomeric polyfunctionalized fullerene derivatives by means of efficient chemical transformation of a specific regioisomeric fullerene polyepoxide. In order to elucidate the reactivity of the carbocationic active species generated from fullerene epoxides, the next part of this section is oriented toward studies on the reaction of fullerene epoxides with nucleophiles other than carbonyl compounds.
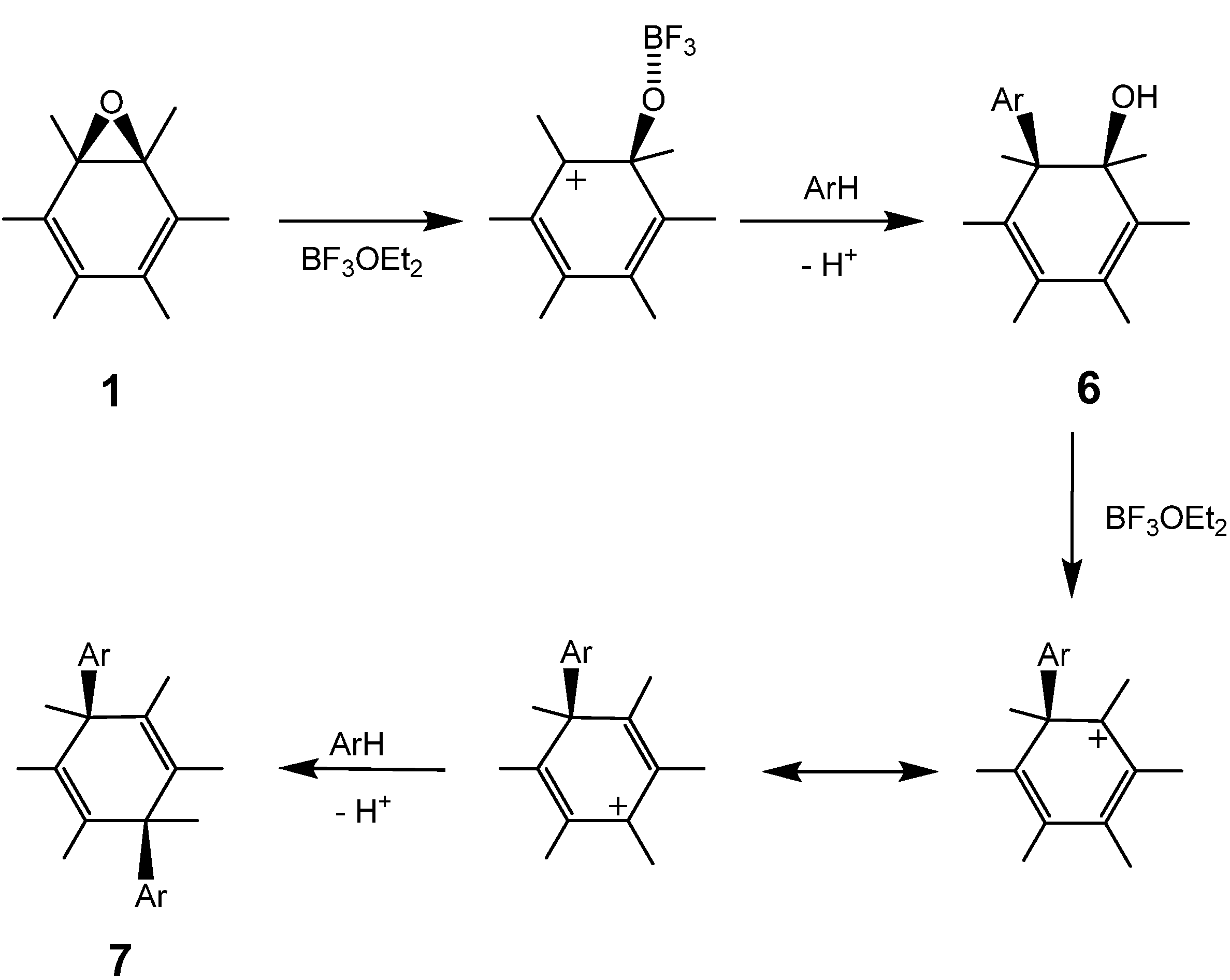
3.2. Nucleophilic Substitution of Fullerene Epoxide with Aromatic Compounds
The reaction of a toluene solution of fullerene epoxide
1a in the presence of 5 equiv. of boron trifluoride etherate (BF
3·OEt
2) at r.t. for 60 min led to the formation of 1,4-bis(
p-tolyl)-1,4-dihydro [60]fullerene (
7a) in 80.8% yield together with a very small amount of pristine fullerene C
60 based on HPLC analysis [
28]. Compound
7a was purified by flash column chromatography on a silica gel with n-hexane as an eluent. A detailed spectral characterization of
7a revealed that two
p-tolyl moieties were introduced to the C
60 core in a 1,4-addition pattern. The FT-IR spectrum was in good agreement with that in an earlier report [
29,
30]. The characteristic vibrations for the 1,4-adduct were found at 1,430, 1,187, 573, and 527 cm
−1, and the C-H vibrations for toluene showed strong vibrations at 2,963 and 810 cm
−1. The atmospheric pressure photochemical ionization (APPI) mass spectrum showed a molecular ion peak at
m/z 902 (
i.e., 166 units more than
1a), corresponding to a 1:2 adduct of C
60 and toluene-H. In the
1H-NMR spectrum, in addition to two doublets at 7.94 (2H), 7.93 (2H), 7.28 (2H), and 7.27 (2H) ppm for eight phenyl protons, one singlet methyl proton was observed at 2.46 (6H) ppm. The
13C-NMR showed the characteristic absorption of aromatic carbons in the
p-tolyl group at 138.25 (2C), 136.91 (2C), 129.52 (4C), and 126.94 (4C) ppm, and two quaternary carbons of the C
60 moiety were observed at 60.86(2C) ppm. The UV-VIS spectrum showed a characteristic absorption band around 450 nm. The 1,4-bisadduct structure for
7a was evidently confirmed by comparing these spectra with FT-IR,
1H-NMR and
13C-NMR spectra for similar compounds [
29,
30].
