New Quinoxaline Derivatives as Potential MT1 and MT2 Receptor Ligands

Abstract
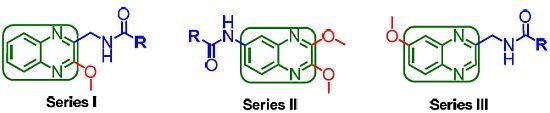
:
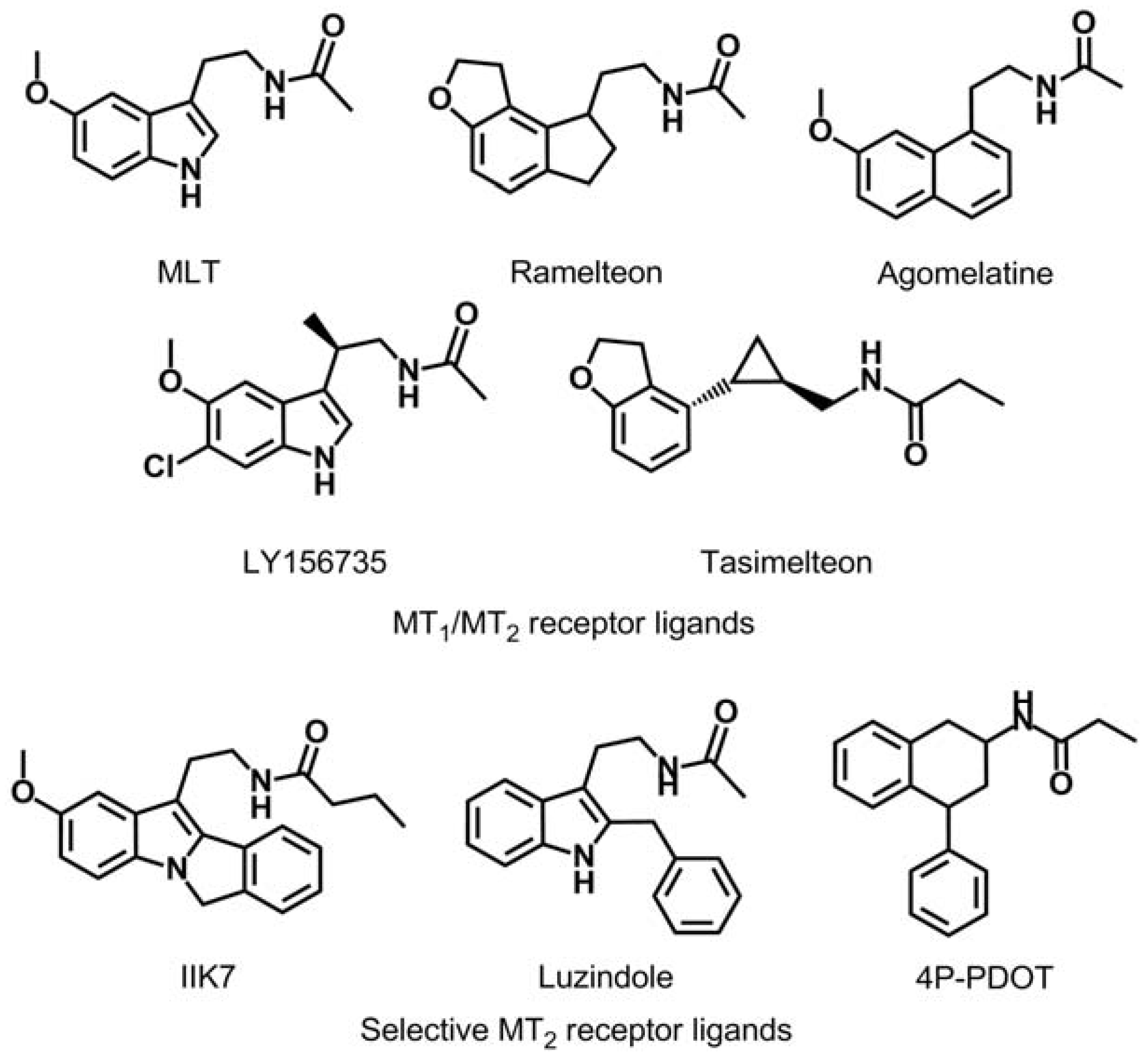
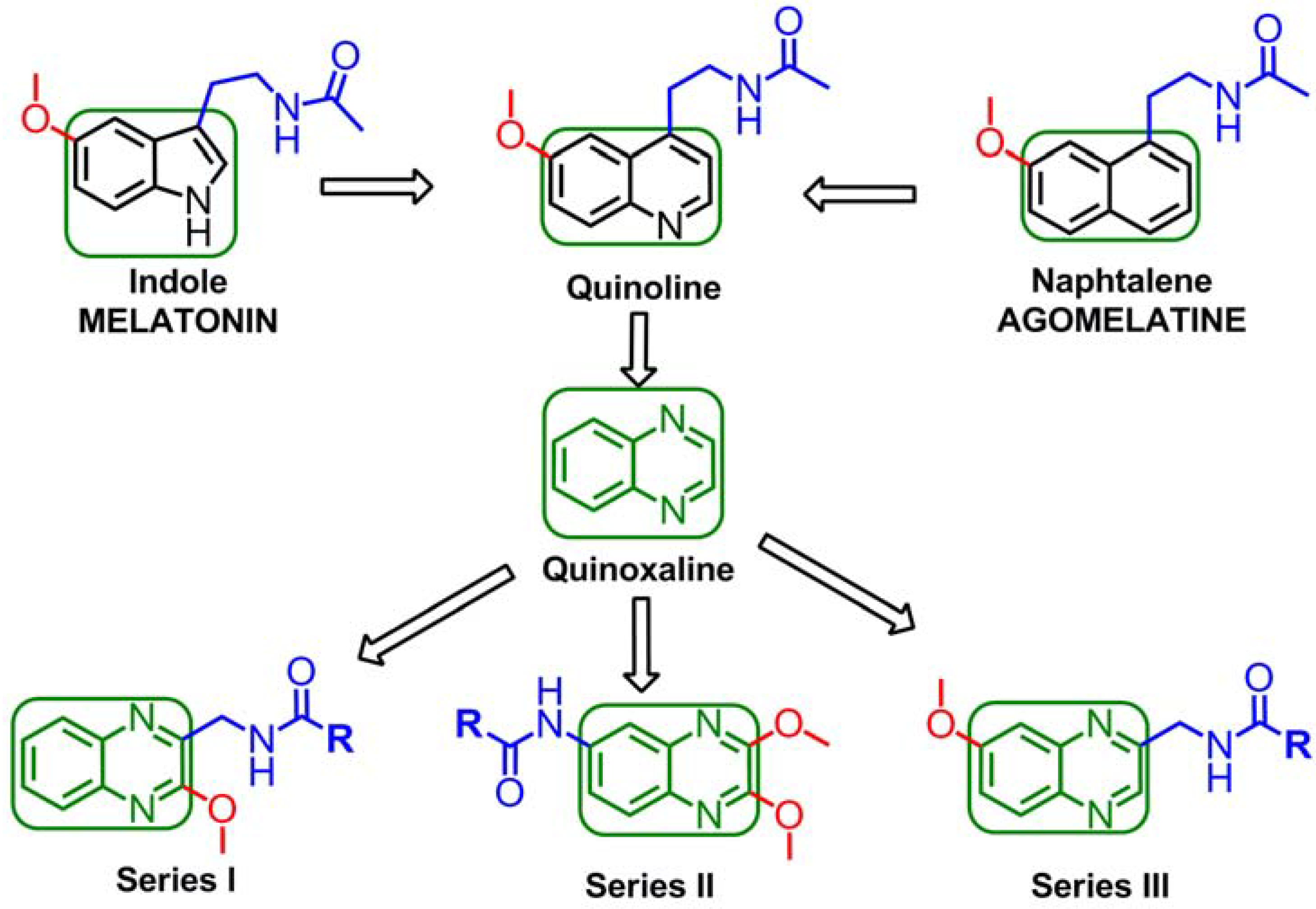
1. Introduction


2. Results and Discussion
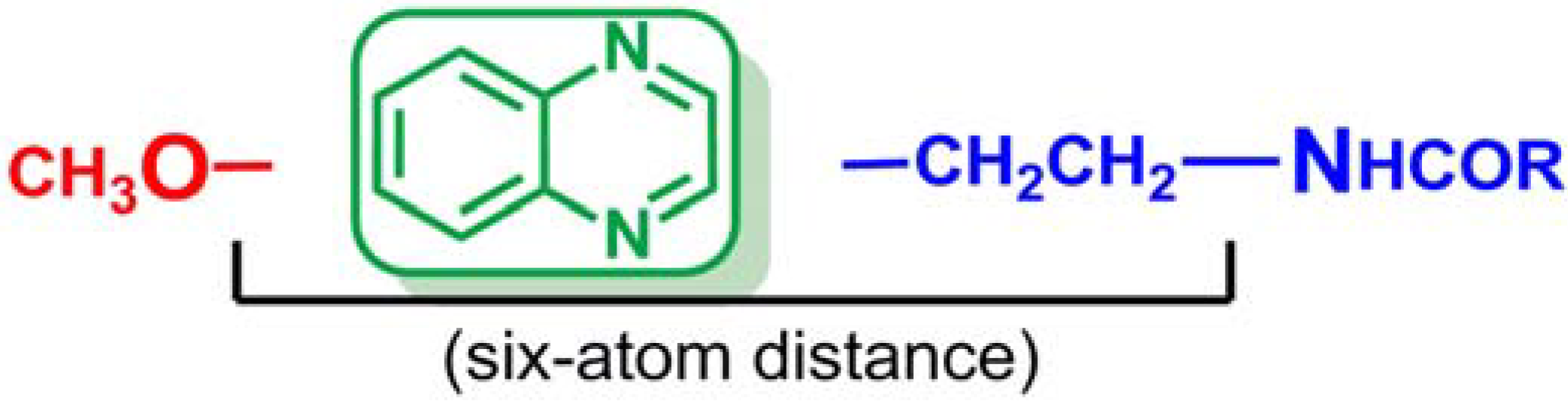
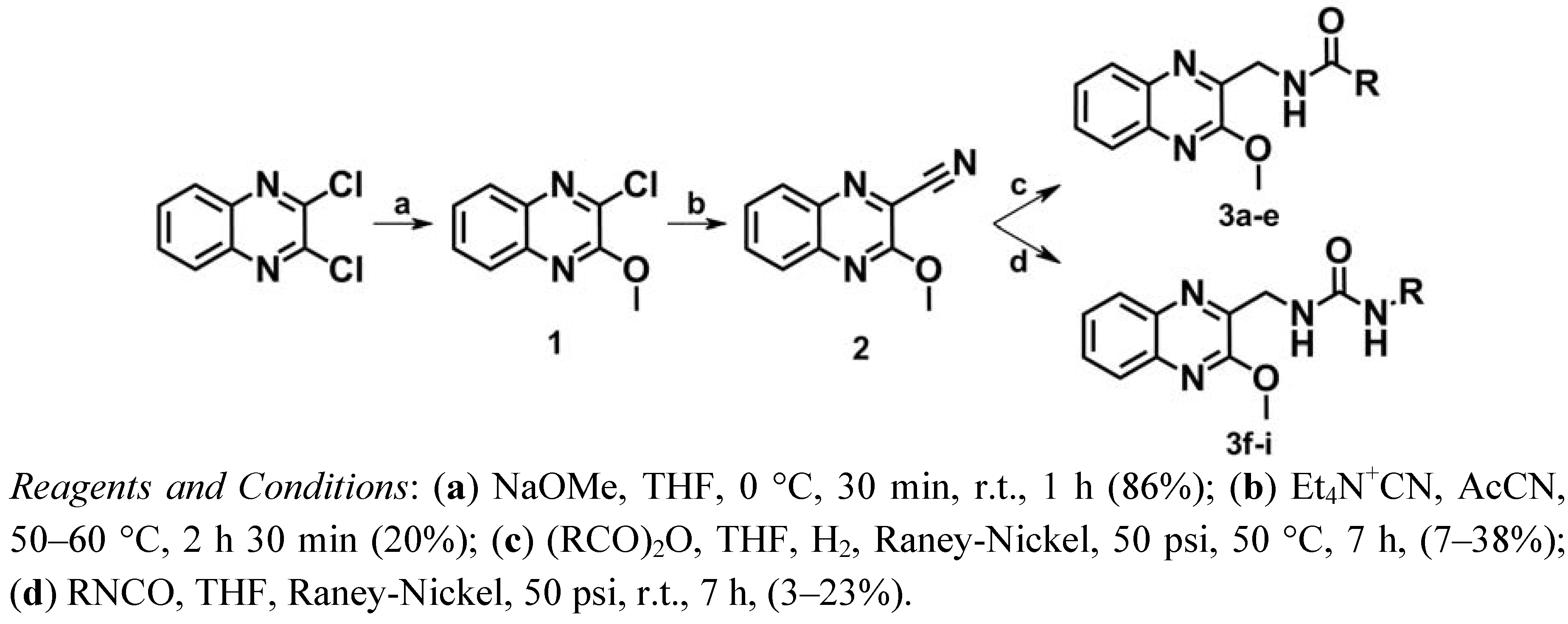
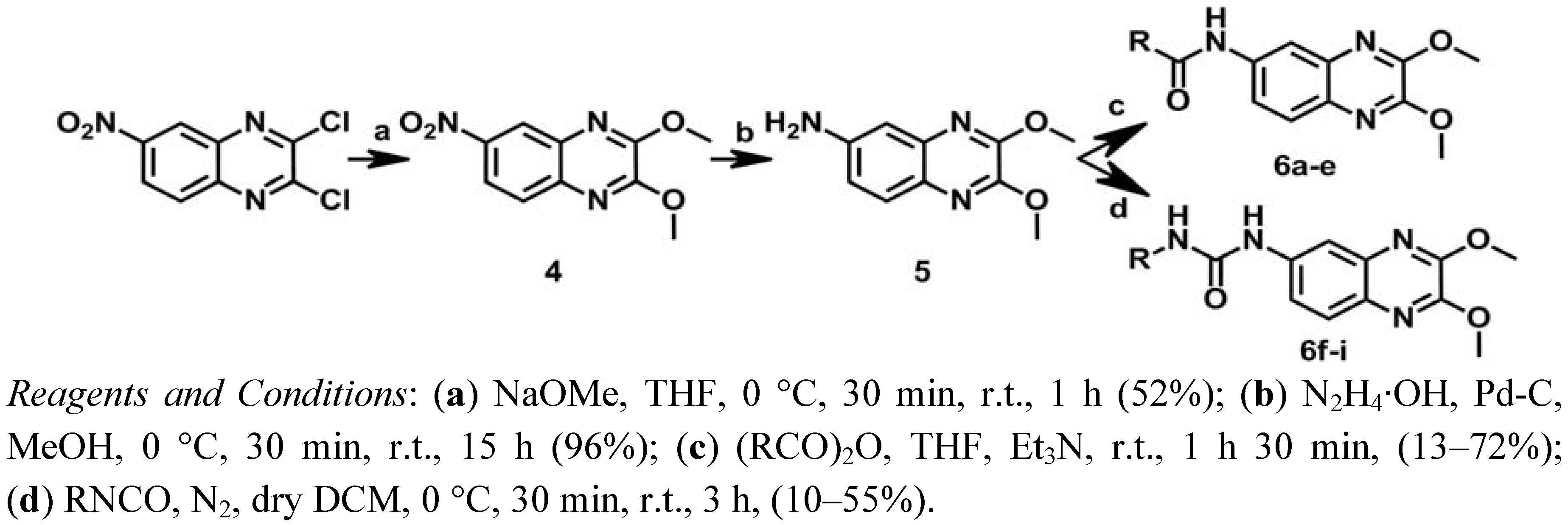
2.1. Chemistry
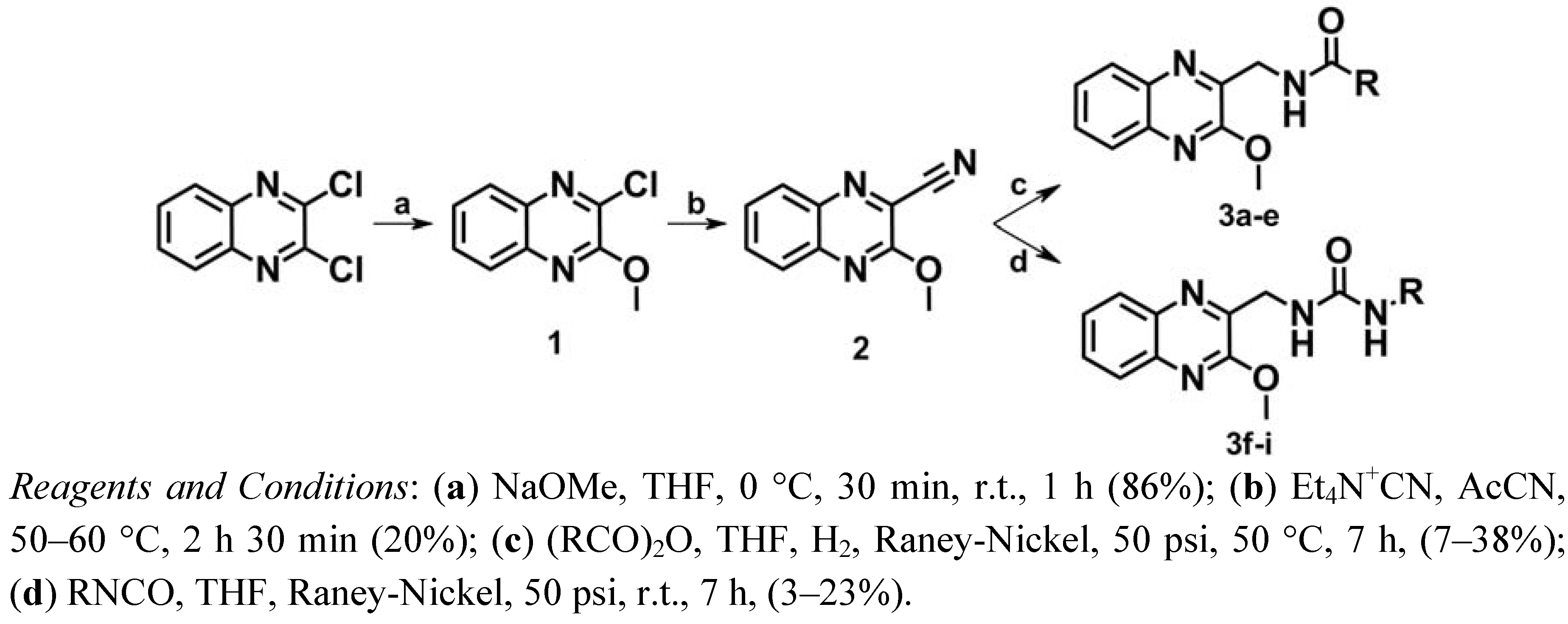


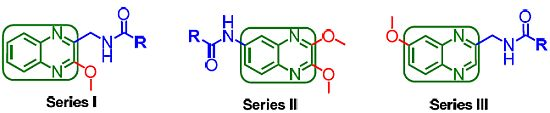
2.2. Pharmacology and Structure-Activity Relationship

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quinoxalines | Comp. | R | MT1 Ki (µM) ± SEM | MT2 Ki (µM) ± SEM |
|---|---|---|---|---|
 | 3a | CH3 | 2.98 ± 0.36 | 0.88 ± 0.30 |
| 3b | CH2CH3 | 2.60 ± 0.23 | 0.66 ± 0.01 | |
| 3c | CH2CH2CH3 | 0.75 ± 0.36 | 1.10 ± 0.03 | |
| 3d | CH(CH3)2 | 1.23 ± 0.32 | 0.47 ± 0.02 | |
| 3e | Ph | >103 | >103 | |
| 3f | NHCH2CH3 | >103 | 0.40 ± 0.13 | |
| 3g | NHCH2CH2CH3 | >103 | 0.44 ± N.D. | |
| 3h | NHCH(CH3)2 | >103 | >103 | |
| 3i | NHPh | >103 | >103 | |
 | 6a | CH3 | 20.00 ± 1.82 | 0.08 ± N.D. |
| 6b | CH2CH3 | 17.60 ± 7.81 | 4.36 ± 1.22 | |
| 6c | CH2CH2CH3 | 11.50 ± 2.80 | 1.35 ± 0.31 | |
| 6d | CH(CH3)2 | 3.40 ± 1.37 | 10.50 ± 2.96 | |
| 6e | Ph | >103 | >103 | |
| 6f | NHCH2CH3 | 3.41 ± 1.89 | 28.80 ± N.D | |
| 6g | NHCH2CH2CH3 | 1.63 ± 0.44 | 0.489 ± 0.07 | |
| 6h | NHCH(CH3)2 | >103 | >103 | |
| 6i | NHPh | >103 | >103 | |
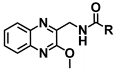
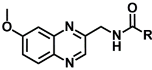 | 10a | CH3 | >103 | >103 |
| 10b | CH2CH3 | >103 | 0.34 ± 0.15 | |
| 10c | CH2CH2CH3 | 0.21 ± 0.11 | 0.10 ± 0.01 | |
| 10d | CH(CH3)2 | 0.32 ± 0.04 | 0.16 ± 0.00 | |
| 10e | Ph | >103 | >103 | |
 | MLT | 0.14·× 10−3 ± 0.03·× 10−3 | 0.41·× 10−3 ± 0.04·× 10−3 |
| Compound | MT2 | |
|---|---|---|
| EC50 ± SEM (μM) | Emax ± SEM (%) | |
| MLT | 0.49 ×·10−3 ± 0.05·× 10−3 | 100 |
| 6a | >10 | |
| 6c | 1.3 ± 0.26 | 84 ± 9.5 |

3. Experimental
3.1. Chemical Synthesis
3.1.1. General Remarks
3.1.2. Synthesis of 2-Chloro-3-methoxyquinoxaline (1)
3.1.3. Synthesis of 3-Methoxyquinoxaline-2-carbonitrile (2)
