Computerized Modeling of Adenosine Triphosphate, Adenosine Triarsenate and Adenosine Trivanadate

Abstract
:1. Introduction

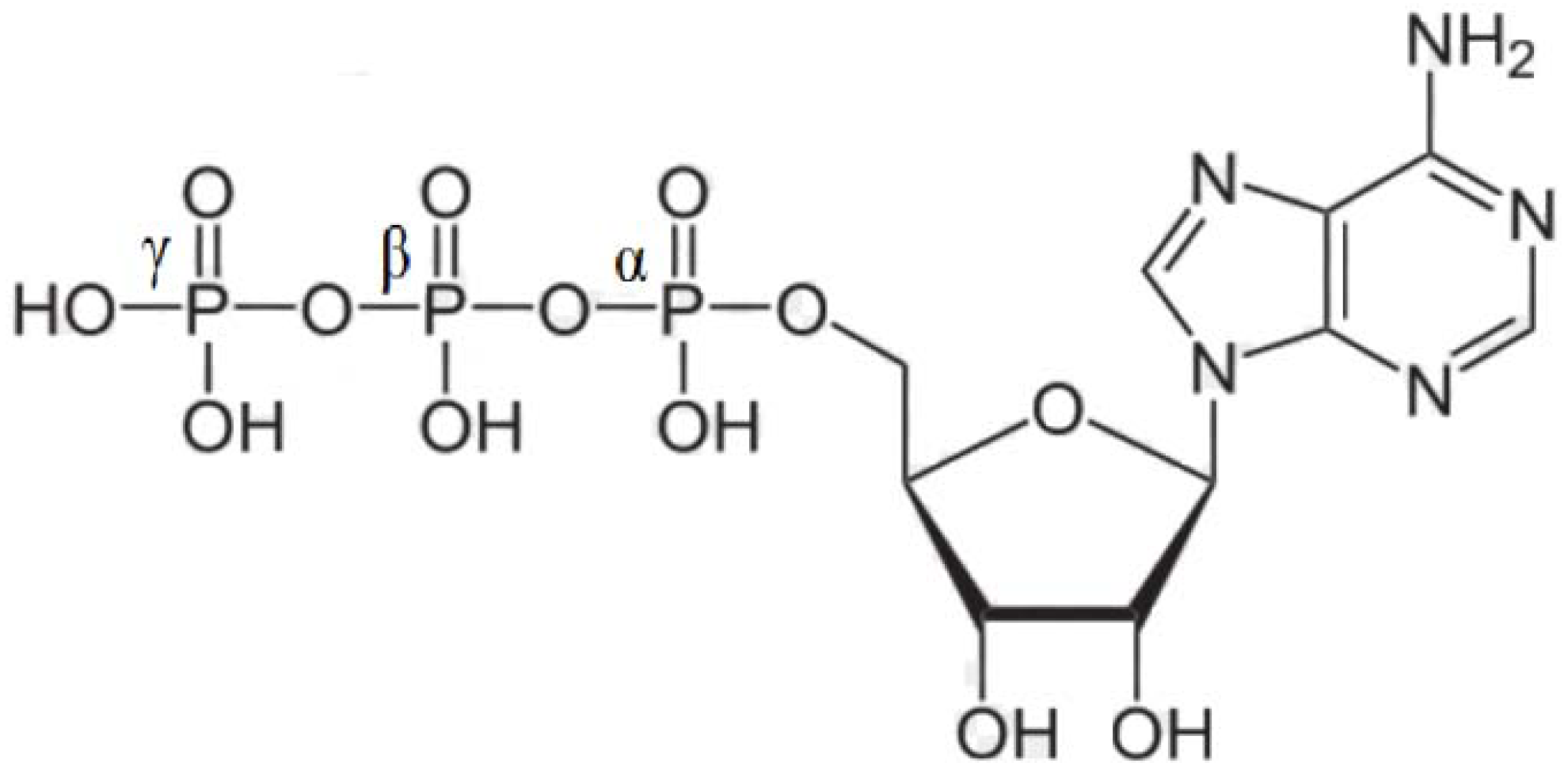{kind=link}
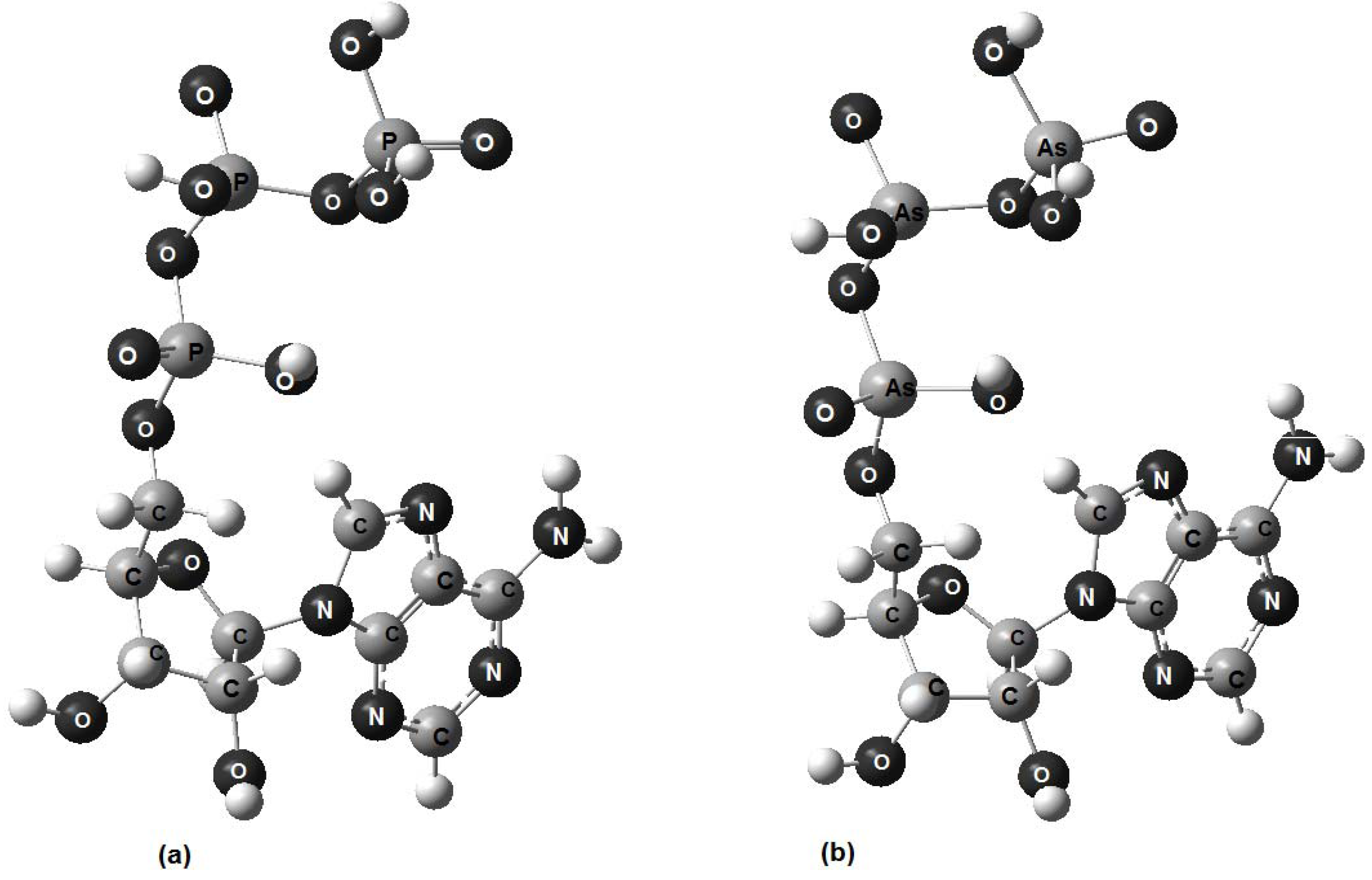{kind=link}
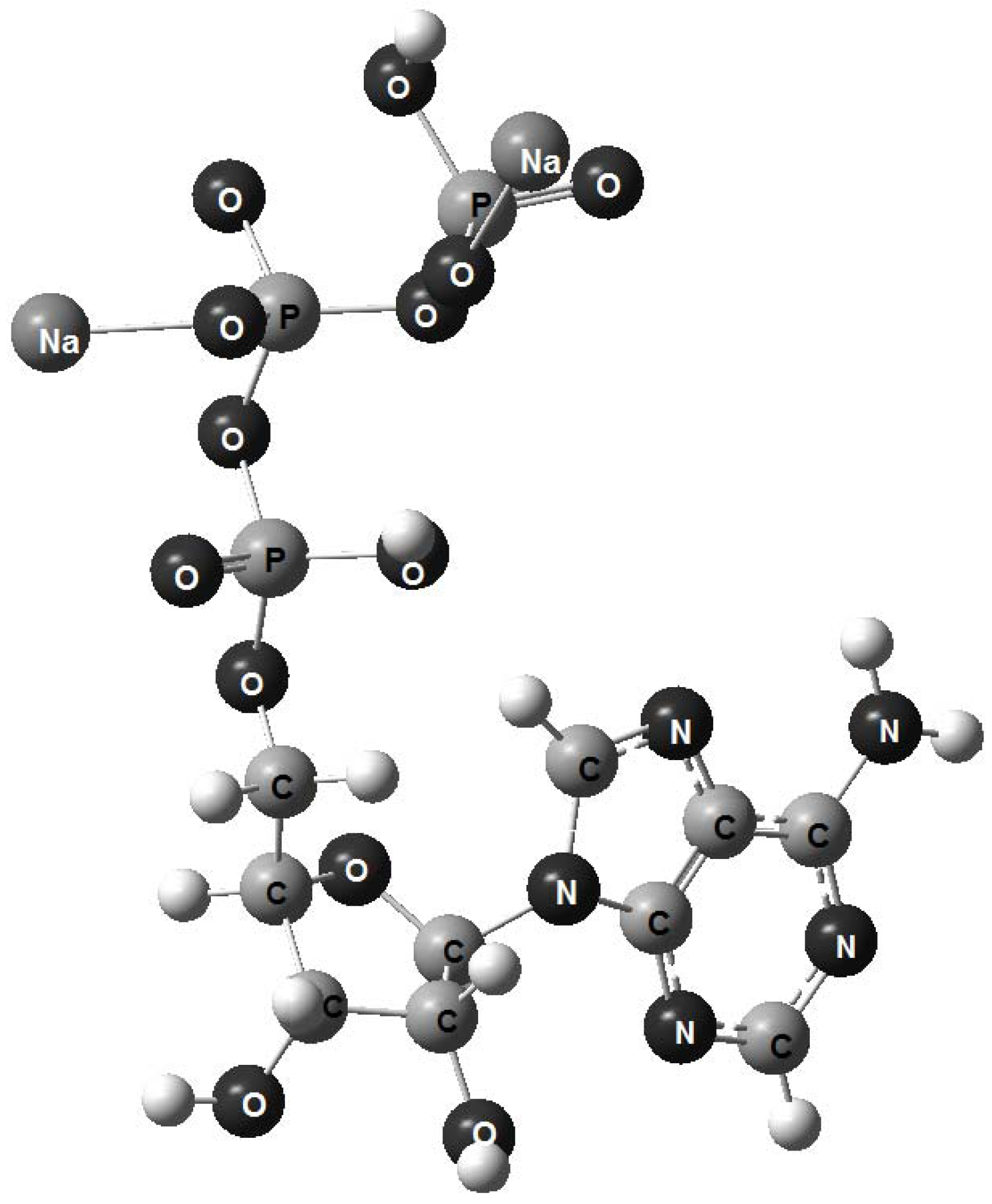{kind=link}
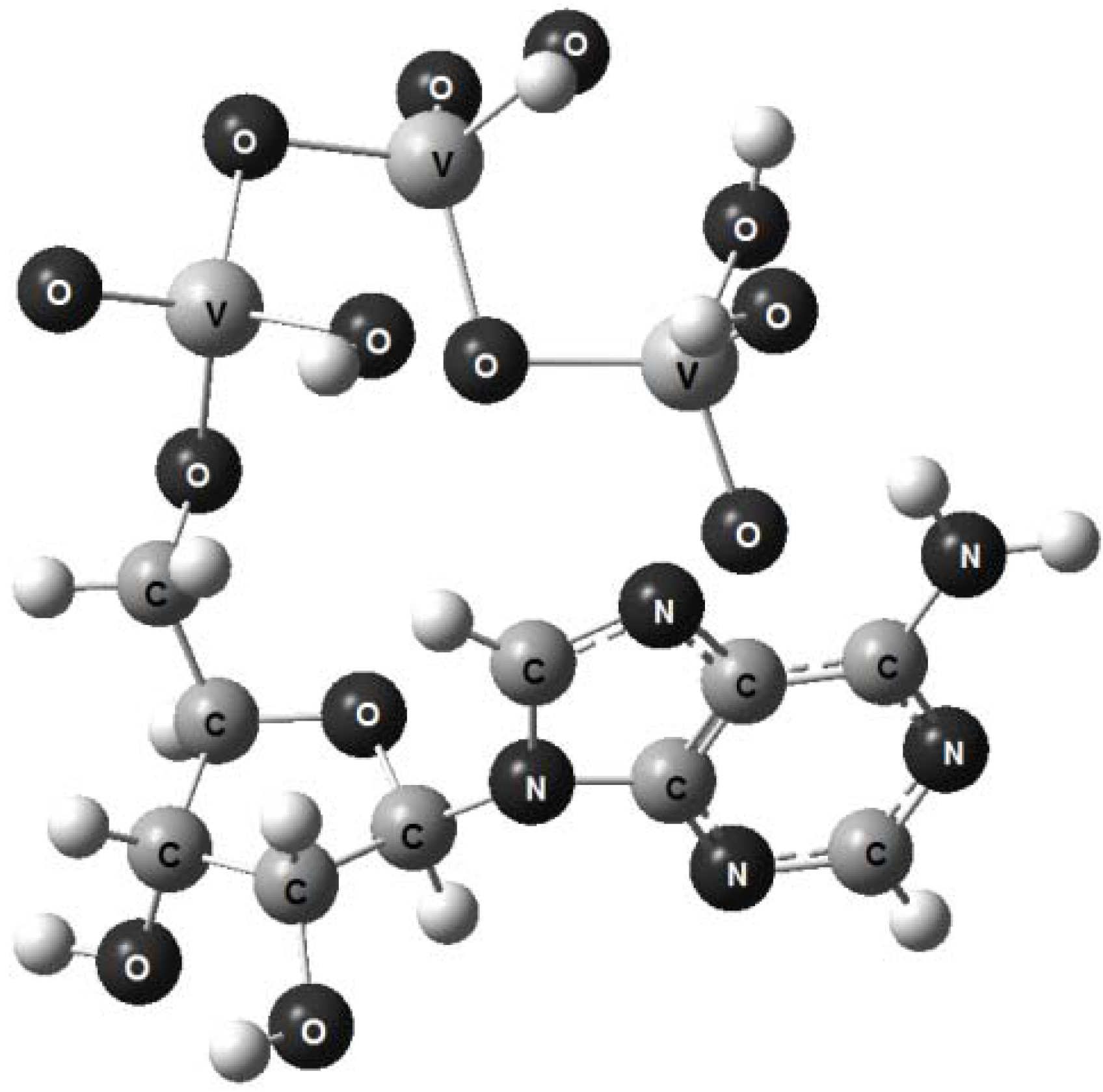{kind=link}