Reactions of
1 with other aromatic compounds were carried out under the same conditions as described above, the results of which are listed in
Table 4. In anisole,
o-xylene and
m-xylene the reaction produced the corresponding 1,4-bisadducts (
7b,
7c and
7d) in 45–95% yields, whereas neither the 1,2-(
6e) nor 1,4-bisadduct (
7e) was produced in
p-xylene.
Table 4.
Substitution of 1 with aromatic compounds.
Table 4.
Substitution of 1 with aromatic compounds.
![Molecules 17 06395 i017]() |
|---|
| | Aromatic Compound
a | Isolated Yield of 6 / %
b | Isolated Yield of 7 / %
c |
|---|
| a | toluene | 0 | 80.8 |
| b | anisole | 0 | 95.8 |
| c | o-xylene | 0 | 76.5 |
| d | m-xylene | Trace | 45.3
d |
| e | p-xylene | 0 | 0 |
| f | 1,3,5-trimethylbenzene | 35.3 | Trace |
| g | chlorobenzene | 0 | 0 |
| h | benzene | 0 | 0 |
Although the purification of 1,4-bisadducts
7a,
7b, and
7c was easily carried out with the HPLC apparatus,
3d was not well separated owing to the formation of structural isomers. In the case of 1,3,5-trimethylbenzene, the corresponding 1-(substituted phenyl)-2-hydroxyl-1,2-dihydro[60]fullerene (
6f) was dominantly formed instead of the 1,4-bisadduct, as identified by UV-VIS, APPI mass,
1H- and
13C-NMR spectra. Usually the sp
3 carbons of the C
60 cage were observed to shift downfield
ca. 10 ppm for 1,2-adducts relative to the 1,4-adducts. The two peaks at 86.9 and 72.0 ppm for the two sp
3 carbons of the fullerene core in
6f indicate a typical structure of 1,2-adduct. This suggests that the formation of 1,4-bisadduct from
1a proceeds stepwise via 1,2-adduct
6 as an intermediate. In fact, the formation of
6d can be observed as a transient intermediate in the course of the reaction in
m-xylene. With respect to the UV-VIS spectrum beyond wavelengths of 300 nm,
6d is identical to
6f. The substitution reaction scarcely proceeded in benzene and chlorobenzene, even above 75 °C, and pristine C
60 was observed instead of 1,4-adducts. The formation of pristine C
60 without epoxy group was observed. Thus, it appears that the progress of the reaction depends considerably on the nucleophilicity of the aromatic compound. In the absence of BF
3℘OEt
2, no reaction was observed in any solvent. From this general behavior, we may conclude that the first substitution of
1a to obtain
6 occurs via a carbocationic intermediate generated with the assistance of the Lewis acid, followed by a nucleophilic attack of an aromatic compound on the cation. In the transformation of
6 into
7, a reasonable assumption is that the substitution of the hydroxyl group proceeded by an S
N1- or S
N2-type mechanism with allylic rearrangement (
Scheme 3).
Scheme 3.
Proposed mechanism for the substitution of C60O with aromatic compounds. Part of the fullerene surface is shown.
Scheme 3.
Proposed mechanism for the substitution of C60O with aromatic compounds. Part of the fullerene surface is shown.
Generally, the Lewis acid-assisted substitution of the epoxy group is assumed to follow a concerted S
N2-like mechanism involving a backside attack of a nucleophilic reagent on an epoxide carbon atom [
26]. However, such an attack is rather unrealistic, because the fullerene cage occupies the entire side opposite the epoxy ring. The first substitution of the epoxy ring on a fullerene, therefore, would occur with an energetically unfavorable S
N1-like attack of a nucleophilic reagent on the carbocation generated by the C−O bond cleavage of the epoxy ring. On the other hand, the second substitution for
6 might occur by an S
N2'-type mechanism, in which the nucleophilic attack on the residual hydroxyl group occurs first, followed by the elimination of -OH on the side with an allylic rearrangement [
31]. The fact that no reaction proceeded in the absence of the Lewis acid catalyst suggests that the second substitution occurs by an S
N1- or S
N2-type mechanism with allylic rearrangement, where
7 is formed via a carbocationic intermediate with an allylic resonance [
32,
33,
34,
35,
36], generated with the assistance of BF
3·OEt
2. In no case was the formation of 1,2-bis(substituted phenyl)adduct observed, presumably because of the steric hindrance between the two substituents.