3.1.4. General Procedure for Synthesis of N-(3-Methoxyquinoxalin-2-ylmethyl)-alkylamidea 3a–e
3.1.5. General Procedure for Synthesis of 1-Alkyl-3-(3-methoxyquinoxalin-2-ylmethyl)ureas 3f–i
3.1.6. Synthesis of 2,3-Dimethoxy-6-nitroquinoxaline (4)
3.1.7. Synthesis of 6-Amino-2,3-dimethoxyquinoxaline (5)
3.1.8. General Procedure for Synthesis of N-(2,3-Dimethoxyquinoxalin-6-yl)alkylamides 6a–e
3.1.9. General Procedure for Synthesis of 1-(2,3-Dimethoxyquinoxaline-6-yl)-3-alkylureas 6f–i
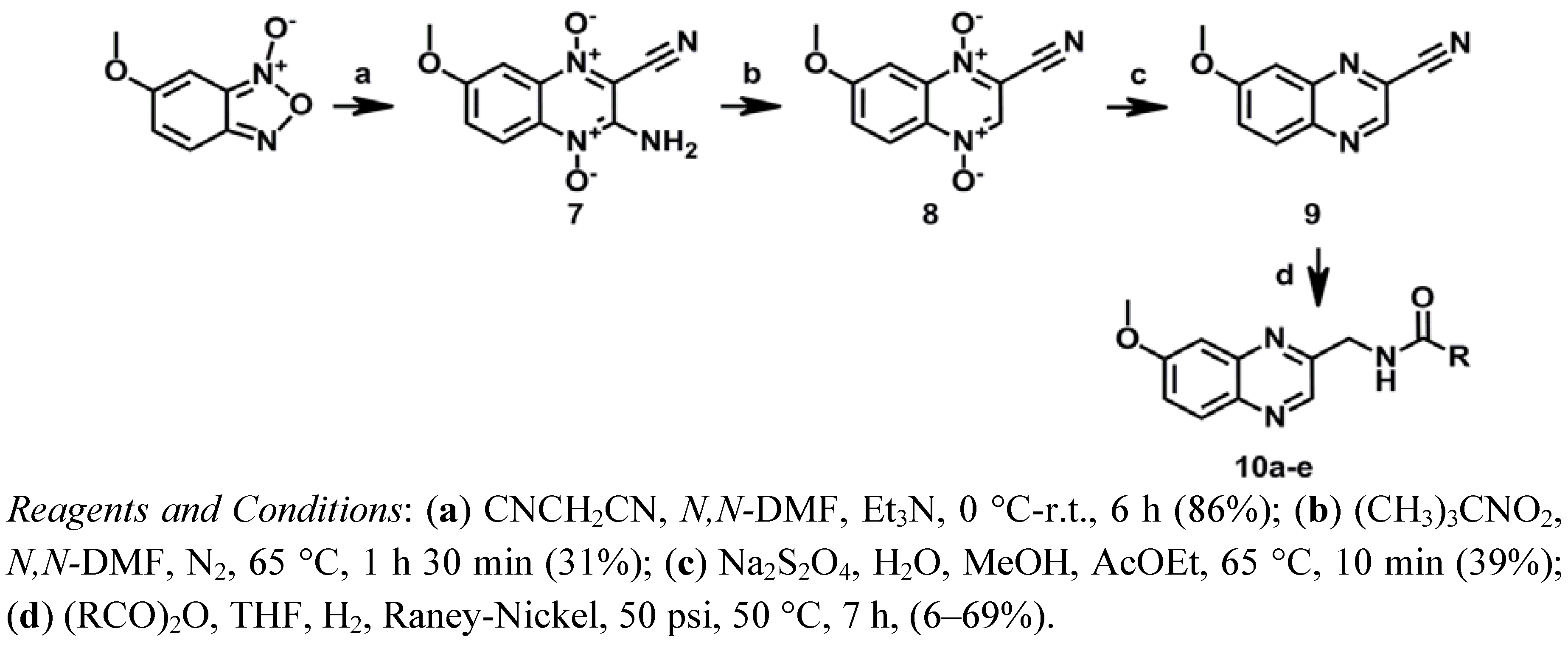
3.1.10. Synthesis of 3-Amino-7-methoxy-1,4-di-N-oxidequinoxaline-2-carbonitrile (7)
3.1.11. Synthesis of 7-Methoxy-1,4-di-N-oxidequinoxaline-2-carbonitrile (8)
3.1.12. Synthesis of 7-Methoxyquinoxaline-2-carbonitrile (9)
3.1.13. General Procedure for Synthesis of N-[(7-Methoxyquinoxalin-2-yl)methyl]alkylamides 10a–e
3.2. Pharmacology
3.2.1. Reagents and Chemicals
3.2.2. Cell Culture
3.2.3. Binding Assays
4. Conclusions
Acknowledgments
References and Notes
- Gállego, J.; Toledo, J.B.; Urrestarazu, E.; Iriarte, J. Clasificación de los trastornos del sueño. An. Sist. Sanit. Navar. 2007, 30, 19–36. [Google Scholar]
- Roth, T. Insomnia: Definition, prevalence, etiology, and consequences. Clin. Sleep Med. 2007, 3, S7–S10. [Google Scholar]
- Sarrais, F.; De Castro, T. El insomnio. Anales del Sistema Sanitario de Navarra 2007, 30, 121–134. [Google Scholar]
- Spadoni, G.; Bedini, A.; Rivara, S.; Mor, M. Melatonin receptor agonists: New options for insomnia and depression treatment. CNS Neurosci. Ther. 2011, 17, 733–741. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; Srinivasan, V.; Spence, D.W.; Cardinali, D.P. Role of the melatonin system in the control of sleep: Therapeutic implications. CNS Drugs 2007, 21, 995–1018. [Google Scholar] [CrossRef]
- Zlotos, D.P. Recent advances in melatonin receptor ligands. Arch. Pharm. 2005, 338, 229–247. [Google Scholar] [CrossRef]
- Dubocovich, M.L. Melatonin receptors: Role on sleep and circadian rhythm regulation. Sleep Med. 2007, 8, S34–S42. [Google Scholar] [CrossRef]
