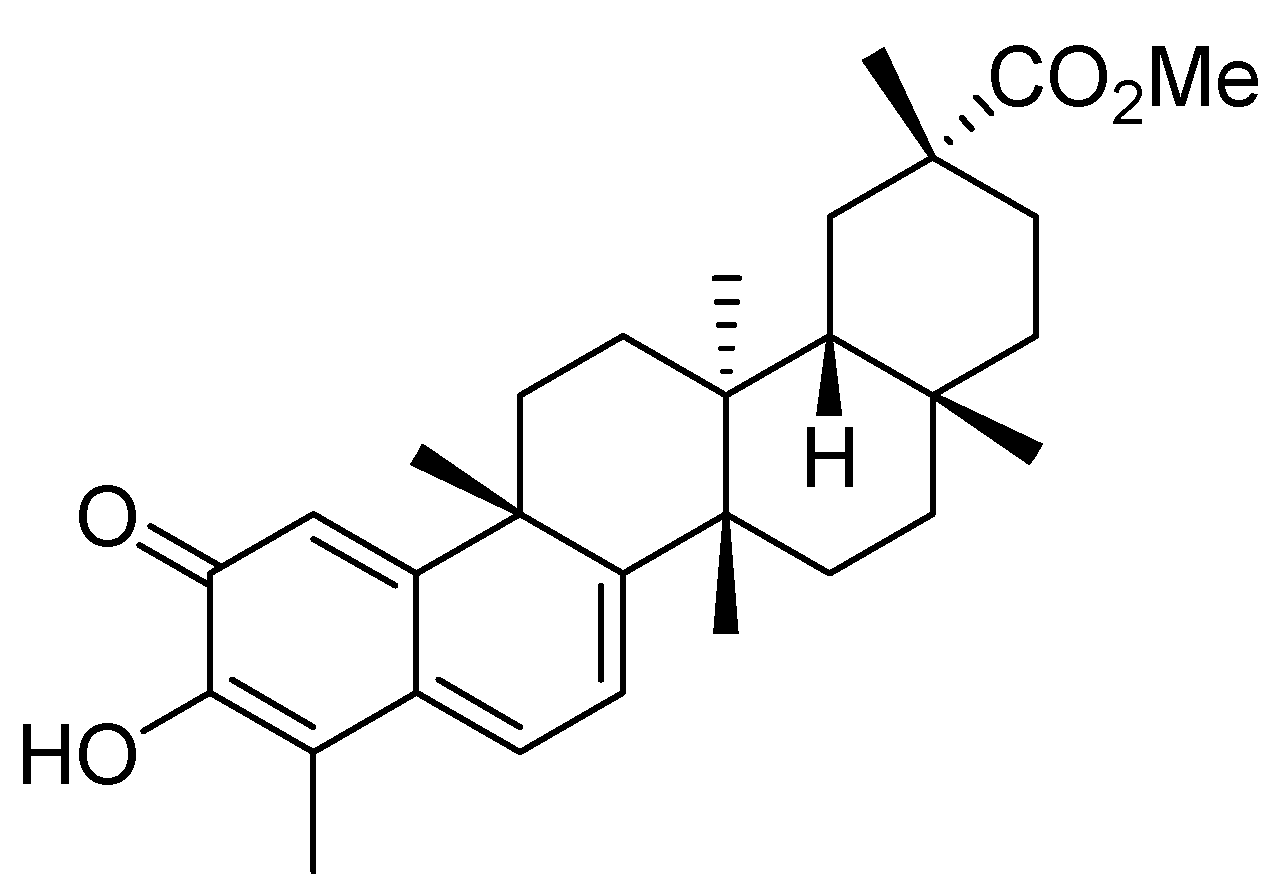
Pristimerin, a Triterpenoid, Inhibits Tumor Angiogenesis by Targeting VEGFR2 Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results and Discussion
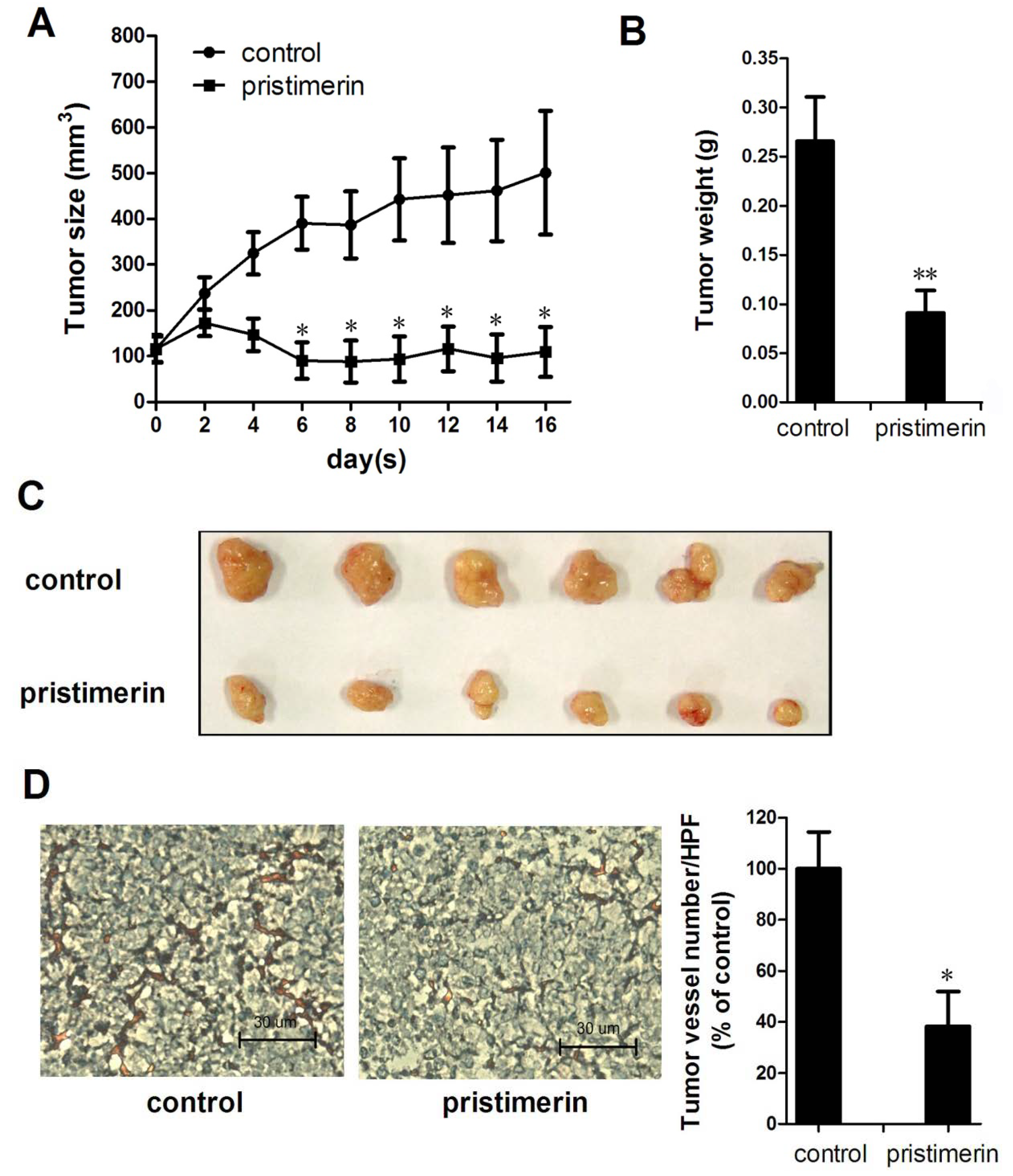
2.1. Pristimerin Inhibits Tumor Growth and Tumor Angiogenesis in a Xenograft Mouse Model

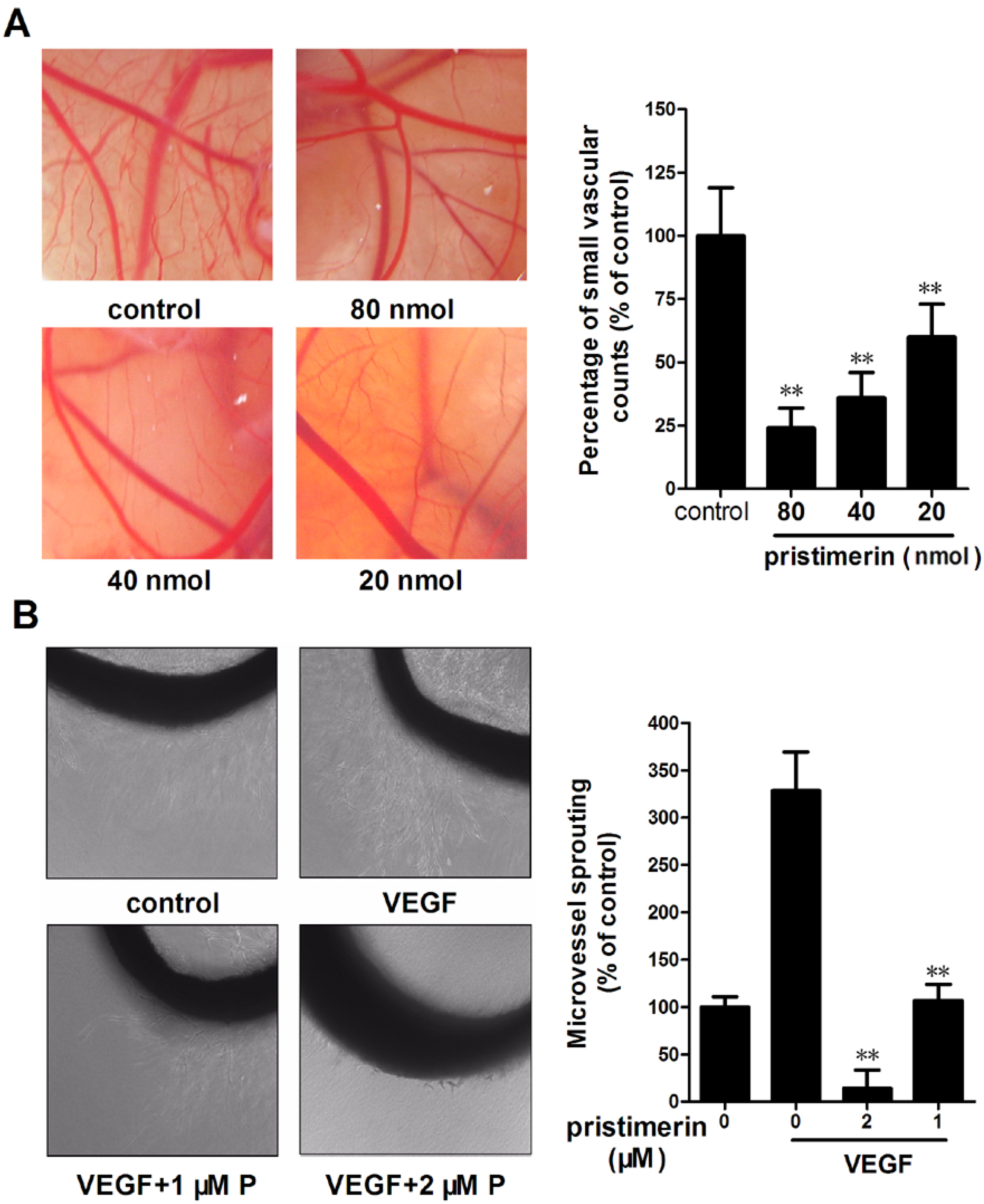
2.2. Pristimerin Inhibits Angiogenesis in Vivo and VEGF-induced Vessel Sprouting ex Vivo

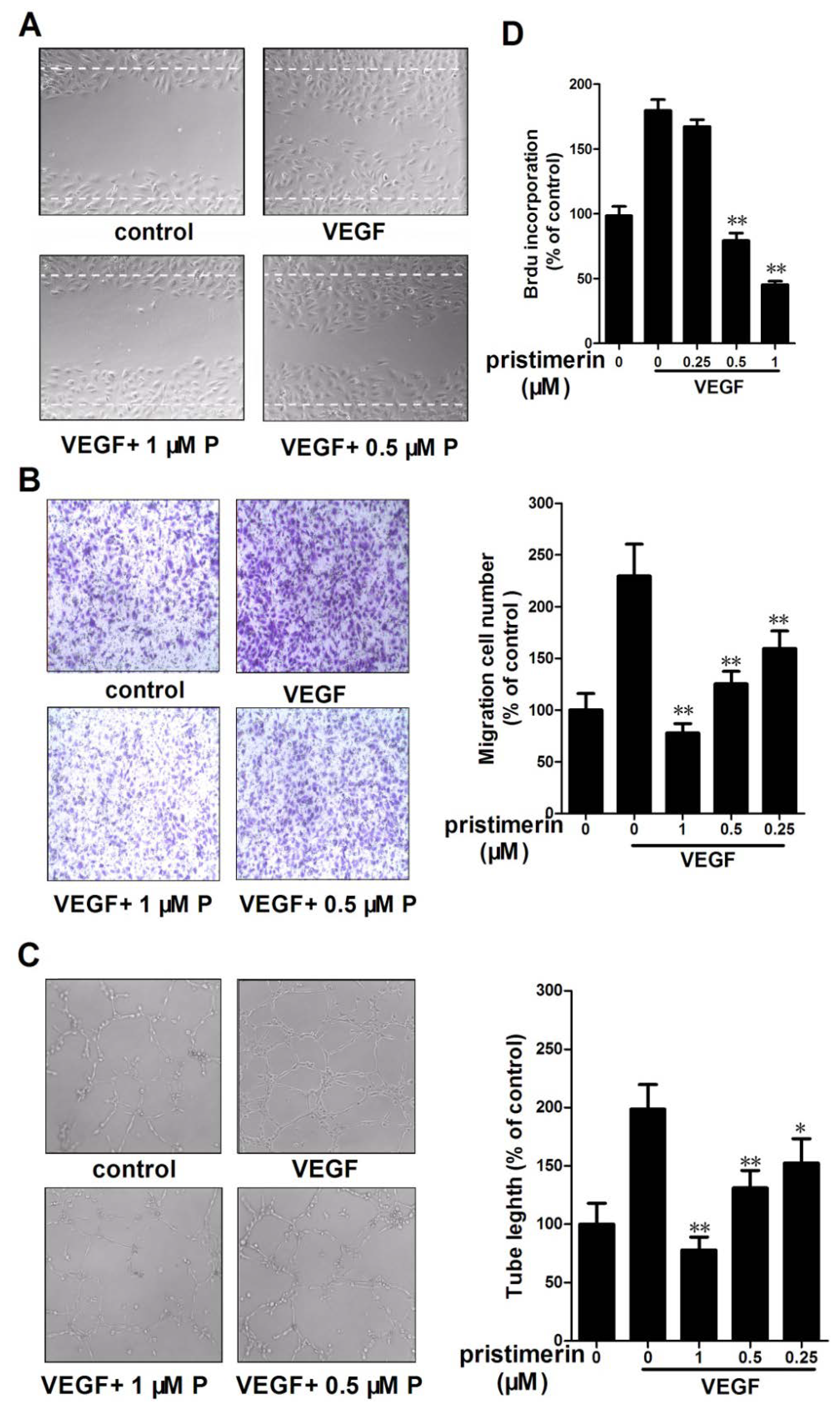
2.3. Pristimerin Inhibits the VEGF-Induced Chemotactic Motility, Capillary Structure Formation and Proliferation of HUVECs in Vitro

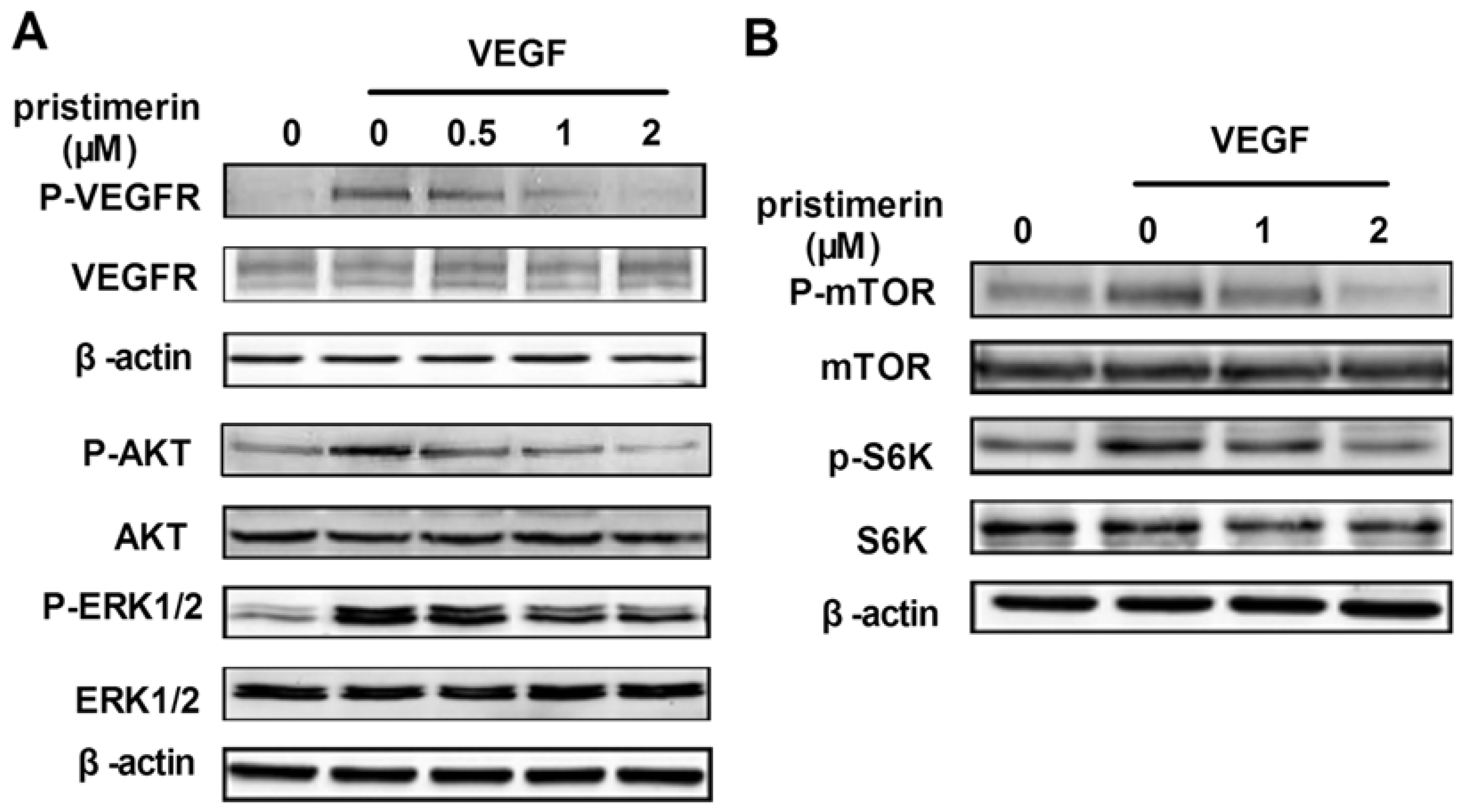
2.4. Influence of Pristimerin on VEGFR2 and Related Signaling Pathways

2.5. Discussion
3. Experimental
3.1. Cell lines and Cell Culture
3.2. Reagents and Antibodies
3.3. Human Breast Tumor Xenograft Mouse Model
3.4. Histology and Immunohistochemistry
3.5. Chicken Chorioallantoic Membrane (CAM) Assay
3.5. Rat Aortic Ring Assay
3.7. Cell Proliferation Assay by Bromodeoxyuridine (BrdU) Incorporation
3.8. Wound-Healing Migration Assay
3.9. Transwell Migration Assay
3.10. Capillary-Like Tube Formation Assay
3.11. Western Immunoblot Analysis
3.12. Statistical Analysis
4. Conclusions