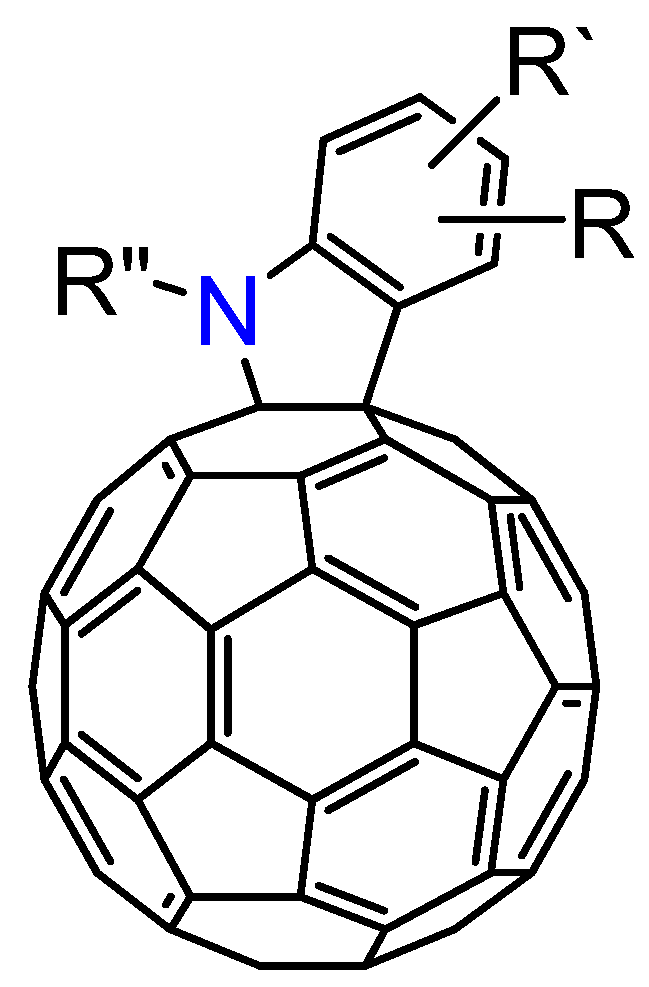
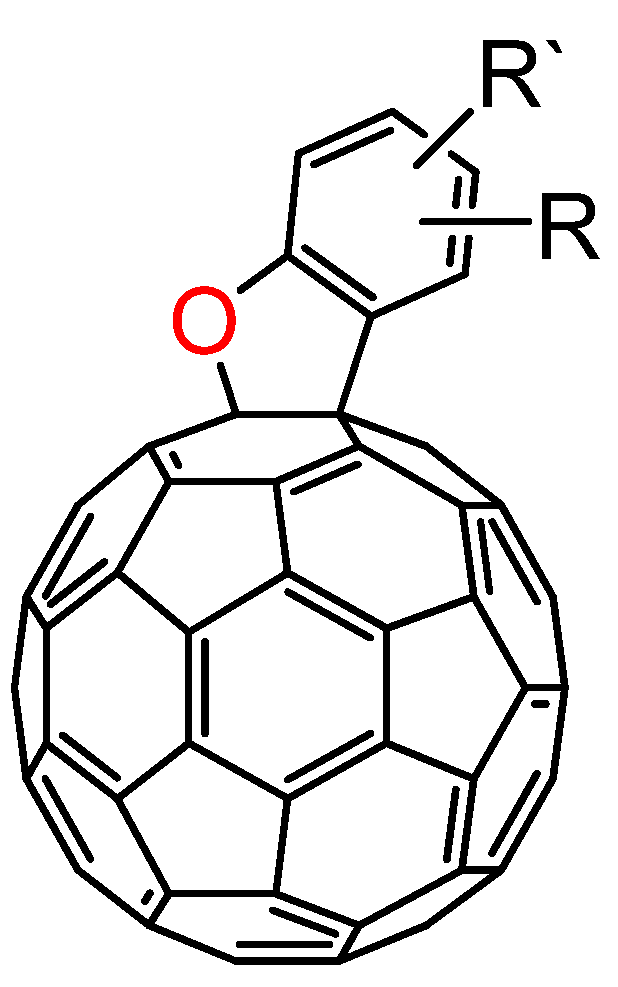
3.3. Formation of Indolino[2',3':1,2][60]fullerene Derivatives from C60O and Aniline Derivatives
As shown in
section 3.1, the reactions of C
60O
x with nucleophiles in the presence of acidic compounds have uncovered a means to achieve the regioselective functionalization of various fullerene derivatives. In extending this procedure, we found that a reaction of C
60O with aniline derivatives in the presence of acidic compounds afforded the indoline-fused fullerene derivatives: indolino[2',3':1,2]-1,2-dihydro[60]fullerene (
Figure 4), in moderate to excellent isolated yields (ca. 60–80%) [
37]. To the best of our knowledge, only a few cases have thus far been reported on the preparation of indolino[60]fullerene analogues [
38,
39,
40,
41,
42]. This method enabled access to a wide variety of indolino[60]fullerene derivatives because numerous anilines are commercially available.
Figure 4.
Molecular structure of indolino[2',3':1,2]-1,2-dihydro[60]fullerene derivatives 8.
Figure 4.
Molecular structure of indolino[2',3':1,2]-1,2-dihydro[60]fullerene derivatives 8.
First, we examined the catalytic activities of different types of acidic compound for this reaction by using 4-dodecylaniline as a model compound. The examined acidic compounds were BiCl
3, BF
3·OEt
2, Montmorillonite K10, Sepiolite, TsOH, and Amberlyst 15
®. The Brønsted acids TsOH and Amberlyst 15
® did not exhibit catalytic activities for the reactions, and all of the C
60O was converted to pristine C
60. Although the Lewis acid compound; BiCl
3 gave products with moderate production yields (−50%), BF
3·OEt
2 did not give products and only the pristine C
60 was obtained. The clay compounds, Montmorillonite K10 and Sepiolite exhibited excellent catalytic activities toward the cyclization reactions (
Table 5).