- Alarma-Estrany, P.; Pintor, J. Melatonin receptors in the eye: Location, second messengers and role in ocular physiology. Pharmacol. Ther. 2007, 113, 507–522. [Google Scholar] [CrossRef]
- El Kazzouli, S.; Griffon du Bellay, A.; Berteina-Raboin, S.; Delagrange, P.; Caignard, D.-H.; Guillaumet, G. Design and synthesis of 2-phenylimidazo[1,2-a]pyridines as a novel class of melatonin receptor ligands. Eur. J. Med. Chem. 2011, 46, 4252–4257. [Google Scholar]
- Markl, C.; Attia, M.I.; Julius, J.; Sethi, S.; Witt-Enderby, P.A.; Zlotos, D.P. Synthesis and pharmacological evaluation of 1,2,3,4-tetrahydropyrazino[1,2-a]indole and 2-[(phenylmethylamino)methyl]-1H-indole analogues as novel melatoninergic ligands. Bioorg. Med. Chem. 2009, 17, 4583–4594. [Google Scholar] [CrossRef]
- Ettaoussi, M.; Sabaouni, A.; Rami, M.; Boutin, J.A.; Delagrange, P.; Renard, P.; Spedding, M.; Caignard, D.-H.; Berthelot, P.; Yous, S. Design, synthesis and pharmacological evaluation of new series of naphthalenic analogues as melatoninergic (MT1/MT2) and serotoninergic 5-HT2C dual ligands (I). Eur. J. Med. Chem. 2012, 49, 310–323. [Google Scholar] [CrossRef]
- Fisher, S.P.; Sugden, D. Sleep-promoting action of IIK7, a selective MT2 melatonin receptor agonist in the rat. Neurosci. Lett. 2009, 457, 93–96. [Google Scholar] [CrossRef]
- Leclerc, V.; Ettaoussi, M.; Rami, M.; Farce, A.; Boutin, J.A.; Delagrange, P.; Caignard, D.-H.; Renard, P.; Berthelot, P.; Yous, S. Design and synthesis of naphthalenic derivatives as new ligands at the melatonin binding site MT3. Eur. J. Med. Chem. 2011, 46, 1622–1629. [Google Scholar] [CrossRef]
- Srinivasan, V.; Brzezinski, A.; Pandi-Perumal, S.R.; Spence, D.W.; Cardinali, D.P.; Brown, G.M. Melatonin agonists in primary insomnia and depression-associated insomnia: Are they superior to sedative-hypnotics? Prog. Neuro-Psychoph. 2011, 35, 913–923. [Google Scholar] [CrossRef]
- Koike, T.; Hoashi, Y.; Takai, T.; Nakayama, M.; Yukuhiro, N.; Ishikawa, T.; Hirai, K.; Uchikawa, O. 1,6-Dihydro-2H-indeno 5,4-b furan Derivatives: Design, Synthesis, and Pharmacological Characterization of a Novel Class of Highly Potent MT2-Selective Agonists. J. Med. Chem. 2011, 54, 3436–3444. [Google Scholar] [CrossRef]