Acknowledgments
References and Notes
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- Kowanetz, M.; Ferrara, N. Vascular endothelial growth factor signaling pathways: Therapeutic perspective. Clin. Cancer Res. 2006, 12, 5018–5022. [Google Scholar] [CrossRef]
- Pyun, B.J.; Choi, S.; Lee, Y.; Kim, T.W.; Min, J.K.; Kim, Y.; Kim, B.D.; Kim, J.H.; Kim, T.Y.; Kim, Y.M.; et al. Capsiate, a nonpungent capsaicin-like compound, inhibits angiogenesis and vascular permeability via a direct inhibition of Src kinase activity. Cancer Res. 2008, 68, 227–235. [Google Scholar] [CrossRef]
- Falcon, B.L.; Barr, S.; Gokhale, P.C.; Chou, J.; Fogarty, J.; Depeille, P.; Miglarese, M.; Epstein, D.M.; McDonald, D.M. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011, 71, 1573–1583. [Google Scholar] [CrossRef]
- Burstein, H.J.; Schwartz, R.S. Molecular origins of cancer. N. Engl. J. Med. 2008, 358, 527. [Google Scholar] [CrossRef]
- Costa, P.M.; Ferreira, P.M.; Bolzani Vda, S.; Furlan, M.; de Freitas Formenton Macedo Dos Santos, V.A.; Corsino, J.; de Moraes, M.O.; Costa-Lotufo, L.V.; Montenegro, R.C.; Pessoa, C. Antiproliferative activity of pristimerin isolated from Maytenus ilicifolia (Celastraceae) in human HL-60 cells. Toxicol. In Vitro 2008, 22, 854–863. [Google Scholar] [CrossRef]