Table 5.
Catalytic activities of acidic compounds on formation of 5-dodecylindolino[60]fullerene.
Table 5.
Catalytic activities of acidic compounds on formation of 5-dodecylindolino[60]fullerene.
| Catalyst | Type | Time | Yield of 8a / % a |
|---|
| Montmorillonite K10 | clay mineral | 6 h | 56.7 |
| Sepiolite | clay mineral | 6 h | 63.8 |
| BiCl3 | Lewis acid | 2 h | 50.3 |
| BF3·OEt2 | Lewis acid | 2 h | 0 (C60 was given) |
| p-TsOH | Brønsted acid | over 5 days | 0 (C60 was given) |
| Amberlyst 15® | Brønsted acid | over 5 days | trace (C60 was given) |
The lack of catalytic activity of the Brønsted acid may be attributed to a neutralization process between the Brønsted acid and anilines. For BiCl3, the Lewis acidity may be slightly strong; thus, the epoxy oxygen may have been removed from the C60 core more quickly than with the nucleophilic addition of anilines. In contrast, BF3·OEt2 forms a Lewis acid-base complex with aniline. The capped aniline may have lost its nucleophilicity, and thus completely prevented the reaction from proceeding. Cray minerals presumably have moderate Lewis acidity relative to BF3, and may thus be appropriate for the reaction.
Subsequently, in order to examine the versatility of this reaction, we carried out the reactions with a wide variety of anilines. The examined anilines and results are summarized in