- Poissonnier-Durieux, S.; Ettaoussi, M.; Pérès, B.; Boutin, J.A.; Audinot, V.; Bennejean, C.; Delagrange, P.; Caignard, D.H.; Renard, P.; Berthelot, P.; et al. Synthesis of 3-phenylnaphthalenic derivatives as new selective MT2 melatoninergic ligands. Bioorg. Med. Chem. 2008, 16, 8339–8348. [Google Scholar] [CrossRef]
- Durieux, S.; Chanu, A.; Bochu, C.; Audinot, V.; Coirnailleau, S.; Boutin, J.A.; Delagrange, P.; Caignard, D.H.; Bennejean, C.; Renard, P.; et al. Design and synthesis of 3-phenyltetrahydronaphthalenic derivatives as new selective MT2 melatoninergic ligands. Part II. Bioorg. Med. Chem. 2009, 17, 2963–2974. [Google Scholar] [CrossRef]
- Rivara, S.; Lodola, A.; Mor, M.; Bedini, A.; Spadoni, G.; Lucini, V.; Pannacci, M.; Fraschini, F.; Scaglione, F.; Ochoa-Sanchez, R.; et al. N-(Substituted anilinoethyl)amides: Design, synthesis and pharmacological characterization of a new class of melatonin receptor ligands. J. Med. Chem. 2007, 50, 6618–6626. [Google Scholar]
- Hu, Y.; Ho, M.K.; Chan, K.H.; New, D.C.; Wong, Y.H. Synthesis of substituted N-[3-(3-methoxyphenyl)propyl]amides as highly potent MT2-selective melatonin ligands. Bioorg. Med. Chem. Lett. 2010, 20, 2582–2585. [Google Scholar] [CrossRef]
- Navajas, C.; Kokkola, T.; Poso, A.; Honka, N.; Gynther, J.; Laitinen, J.T. A rhodopsin-based model for melatonin recognition at its G protein-coupled receptor. Eur. J. Pharmacol. 1996, 304, 173–183. [Google Scholar] [CrossRef]
- Sugden, D.; Chong, N.W.S.; Lewis, D.F.W. Structural requirements at the melatonin receptor. Br. J. Pharmacol. 1995, 114, 618–623. [Google Scholar] [CrossRef]
- Spadoni, G.; Balsamini, C.; Diamantini, G.; Di Giacomo, B.; Tarzia, G.; Mor, M.; Plazzi, P.V.; Rivara, S.; Lucini, V.; Nonno, R.; et al. Conformationally restrained melatonin analogues: Synthesis, binding affinity for the melatonin receptor, evaluation of the biological activity, and molecular modeling study. J. Med. Chem. 1997, 40, 1990–2002. [Google Scholar]
