Synthesis and Biological Evaluation of Novel Furozan-Based Nitric Oxide-Releasing Derivatives of Oridonin as Potential Anti-Tumor Agents

Abstract
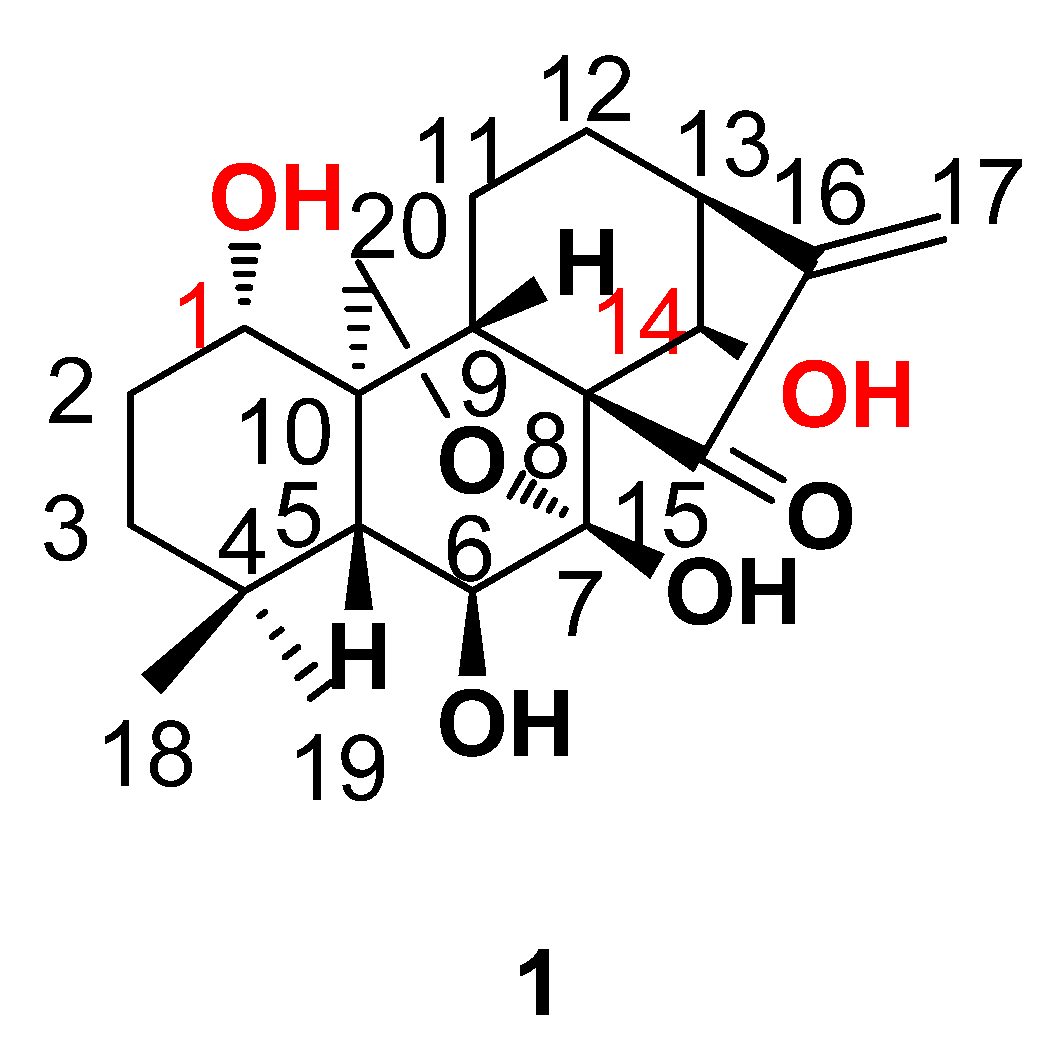
:1. Introduction

2. Results and Discussion
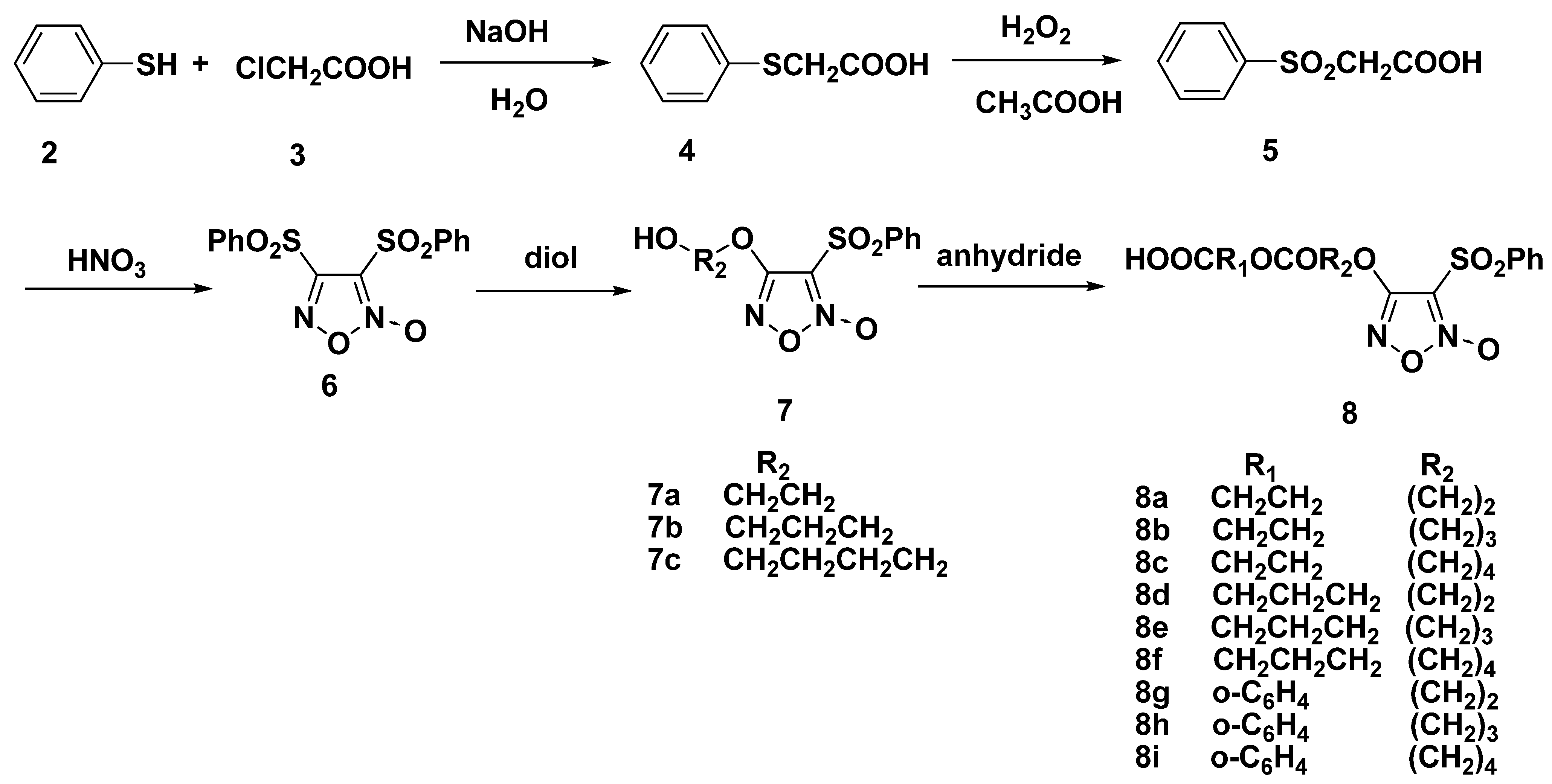
2.1. Synthesis of Furoxan-Based NO Donor

2.2. Synthesis of Furoxan/Oridonin Hybrids


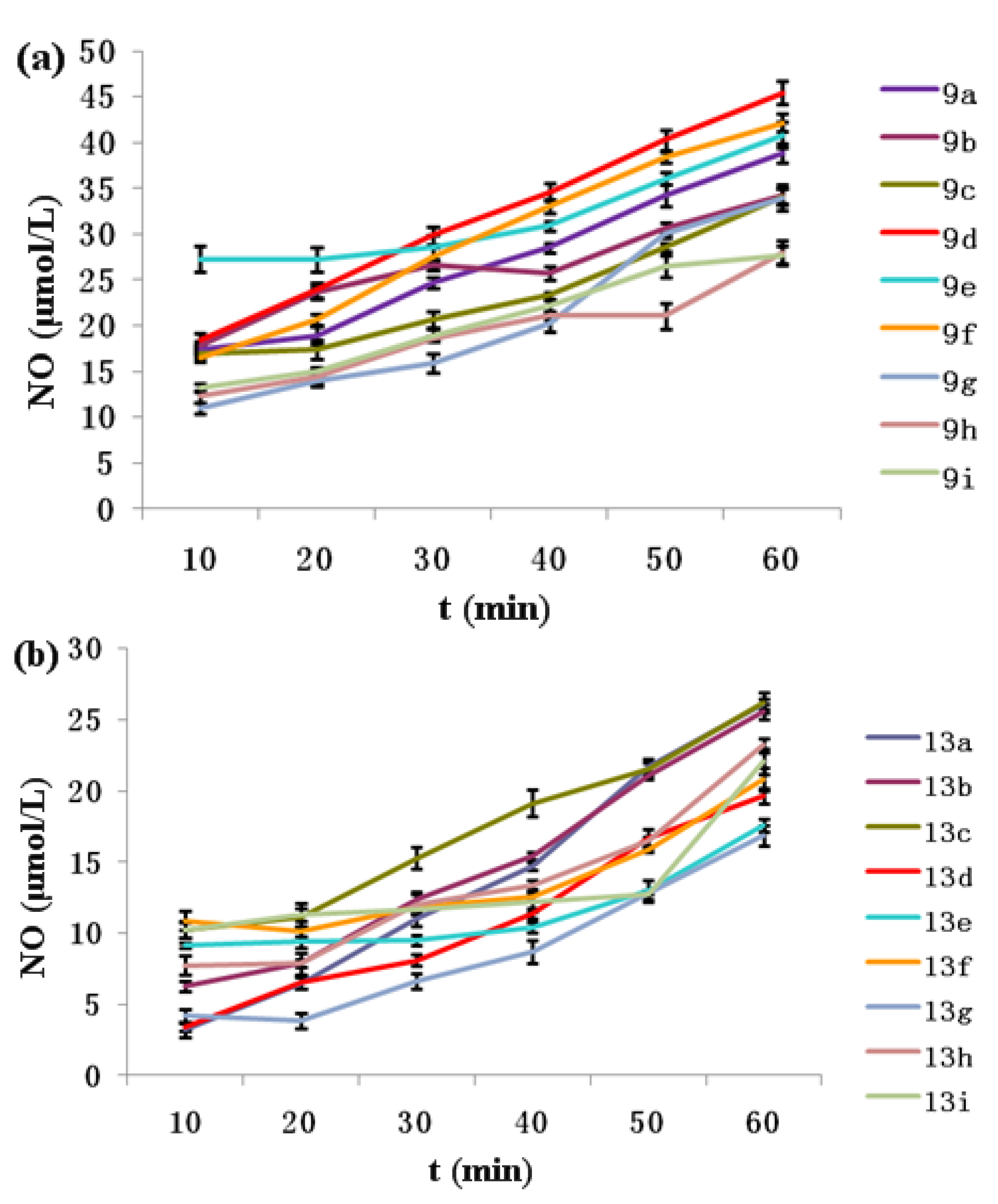
2.3. NO-Releasing Test of Hybrids 9a–i and 13a–i in Vitro

2.4. Anti-proliferative Activity in Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Bel-7402 | K562 | MGC-803 | CaEs-17 |
|---|---|---|---|---|
| Taxol b | 1.89 ± 0.09 | 0.41 ± 0.02 c | 0.85 ± 0.06 c | 0.43 ± 0.03 d |
| Oridonin | 7.48 ± 0.53 | 4.76 ± 0.32 | 5.69 ± 0.39 | 11.03 ± 1.02 |
| 9a | 2.37 ± 0.85 | 4.33 ± 0.14 | 3.22 ± 0.19 | 8.46 ± 0.05 |
| 9b | 1.91 ± 0.09 | 3.46 ± 0.60 | 2.57 ± 0.07 | 6.98 ± 0.20 |
| 9c | 2.23 ± 0.04 | 4.02 ± 0.05 | 3.46 ± 0.23 | 8.17 ± 1.01 |
| 9d | 1.89 ± 0.22 | 3.78 ± 0.19 | 3.08 ± 0.47 | 8.04 ± 0.18 |
| 9e | 1.33 ± 0.15 | 2.85 ± 0.03 | 2.21 ± 0.16 | 6.77 ± 0.32 |
| 9f | 1.97 ± 0.04 | 3.72 ± 0.26 | 3.23 ± 0.25 | 8.09 ± 0.47 |
| 9g | 0.95 ± 0.21 c | 1.94 ± 0.14 | 1.98 ± 0.13 | 4.81 ± 0.10 c |
| 9h | 0.86 ± 0.08 c | 1.82 ± 0.07 | 1.81 ± 0.20 | 4.56 ± 0.32 c |
| 9i | 0.97 ± 0.10 c | 1.92 ± 0.34 | 1.90 ± 0.11 | 5.24 ± 0.18 |
| 12 | 3.21 ± 0.25 | 5.06 ± 0.18 | 4.05 ± 0.04 | 7.24 ± 0.41 |
| 13a | 2.85 ± 0.14 | 4.65 ± 0.07 | 3.77 ± 0.31 | 5.30 ± 0.28 |