Table 6. Each aniline examined gave the corresponding indolino[60]fullerene derivative in good yield except for the reaction with
N-Me-
p-toluidine (Entry
8f). For
8f, the steric hindrance of the methyl group on the nitrogen atom might inhibit nucleophilic addition to the carbon atom of the fullerene core.
Table 6.
Isolated yields of indolino[60]fullerene derivatives.
Table 6.
Isolated yields of indolino[60]fullerene derivatives.
| Entry | Aromatic Amine | 5- | N- | Yield / % a |
|---|
| 8a | 4-Dodecylaniline | n-C12H25 | H | 77.7 |
| 8b | 4-
n-Butylaniline | n-C4H9 | H | 64.1 |
| 8c | p-Toluidine | Me | H | 84.0 |
| 8d | 4-Fluoroaniline | F | H | 75.7 |
| 8e | Aniline | H | H | 78.9 |
| 8f | N-Me-p-toluidine | Me | Me | 29.3 |
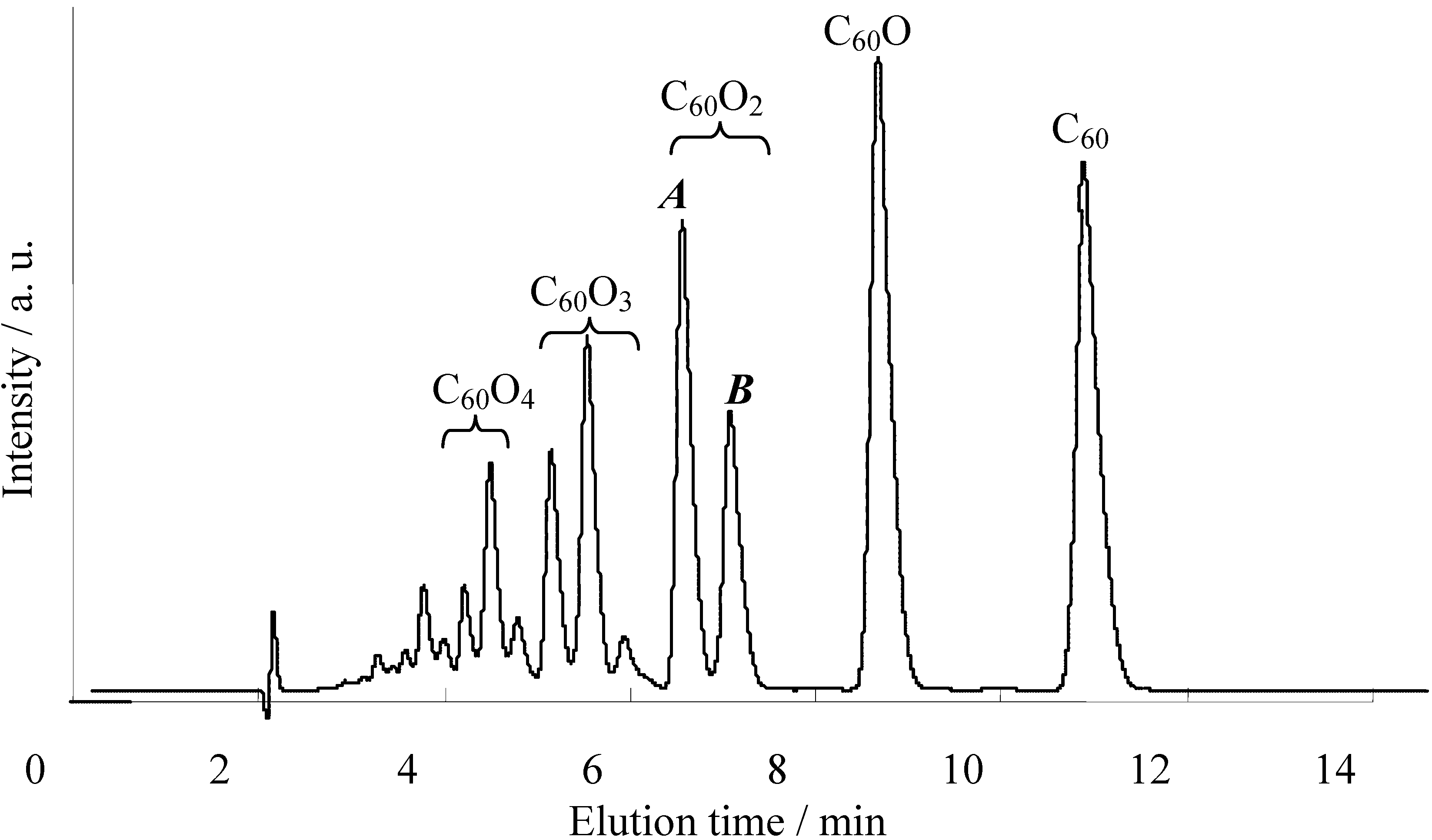
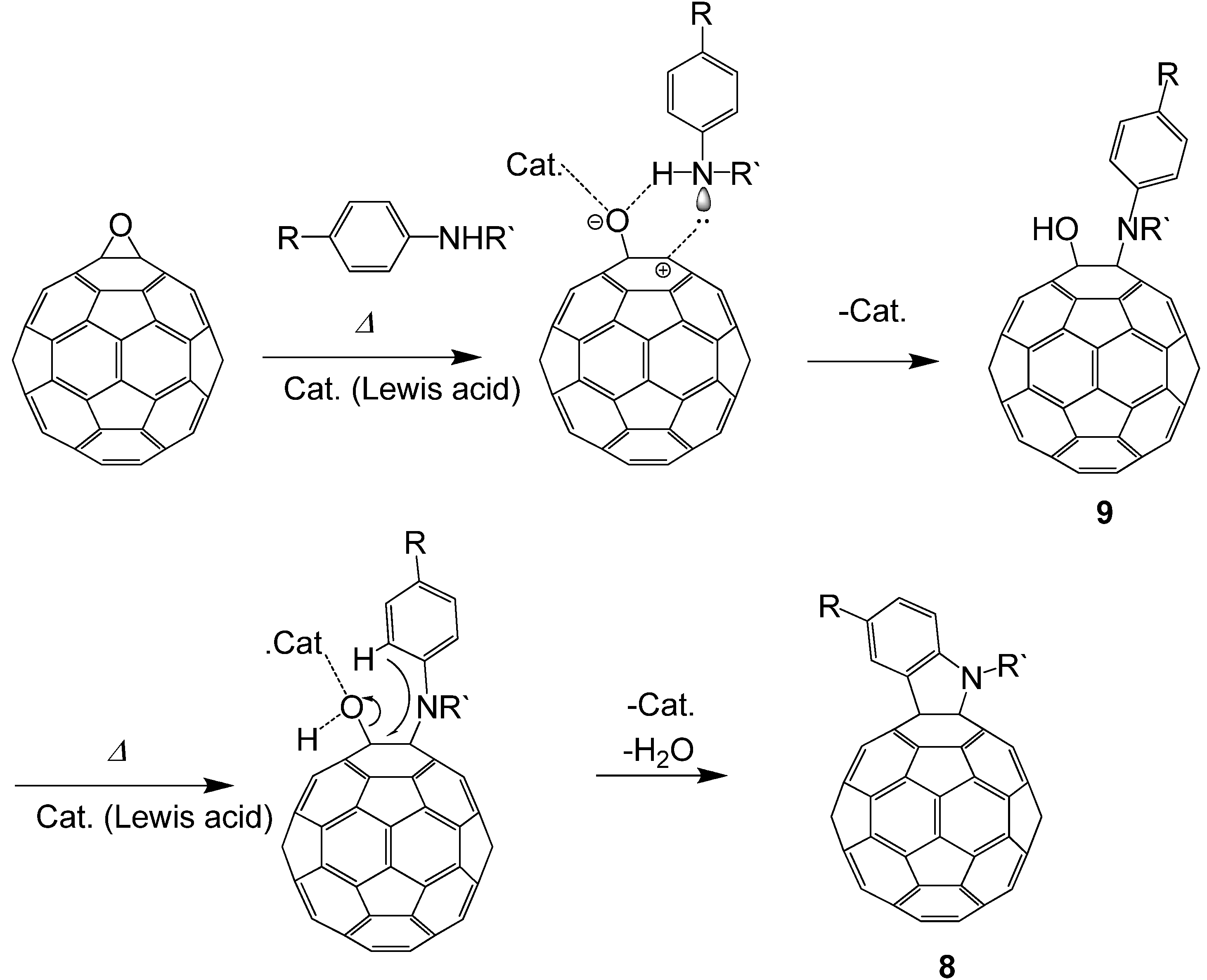
Next, we attempted to reveal the reaction mechanism of the formation of indolino[60]fullerene from C
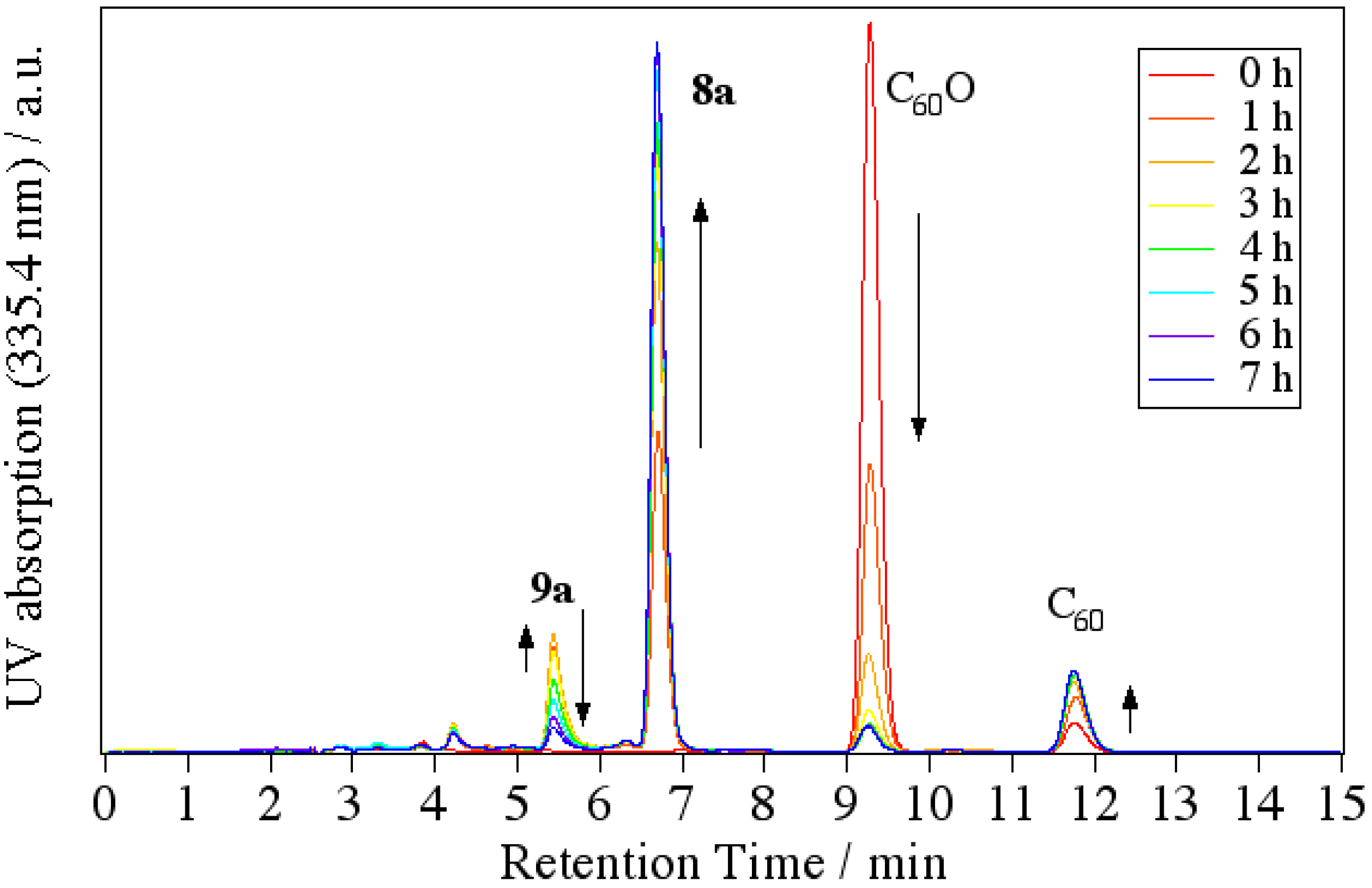
60O and anilines. By tracing the reaction with an HPLC-MS system, we revealed that the cyclization reaction proceeded via 1,2-anilinoalcohol fullerene derivative. The time course of the HPLC chart and the change in the relative peak area for each compound are shown in
Figure 5.
Figure 5.
Time course of the HPLC chart for the reaction of C60O with 4-dodecylaniline.
Figure 5.
Time course of the HPLC chart for the reaction of C60O with 4-dodecylaniline.