- Sicsic, S.; Serraz, I.; Andrieux, J.; Bremont, B.; Mathe-Allainmat, M.; Poncet, A.; Shen, S.; Langlois, M. Three-dimensional quantitative structure-activity relationship of melatonin receptor ligands: a comparative molecular field analysis study. J. Med. Chem. 1997, 40, 739–748. [Google Scholar] [CrossRef]
- Grol, C.J.; Jansen, J.M. The high affinity melatonin binding site probed with conformationally restricted ligands. II. Homology modeling of the receptor. Bioorg. Med. Chem. 1996, 4, 1333–1339. [Google Scholar] [CrossRef]
- Mor, M.; Rivara, S.; Silva, C.; Bordi, F.; Plazzi, P.V.; Spadoni, G.; Diamantini, G.; Balsamini, C.; Tarzia, G.; Fraschini, F.; et al. Melatonin receptor ligands: Synthesis of new melatonin derivatives and comprehensive comparative molecular field analysis (CoMFA) study. J. Med. Chem. 1998, 41, 3831–3844. [Google Scholar]
- Jansen, J.M.; Copinga, S.; Gruppen, G.; Molinari, E.J.; Dubocovich, M.L.; Grol, C.J. The high affinity melatonin binding site probed with conformationally restricted ligand. I. POharmacophore and minireceptor models. Bioorg. Med. Chem. 1996, 4, 1321–1332. [Google Scholar] [CrossRef]
- Marot, C.; Chavatte, P.; Morin-Allory, L.; Viaud, M.C.; Guillaumet, G.; Renard, P.; Lesiur, D.; Michel, A. Pharmacophoric search and 3D-QSAR comparative molecular field analysis studies on agonists of melatonin sheep receptors. J. Med. Chem. 1998, 41, 4453–4465. [Google Scholar] [CrossRef]
- Spadoni, G.; Bedini, A.; Rivara, S.; Mor, M. Melatonin Receptor Agonists: New Options for Insomnia and Depression Treatment. CNS Neurosci. Ther. 2011, 17, 733–741. [Google Scholar] [CrossRef]
- Li, P.-K.; Chu, G.-H.; Gillen, M.L.; Witt-Enderby, P.A. Synthesis and receptor binding studies of quinolinic derivatives as melatonin receptor ligands. Bioorg. Med. Chem. Lett. 1997, 7, 2177–2180. [Google Scholar] [CrossRef]
- Yous, S.; Andrieux, J.; Howell, H.E.; Morgan, P.J.; Renard, P.; Pfeiffer, B.; Lesieur, D.; Guardiola-Lemaitre, B. Novel naphthalenic ligands with high affinity for the melatonin receptor. J. Med. Chem. 1992, 35, 1484–1486. [Google Scholar] [CrossRef]