| Tripolyions | ||||
|---|---|---|---|---|
| P3O105− | As3O105− | |||
| Distances (Å) | P-Ob * | 1.52 | As-Ob * | 1.67 |
| P OH | 1.61 | As OH | 1.74 | |
| O-P-O | 96 | O-As-O | 93 | |
| Angles (°) | P-Ob-P | 156 | As-Ob-As | 120 |

2. Molecular Modeling
3. Results and Discussion



| Parameters | Compounds | Published data | ||||
|---|---|---|---|---|---|---|
| ATP | Na2ATP | AT-As | AT-V | calc. | exper. | |
| Distances | ||||||
| E *** = O | 1.51 | 1.49 | 1.64 | 1.75 | 1.50 | 1.50 |
| E-O | 1.68/1.69 | 1.60 | 1.83 | 1.96 | 1.57 | 1.50 |
| E-Ob | 1.70 | 1.60 | 1.85 | 1.96 | 1.58/1.65 | 1.61 |
| C-Ob | 1.40 | 1.42 | 1.40 | 1.39 | 1.48 | 1.42 |
| H. . .O | 0.99 | 0.97 | 0.99 | 0.99 | 0.98/1.04 | - |
| Angles | ||||||
| E-Ob-E | 107.6 | 112.6/114.6 | 105.7 | 104.6 | 119.5 | 125.6 |
| E-Ob-C | 108.6 | 124.1 | 107.8 | 105.4 | 120 | 122.9 |
4. Conclusions
- 1. Computerized molecular models of adenosine triphosphate, adenosine triarsenate and adenosine trivanadate have been proposed.