- Tiedemann, R.E.; Schmidt, J.; Keats, J.J.; Shi, C.X.; Zhu, Y.X.; Palmer, S.E.; Mao, X.; Schimmer, A.D.; Stewart, A.K. Blood 2009, 113, 4027–4037. [CrossRef]
- Byun, J.Y.; Kim, M.J.; Eum, D.Y.; Yoon, C.H.; Seo, W.D.; Park, K.H.; Hyun, J.W.; Lee, Y.S.; Lee, J.S.; Yoon, M.Y.; et al. Reactive oxygen species-dependent activation of Bax and poly(ADP-ribose) polymerase-1 is required for mitochondrial cell death induced by triterpenoid pristimerin in human cervical cancer cells. Mol. Pharmacol. 2009, 76, 734–744. [Google Scholar] [CrossRef]
- Yang, H.; Landis-Piwowar, K.R.; Lu, D.; Yuan, P.; Li, L.; Reddy, G.P.; Yuan, X.; Dou, Q.P. Pristimerin induces apoptosis by targeting the proteasome in prostate cancer cells. J. Cell Biochem. 2008, 103, 234–244. [Google Scholar] [CrossRef]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef]
- Wu, C.C.; Chan, M.L.; Chen, W.Y.; Tsai, C.Y.; Chang, F.R.; Wu, Y.C. Pristimerin induces caspase-dependent apoptosis in MDA-MB-231 cells via direct effects on mitochondria. Mol. Cancer Ther. 2005, 4, 1277–1285. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, N.; Ling, Y.; Wang, L.; You, Q.; Li, Z.; Guo, Q. J. Pharmacol. Sci. 2010, 112, 37–45. [CrossRef]