| 13b | 2.19 ± 0.19 | 3.85 ± 0.06 | 2.90 ± 0.12 | 4.11 ± 0.07 c |
| 13c | 2.76 ± 0.42 | 4.11 ± 0.15 | 3.65 ± 0.40 | 5.22 ± 0.12 |
| 13d | 2.70 ± 0.09 | 4.08 ± 0.30 | 3.64 ± 0.12 | 5.38 ± 0.24 |
| 13e | 2.13 ± 0.17 | 3.04 ± 0.21 | 2.79 ± 0.10 | 4.00 ± 0.31 c |
| 13f | 2.66 ± 0.30 | 3.97 ± 0.16 | 3.42 ± 0.27 | 5.11 ± 0.39 |
| 13g | 1.94 ± 0.13 | 2.22 ± 0.29 | 2.45 ± 0.51 | 3.28 ± 0.06 c |
| 13h | 1.72 ± 0.08 | 2.08 ± 0.34 | 2.22 ± 0.29 | 3.24 ± 0.23 c |
| 13i | 1.86 ± 0.15 | 2.65 ± 0.08 | 2.41 ± 0.16 | 3.13 ± 0.21 c |
3. Experimental
3.1. Chemistry
3.1.1. General Procedure for the Preparation of 8a–i
3.1.2. General Procedure for the Preparation of 9a–i and 13a–i
3.2. In Vitro MTT Assay
3.3. NO-Releasing Test: Nitrate/Nitrite Measurement in Vitro
4. Conclusions
Acknowledgments
References and Notes
- Mocellin, S. Nitric oxide: Cancer target or anticancer agent? Curr. Cancer Drug Targets 2009, 9, 214–236. [Google Scholar] [CrossRef]
- Ignarro, L.J. Signal transduction mechanisms involving nitric oxide. Biochem. Pharmacol. 1991, 41, 485–490. [Google Scholar] [CrossRef]
- Fukuto, J.M.; Wink, D.A. Nitric oxide (NO): Formation and biological roles in mammalian systems. Met. Ions Biol. Syst. 1999, 36, 547–595. [Google Scholar]
- Wink, D.A.; Ridnour, L.A.; Hussain, S.P.; Harris, C.C. The reemergence of nitric oxide and cancer. Nitric Oxide 2008, 19, 65–67. [Google Scholar]