At the start time, peaks corresponding to C60O at 9.3 min and slightly impure C60 at 11.8 min appeared. At first, a new peak (9a) appeared at 5.4 min in the LC chart, and then, a peak corresponding to 8a at 6.8 min increased as the peak area of 9a decreased. Finally, the peaks of the starting C60O and 9a were completely consumed and the peak 8a and a small peak corresponding to pristine C60 as a decomposed compound at 11.8 min were observed. Therefore, we surmised that 9a was an intermediate of the indolino[60]fullerene derivative. To confirm our assumption, isolated 9a with the catalyst under similar reaction conditions, whereupon the reaction gave 8a.
In order to assign each peak, we isolated and characterized compound
9a by means of IR, APPI-MS, and NMR spectra. In the IR spectrum, an absorption corresponding to O–H stretching was observed at 3,793 cm
–1. The MS spectrum of
9a showed a peak at
m/
z = 998. The MS of the peak corresponds to the summation of C
60O (mw = 736) and dodecylaniline (mw = 262). As a result of
1H-NMR, we observed in the range of 0.87–2.60 ppm, two doublet peaks of aryl protons corresponding to four aniline protons at 7.19 and 7.61 ppm, and one singlet peak of NH at 5.93 ppm. Taking these results into consideration, we assigned
9a to a 1-anilino-2-hydroxy[60]fullerene derivative (
Scheme 4).
Scheme 4.