- Taraboletti, G.; Giavazzi, R. Modelling approaches for angiogenesis. Eur. J. Cancer 2004, 40, 881–889. [Google Scholar] [CrossRef]
- Sheng, H.; Sun, H. Synthesis, biology and clinical significance of pentacyclic triterpenes: A multi-target approach to prevention and treatment of metabolic and vascular diseases. Nat. Prod. Rep. 2011, 28, 543–593. [Google Scholar] [CrossRef]
- Salminen, A.; Lehtonen, M.; Suuronen, T.; Kaarniranta, K.; Huuskonen, J. Terpenoids: Natural inhibitors of NF-kappaB signaling with anti-inflammatory and anticancer potential. Cell Mol. Life Sci. 2008, 65, 2979–2999. [Google Scholar] [CrossRef]
- Pang, X.; Yi, Z.; Zhang, J.; Lu, B.; Sung, B.; Qu, W.; Aggarwal, B.B.; Liu, M. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010, 70, 1951–1959. [Google Scholar] [CrossRef]
- Yadav, V.R.; Prasad, S.; Sung, B.; Kannappan, R.; Aggarwal, B.B. Targeting inflammatory pathways by triterpenoids for prevention and treatment of cancer. Toxins (Basel) 2010, 2, 2428–2466. [Google Scholar]
- Napoleone Ferrara, R.S.K. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef]
- Huang, D.; Ding, Y.; Luo, W.M.; Bender, S.; Qian, C.N.; Kort, E.; Zhang, Z.F.; VandenBeldt, K.; Duesbery, N.S.; Resau, J.H.; Teh, B.T. Inhibition of MAPK kinase signaling pathways suppressed renal cell carcinoma growth and angiogenesis in vivo. Cancer Res. 2008, 68, 81–88. [Google Scholar]