- Hirst, D.; Robson, T. Nitric oxide in cancer therapeutics: Interaction with cytotoxic chemotherapy. Curr. Pharm. Des. 2010, 16, 411–420. [Google Scholar] [CrossRef]
- Millet, A.; Bettaieb, A.; Renaud, F.; Prevotat, L.; Hammann, A.; Solary, E.; Mignotte, B.; Jeannin, J.F. Influence of the nitric oxide donor glyceryl trinitrate on apoptotic pathways in human colon cancer cells. Gastroenterology 2002, 123, 235–246. [Google Scholar] [CrossRef]
- Postovit, L.M.; Adams, M.A.; Lash, G.E.; Heaton, J.P.; Graham, C.H. Nitric oxide-mediated regulation of hypoxia-induced B16F10 melanoma metastasis. Int. J. Cancer 2004, 108, 47–53. [Google Scholar] [CrossRef]
- Findlay, V.J.; Townsend, D.M.; Saavedra, J.E.; Buzard, G.S.; Citro, M.L.; Keefer, L.K.; Ji, X.; Tew, K.D. Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Mol. Pharmacol. 2004, 65, 1070–1079. [Google Scholar] [CrossRef]
- Yasuda, H.; Yamaya, M.; Nakayama, K.; Sasaki, T.; Ebihara, H.; Kanda, A.; Asada, M.; Inoue, D.; Suzuki, T.; Okazaki, T.; et al. Randomized phase II trial comparing nitroglycerin plus vinorelbine and cisplatin with vinorelbine and cisplatin alone in previously untreated stage IIIB/IV non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 688–694. [Google Scholar] [CrossRef]
- Siemens, D.R.; Heaton, J.P.; Adams, M.A.; Kawakami, J.; Graham, C.H. Phase II study of nitric oxide donor for men with increasing prostate-specific antigen level after surgery or radiotherapy for prostate cancer. Urology 2009, 74, 878–883. [Google Scholar] [CrossRef]