Plausible reaction mechanism of indolino[60]fullerene derivative formation from C60O and anilines.
Scheme 4.
Plausible reaction mechanism of indolino[60]fullerene derivative formation from C60O and anilines.
Based on the experimental results, we proposed a plausible mechanism for the formation of indolino[60]fullerene (8) via 1,2-anilinohydroxy fullerene (9) (
Scheme 4). At first, the epoxy ring is opened by adding a Lewis acid to the oxygen of the epoxide. Then, nucleophilic addition of anilines to the carbon atom underlying the epoxy oxygen forms a 1,2-anilinohydroxy fullerene derivatives. Consequently, the Lewis acid again adds to the oxygen atom of the hydroxyl group, and a C–C bond forms between the carbon of C
60 and the
ortho-position of the anilino group to give the indoline-fused fullerene derivative.
3.4. Formation of Benzo[b]furano[2',3':1,2][60]fullerene Derivatives from C60O and Phenols
A similar nucleophilic addition to the fullerene core and the consequent cascade cyclization reaction with phenols via a 1-hydroxy-2-phenolic fullerene derivative as an intermediate was also observed. This reaction gave benzo[
b]furano[2',3':1,2][60]fullerene derivatives in good yields as well (
Figure 6) [
43].
Figure 6.
Molecular structure of benzo[b]furano[2',3':1,2][60]fullerene derivatives.
Figure 6.
Molecular structure of benzo[b]furano[2',3':1,2][60]fullerene derivatives.
Unlike in the case of anilines, BF3·OEt2 exhibited superior catalytic activity for the reaction of C60O with phenols. The reaction of C60O with phenol derivatives in the presence of BF3·OEt2 was complete within 30 min for nearly all examined phenols. The different catalytic activities of the Lewis acids may be attributed to the Lewis acidity of a catalyst, the Lewis basicity of a nucleophile, or both. For the reaction with anilines, a complexation between BF3 and the amine occurs preferentially over the addition of BF3 to the epoxy oxygen. Therefore, either BF3 or the aniline is completely consumed, and the reaction cannot proceed at all. On the other hand, for benzo[b]furano[60]fullerenes, the Lewis basicity of nearly all phenols is weaker than that of anilines. BF3 thus effectively catalyzes the reaction of epoxy ring opening, nucleophilic addition, and cyclization.
The production yields of the benzo[