- Hers, I.; Vincent, E.E.; Tavare, J.M. Akt signalling in health and disease. Cell Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- Pang, X.; Zhang, L.; Wu, Y.; Lin, L.; Li, J.; Qu, W.; Safe, S.; Liu, M. Methyl 2-cyano-3,11-dioxo-18-olean-1,12-dien-30-oate (CDODA-Me), a derivative of glycyrrhetinic acid, functions as a potent angiogenesis inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 172–179. [Google Scholar] [CrossRef]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updat. 2008, 11, 32–50. [Google Scholar] [CrossRef]
- Berven, L.A.; Willard, F.S.; Crouch, M.F. Role of the p70(S6K) pathway in regulating the actin cytoskeleton and cell migration. Exp. Cell Res. 2004, 296, 183–195. [Google Scholar] [CrossRef]
- Lu, Z.; Jin, Y.; Chen, C.; Li, J.; Cao, Q.; Pan, J. Pristimerin induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation by blocking NF-kappaB signaling and depleting Bcr-Abl. Mol. Cancer 2010, 9, 112. [Google Scholar] [CrossRef]
- Garcia-Maceira, P.; Mateo, J. Silibinin inhibits hypoxia-inducible factor-1alpha and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene 2009, 28, 313–324. [Google Scholar] [CrossRef]