- Moharram, S.; Zhou, A.; Wiebe, L.I.; Knaus, E.E. Design and synthesis of 3'- and 5'-O-(3-benzenesulfonylfuroxan-4-yl)-2'-deoxyuridines: Biological evaluation as hybrid nitric oxide donor-nucleoside anticancer agents. J. Med. Chem. 2004, 47, 1840–1846. [Google Scholar] [CrossRef]
- Maksimovic-Ivanic, D.; Mijatovic, S.; Harhaji, L.; Miljkovic, D.; Dabideen, D.; Fan Cheng, K.; Mangano, K.; Malaponte, G.; Al-Abed, Y.; Libra, M.; et al. Anticancer properties of the novel nitric oxide-donating compound (S,R)-3-phenyl-4,5-dihydro-5-isoxazole acetic acid-nitric oxide in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 510–520. [Google Scholar]
- Chen, L.; Zhang, Y.; Kong, X.; Lan, E.; Huang, Z.; Peng, S.; Kaufman, D.L.; Tian, J. Design, synthesis, and antihepatocellular carcinoma activity of nitric oxide releasing derivatives of oleanolic acid. J. Med. Chem. 2008, 51, 4834–4838. [Google Scholar]
- Lai, Y.; Shen, L.; Zhang, Z.; Liu, W.; Zhang, Y.; Ji, H.; Tian, J. Synthesis and biological evaluation of furoxan-based nitric oxide-releasing derivatives of glycyrrhetinic acid as anti-hepatocellular carcinoma agents. Bioorg. Med. Chem. Lett. 2010, 20, 6416–6420. [Google Scholar] [CrossRef]
- Ling, Y.; Ye, X.; Ji, H.; Zhang, Y.; Lai, Y.; Peng, S.; Tian, J. Synthesis and evaluation of nitric oxide-releasing derivatives of farnesylthiosalicylic acid as anti-tumor agents. Bioorg. Med. Chem. 2010, 18, 3448–3456. [Google Scholar] [CrossRef]