- Waring, M.; Ben-Hadda, T.; Kotchevar, A.; Ramdani, A.; Touzani, R.; Elkadiri, S.; Hakkou, A.; Bouakka, M.; Ellis, T. 2,3-Bifunctionalized quinoxalines: Synthesis, DNA interactions and evaluation of anticancer, anti-tuberculosis and antifungal activity. Molecules 2002, 7, 641–656. [Google Scholar] [CrossRef]
- Burguete, A.; Pontiki, E.; Hadjipavlou-Litina, D.; Ancizu, S.; Villar, R.; Solano, B.; Moreno, E.; Torres, E.; Pérez, S.; Aldana, I.; et al. Synthesis and Biological Evaluation of New Quinoxaline Derivatives as Antioxidant and Anti-Inflammatory Agents. Chem. Biol. Drug Des. 2011, 77, 255–267. [Google Scholar] [CrossRef]
- Ancizu, S.; Moreno, E.; Solano, B.; Villar, R.; Burguete, A.; Torres, E.; Pérez-Silanes, S.; Aldana, I.; Monge, A. New 3-methylquinoxaline-2-carboxamide 1,4-di-N-oxide derivatives as anti-mycobacterium tuberculosis agents. Bioorg. Med. Chem. 2010, 18, 2713–2719. [Google Scholar] [CrossRef]
- Olayiwola, G.; Obafemi, C.A.; Taiwo, F.O. Synthesis and neuropharmacological activity of some quinoxalinone derivatives. Afr. J. Biotechnol. 2007, 6, 777–786. [Google Scholar]
- Mathé-Allainmat, M.; Andrieux, J.; Langlois, M. Recent developments in melatonin receptor ligands. Expert Opin. Ther. Pat. 1997, 7, 1447–1458. [Google Scholar] [CrossRef]
- Garratt, P.J.; Jones, R.; Rowe, S.J.; Sugden, D. Mapping the Melatonin Receptor. 1. The 5-Methoxy group is not an essential requirement for biological activity. Bioorg. Med. Chem. Lett. 1994, 4, 1555–1558. [Google Scholar] [CrossRef]
- Garratt, P.J.; Travard, S.; Vonhoff, S.; Tsotinis, A.; Sugden, D. Mapping the melatonin receptor. 4. Comparison of the binding affinities of a series of substituted phenylalkyl amides. J. Med. Chem. 1996, 39, 1797–1805. [Google Scholar] [CrossRef]
- Davies, D.J.; Faust, R.; Garratt, P.J.; Marivingt-Mounir, C.; Davidson, K.; Teh, M.T.; Sugden, D. Binding affinity and biological activity of oxygen and sulfur isosteres ata melatonin receptors as a function of their hydrogen bonding capability. Bioorg. Chem. 2004, 32, 1–12. [Google Scholar] [CrossRef]
- Langlois, M.; Bremont, B.; Shen, S.; Poncet, A.; Andrieux, J.; Sicsic, S.; Serraz, I.; Mathé-Allainmat, M.; Renard, P.; Delagrange, P. Design and synthesis of new naphthalenic derivatives as ligands for 2-[125I]iodomelatonin binding sites. J. Med. Chem. 1995, 38, 2050–2060. [Google Scholar] [CrossRef]