- Weng, Q.; Zhang, J.; Cao, J.; Xia, Q.; Wang, D.; Hu, Y.; Sheng, R.; Wu, H.; Zhu, D.; Zhu, H.; He, Q.; Yang, B. Q39, a quinoxaline 1,4-Di-N-oxide derivative, inhibits hypoxia-inducible factor-1alpha expression and the Akt/mTOR/4E-BP1 signaling pathway in human hepatoma cells. Invest. New Drugs 2011, 29, 1177–1187. [Google Scholar] [CrossRef]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.G.; Lucas, J.; Shor, B.; Kim, J.; Verheijen, J.; Curran, K.; Malwitz, D.J.; et al. Cancer Res. 2009, 69, 6232–6240. [CrossRef]
- Pang, X.; Yi, T.; Yi, Z.; Cho, S.G.; Qu, W.; Pinkaew, D.; Fujise, K.; Liu, M. Morelloflavone, a biflavonoid, inhibits tumor angiogenesis by targeting rho GTPases and extracellular signal-regulated kinase signaling pathway. Cancer Res. 2009, 69, 518–525. [Google Scholar] [CrossRef]
- Inao, T.; Harashima, N.; Monma, H.; Okano, S.; Itakura, M.; Tanaka, T.; Tajima, Y.; Harada, M. Antitumor effects of cytoplasmic delivery of an innate adjuvant receptor ligand, poly(I:C), on human breast cancer. Breast Cancer Res. Treat. 2011. [Google Scholar] [CrossRef]