b]furano[60]fullerene derivatives were subjected to the electron-donating and withdrawing properties of the substituent of the phenols. The isolated yields are summarized in
Table 7.
Table 7.
Isolated yields of benzo[b]furano[60]fullerene derivatives.
Table 7.
Isolated yields of benzo[b]furano[60]fullerene derivatives.
| Entry | Phenols | Yield / %
a |
|---|
| 10b | 4
-n-octylphenol | 91.5 |
| 10c | 4-
n-butylphenol | 88.8 |
| 10d | 4-methylphenol | 91.5 |
| 10e | 4-methoxyphenol | 72.9 |
| 10g | 4-fluorophenol | 30.8 |
| 10h | phenol | 41.1 |
| 10i | 3,5-di-methoxyphenol | 82.2 |
| 10l | 4-nitrophenol | 0 |
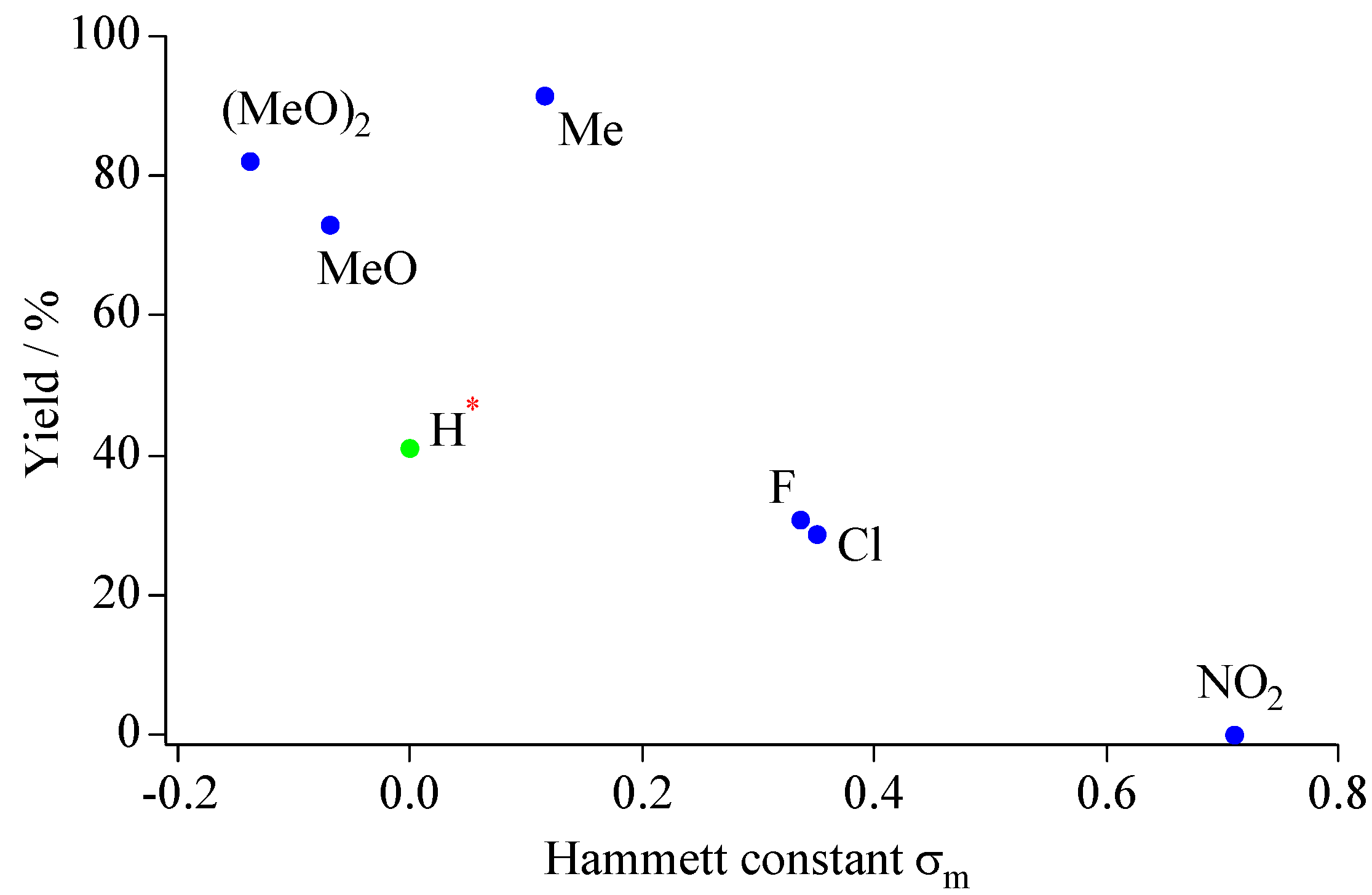
Unlike the case of anilines, the yields of benzo[
b]furano[60]fullerene derivatives were strongly influenced by the type of substituent of the phenols.
Figure 7 shows a plot of the isolated yields for benzo[
b]furano[60]fullerene derivatives as a function of the Hammett constant for the 2-position of the precursor 4-substituted phenols [
44]. Negative Hammett constants correspond to electron-withdrawing (EW) property, and positive ones correspond to the electron-donating (ED) property. The stronger ED property of the substituent increases the nucleophilicity of the oxygen atom of phenols and the carbon atoms of
ortho-position of the phenols. In contrast, EW groups reduce the nucleophilicity of the phenolic oxygen and the carbon atoms of
ortho-position. Therefore, ED functional groups improve the production yields of nucleophilic addition and cascade cyclization reactions. Conversely, 4-nitorophenol could not produce the benzo[
b]furano[60]fullerene owing to the strong electron-withdrawing property of the nitro group. The low yield with pristine phenol (entry
10h) is presumably due to the competitive formation of the 1,4-bisadduct (
Section 3.2), because the
para-position of the phenol is not substituted.
Figure 7.
A plot of Hammett constant vs. production yields forbenzo[b]furano[60]fullerene derivatives.
Figure 7.
A plot of Hammett constant vs. production yields forbenzo[b]furano[60]fullerene derivatives.






{kind=link}
{kind=link}
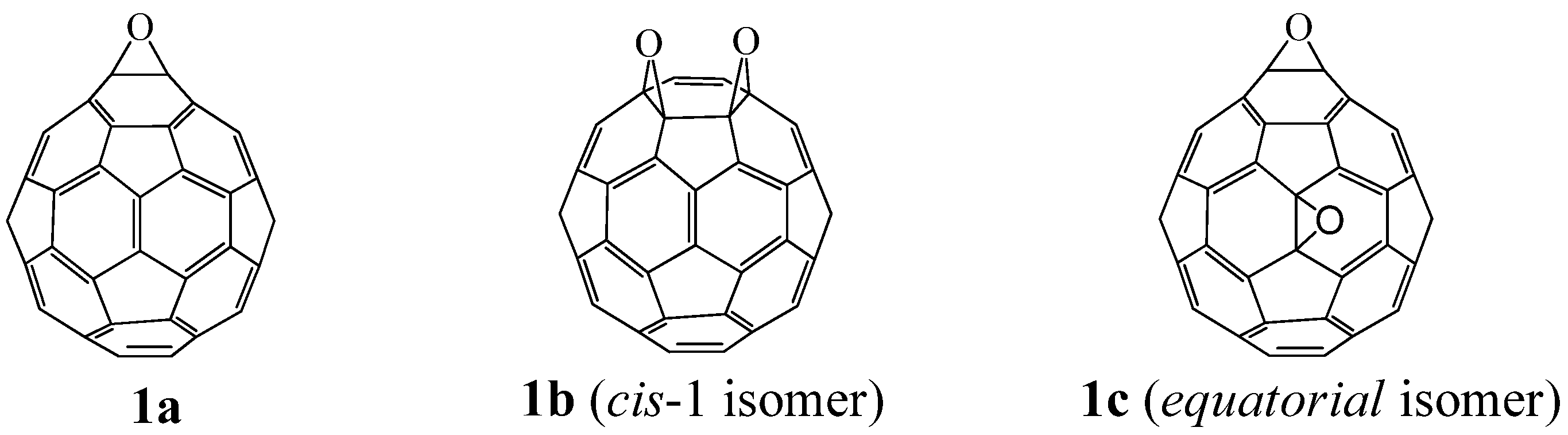{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




 4a (0.29)
4a (0.29)
 4b (0.28)
4b (0.28)







 4b (0.28)
4b (0.28) 4a (0.29)
4a (0.29)