- Beavers, M. P.; Dudash, J.; Zhang, Y. 3-Oxo-3,4-dihydro-quinoxalin-2-yl-amino)-benzamide Derivatives and Related Compound as Glycogen Phosphorylase Inhibitors for the Treatment of Diabetes and Obesity. WO/2005/067932 2005. [Google Scholar]
- Ellis, G.P.; Romney-Alexander, T.M. Cyanation of aromatic halides. Chem. Rev. 1987, 87, 779–794. [Google Scholar] [CrossRef]
- Hermann, K.; Simchen, G. Synthese cyan-substituierter heterocyclen mit tetraethyl-ammoniumcyanid. Liebigs Ann. Chem. 1981, 1981, 333–341. [Google Scholar] [CrossRef]
- Tarzia, G.; Diamantini, G.; Di Giacomo, B.; Spadoni, G.; Esposti, D.; Nonno, R.; Lucini, V.; Pannacci, M.; Fraschini, F.; Stankov, B.M. 1-(2-Alkanamidoethyl)-6-methoxyindole derivatives: A new class of potent iIndole melatonin analogues. J. Med. Chem. 1997, 40, 2003–2010. [Google Scholar] [CrossRef]
- Mor, M.; Spadoni, G.; Di Giacomo, B.; Diamantini, G.; Bedini, A.; Tarzia, G.; Plazzi, P.V.; Rivara, S.; Nonno, R.; Lucini, V.; et al. Synthesis, pharmacological characterization and QSAR studies on 2-substituted indole melatonin receptor ligands. Bioorg. Med. Chem. 2001, 9, 1045–1057. [Google Scholar] [CrossRef]
- Galiano, S.; Ceras, J.; Cirauqui, N.; Pérez, S.; Juanenea, L.; Rivera, G.; Aldana, I.; Monge, A. Novel series of substituted biphenylmethyl urea derivatives as MCH-R1 antagonists for the treatment of obesity. Bioorg. Med. Chem. 2007, 15, 3896–3911. [Google Scholar] [CrossRef]
- Koyuncu, S.; Zafer, C.; Koyuncu, F.B.; Aydin, B.; Can, M.; Sefer, E.; Ozdemir, E.; Icli, S. A New Donor-acceptor Double-cable Carbazole Polymer with Perylene Bisimide Pendant Group: Synthesis, Electrochemical, and Photovoltaic Properties. J. Polym. Sci. A Polym. Chem. 2009, 47, 6280–6291. [Google Scholar] [CrossRef]
- Wallez, V.; Durieux-Poissonnier, S.; Chavatte, P.; Boutin, J.A.; Audinot, V.; Nicolas, J.-P.; Bennejean, C.; Delagrange, P.; Renard, P.; Lesieur, D. Synthesis and structure-affinity-activity relationships of novel benzofuran derivatives as MT2 melatonin receptor selective ligands. J. Med. Chem. 2002, 45, 2788–2800. [Google Scholar] [CrossRef]