- Abdel-Malak, N.A.; Mofarrahi, M.; Mayaki, D.; Khachigian, L.M.; Hussain, S.N. Early growth response-1 regulates angiopoietin-1-induced endothelial cell proliferation, migration, and differentiation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 209–216. [Google Scholar] [CrossRef]
- Pisanti, S.; Picardi, P.; Prota, L.; Proto, M.C.; Laezza, C.; McGuire, P.G.; Morbidelli, L.; Gazzerro, P.; Ziche, M.; Das, A.; Bifulco, M. Genetic and pharmacologic inactivation of cannabinoid CB1 receptor inhibits angiogenesis. Blood 2011, 117, 5541–5550. [Google Scholar] [CrossRef]
- Samples Availability: Samples of the compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mu, X.; Shi, W.; Sun, L.; Li, H.; Jiang, Z.; Zhang, L. Pristimerin, a Triterpenoid, Inhibits Tumor Angiogenesis by Targeting VEGFR2 Activation. Molecules 2012, 17, 6854-6868. https://doi.org/10.3390/molecules17066854
Mu X, Shi W, Sun L, Li H, Jiang Z, Zhang L. Pristimerin, a Triterpenoid, Inhibits Tumor Angiogenesis by Targeting VEGFR2 Activation. Molecules. 2012; 17(6):6854-6868. https://doi.org/10.3390/molecules17066854
Chicago/Turabian StyleMu, Xianmin, Wei Shi, Lixin Sun, Han Li, Zhenzhou Jiang, and Luyong Zhang. 2012. "Pristimerin, a Triterpenoid, Inhibits Tumor Angiogenesis by Targeting VEGFR2 Activation" Molecules 17, no. 6: 6854-6868. https://doi.org/10.3390/molecules17066854