- Ling, Y.; Ye, X.; Zhang, Z.; Zhang, Y.; Lai, Y.; Ji, H.; Peng, S.; Tian, J. Novel nitric oxide-releasing derivatives of farnesylthiosalicylic acid: Synthesis and evaluation of antihepatocellular carcinoma activity. J. Med. Chem. 2011, 54, 3251–3259. [Google Scholar] [CrossRef]
- Tang, X.; Gu, X.; Ai, H.; Wang, G.; Peng, H.; Lai, Y.; Zhang, Y. Synthesis and evaluation of nitric oxide-releasing DDB derivatives as potential Pgp-mediated MDR reversal agents in MCF-7/Adr cells. Bioorg. Med. Chem. Lett. 2012, 22, 801–805. [Google Scholar]
- Kang, N.; Zhang, J.H.; Qiu, F.; Chen, S.; Tashiro, S.; Onodera, S.; Ikejima, T. Induction of G(2)/M phase arrest and apoptosis by oridonin in human laryngeal carcinoma cells. J. Nat. Prod. 2010, 73, 1058–1063. [Google Scholar] [CrossRef]
- Xu, J.; Yang, J.; Ran, Q.; Wang, L.; Liu, J.; Wang, Z.; Wu, X.; Hua, W.; Yuan, S.; Zhang, L.; et al. Synthesis and biological evaluation of novel 1-O- and 14-O-derivatives of oridonin as potential anticancer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 4741–4744. [Google Scholar] [CrossRef]
- Wang, L.; Ran, Q.; Li, D.; Yao, H.; Zhang, Y.; Yuan, S.; Zhang, L.; Shen, M.; Xu, J. Synthesis and anti-tumor activity of 14-O-derivatives of natural oridonin. Chin. J. Nat. Med. 2011, 9, 194–198. [Google Scholar]