- Monge, A.; Martinez-Crespo, F.J.; Lopez de Cerain, A.; Palop, J.A.; Narro, S.; Senador, V.; Marin, A.; Sainz, Y.; Gonzalez, M. Hypoxia-selective agents derived from 2-quinoxalinecarbonitrile 1,4-di-N-oxides. 2. J. Med. Chem. 1995, 38, 4488–4494. [Google Scholar]
- Solano, B.; Junnotula, V.; Marín, A.; Villar, R.; Burguete, A.; Vicente, E.; Pérez-Silanes, S.; Aldana, I.; Monge, A.; Dutta, S.; et al. Synthesis and biological evaluation of new 2-arylcarbonyl-3-trifluoromethylquinoxaline 1,4-di-N-oxide derivatives and their reduced analogues. J. Med. Chem. 2007, 50, 5485–5492. [Google Scholar] [CrossRef]
- Audinot, V.; Mailliet, F.; Lahaye-Brasseur, C.; Bonnaud, A.; Le Gall, A.; Amossé, C.; Dromaint, S.; Rodriguez, M.; Nagel, N.; Galizzi, J.P.; et al. New selective ligands of human cloned melatonin MT1 and MT2 receptors. Naunyn-Schmiedebergs Arch. Pharmacol. 2003, 367, 553–561. [Google Scholar] [CrossRef]
- Farce, A.; Chugunov, A.O.; Logé, C.; Sabaouni, A.; Yous, S.; Dilly, S.; Renault, N.; Vergoten, G.; Efremov, R.G.; Lesieur, D.; et al. Homology modeling of MT1 and MT2 receptors. Eur. J. Med. Chem. 2008, 43, 1926–1944. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ancizu, S.; Castrillo, N.; Pérez-Silanes, S.; Aldana, I.; Monge, A.; Delagrange, P.; Caignard, D.-H.; Galiano, S. New Quinoxaline Derivatives as Potential MT1 and MT2 Receptor Ligands. Molecules 2012, 17, 7737-7757. https://doi.org/10.3390/molecules17077737
Ancizu S, Castrillo N, Pérez-Silanes S, Aldana I, Monge A, Delagrange P, Caignard D-H, Galiano S. New Quinoxaline Derivatives as Potential MT1 and MT2 Receptor Ligands. Molecules. 2012; 17(7):7737-7757. https://doi.org/10.3390/molecules17077737
Chicago/Turabian StyleAncizu, Saioa, Nerea Castrillo, Silvia Pérez-Silanes, Ignacio Aldana, Antonio Monge, Philippe Delagrange, Daniel-Henry Caignard, and Silvia Galiano. 2012. "New Quinoxaline Derivatives as Potential MT1 and MT2 Receptor Ligands" Molecules 17, no. 7: 7737-7757. https://doi.org/10.3390/molecules17077737