- Wang, L.; Li, D.; Wang, C.; Zhang, Y.; Xu, J. Recent progress in the development of natural ent-kaurane diterpenoids with anti-tumor activity. Mini-Rev. Med. Chem. 2011, 11, 910–919. [Google Scholar]
- Wang, L.; Li, D.; Xu, S.; Cai, H.; Yao, H.; Zhang, Y.; Jiang, J.; Xu, J. The conversion of oridonin to spirolactone-type or enmein-type diterpenoid: Synthesis and biological evaluation of ent-6,7-seco-oridonin derivatives as novel potential anticancer agents. Eur. J. Med. Chem. 2012, 52, 242–250. [Google Scholar] [CrossRef]
- Sample Availability: Contact the first authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, D.; Wang, L.; Cai, H.; Zhang, Y.; Xu, J. Synthesis and Biological Evaluation of Novel Furozan-Based Nitric Oxide-Releasing Derivatives of Oridonin as Potential Anti-Tumor Agents. Molecules 2012, 17, 7556-7568. https://doi.org/10.3390/molecules17067556
Li D, Wang L, Cai H, Zhang Y, Xu J. Synthesis and Biological Evaluation of Novel Furozan-Based Nitric Oxide-Releasing Derivatives of Oridonin as Potential Anti-Tumor Agents. Molecules. 2012; 17(6):7556-7568. https://doi.org/10.3390/molecules17067556
Chicago/Turabian StyleLi, Dahong, Lei Wang, Hao Cai, Yihua Zhang, and Jinyi Xu. 2012. "Synthesis and Biological Evaluation of Novel Furozan-Based Nitric Oxide-Releasing Derivatives of Oridonin as Potential Anti-Tumor Agents" Molecules 17, no. 6: 7556-7568. https://doi.org/10.3390/molecules17067556