- 2. The analysis of structural parameters shows that ATP and ATAs are stereochemically equivalent.
- 3. The structural arrangement of adenosine trivanadate does not seem to be capable of withstanding a swift hydrolytical splitting in aqueous milieu.
- 4. The universal force field as implemented in the Gaussian software packages is an appropriate tool for the optimization of less-common bioactive compositions.
Acknowledgments
References
- Pratt, A.J. Why Nature chose phosphates over arsenates? J. Cosmol. 2010, 13, 3601–3608. [Google Scholar]
- Moore, S.A.; Moennich, D.M.C.; Gresser, M.J. Synthesis and hydrolysis of ADP-arsenate by beef heart submitochondrial particles. J. Biol. Chem. 1983, 258, 6266–6271. [Google Scholar]
- Wolfe-Simon, F.; Blum, J.S.; Kulp, T.R.; Gordon, G.W.; Hoeft, S.E.; Pett-Ridge, J.; Stolz, J.F.; Webb, S.M.; Weber, P.K.; Davies, P.C.W.; et al. A bacterium that can grow by using arsenic instead of phosphorus. Science 2011, 332, 11663–11666. [Google Scholar]
- Wolfe-Simon, F.; Switzer Blum, J.; Kulp, T.R.; Gordon, G.W.; Hoeft, S.E.; Pett-Ridge, J.; Stolz, J.F.; Webb, S.M.; Webb, P.K.; Davies, P.C.W.; et al. Response to Comments on “A Bacterium That Can Grow Using Arsenic Instead of Phosphorus”. Science 2011. [Google Scholar] [CrossRef]
- Huang, C.H.; Mitchell, R.A. Arsenate and phosphate as modifiers of adenosine triphosphate driven energy-linked reduction. Kinetic study of the effects of modifiers on inhibition by adenosine diphosphate. Biochemistry 1972, 11, 2278–2283. [Google Scholar] [CrossRef]
- Quist, E.E.; Hokin, L.E. The presence of two (Na+ + K+)-ATPase inhibitors in equine muscle ATP: Vanadate and a dithiorythritol-dependent inhibitor. Biochim. Biophys. Acta 1978, 511, 201–212. [Google Scholar]
- Wells, A.F. Structural Inorganic Chemistry, 5th ed; Oxford University Press: New York, NY, USA, 1984. [Google Scholar]
- Jost, K.H.; Worzala, H.; Thilo, E. Structure of As2O5·5/3H2O. Acta Crystallogr. 1966, 21, 808–813. [Google Scholar] [CrossRef]
- Tracey, A.S.; Crans, D.C. Vanadium Compounds: Chemistry, Biochemistry and Therapeutic Applications; American Chemical Society: Washington, DC, USA, 1998. [Google Scholar]
- Wolfe-Simon, F.; Davies, P.C.W.; Anbar, A.D. Did nature also choose arsenic? Int. J. Astrobiol. 2009, 8, 69–74. [Google Scholar] [CrossRef]
- Akola, J.; Jones, R.O. ATP hydrolysis in water-a density functional study. J. Phys. Chem. B 2003, 107, 11774–11783. [Google Scholar] [CrossRef]
- Kustin, K.; Costa Pessoa, J.; Crans, D.C. Vanadium: The Versatile Metal, 1st ed; American Chemical Society: Washington, DC, USA, 2007. [Google Scholar]
- Frisch, A.M.J.; Trucks, G.W.; Shelelgel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03 ; Gaussian, Inc.: Wallingford, CT, USA, 2004; revision E.0. [Google Scholar]
- Frisch, A.M.J.; Dennington, R.D., II; Keith, T.A.; Millam, J.; Nielsen, A.B.; Holder, A.J.; Hiscocks, J. Gaussview; Gaussian, Inc.: Wallingford, CT, USA, 2004; 4.1.2. [Google Scholar]
- Young, D.C. Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems; Wiley-Interscience: New York, NY, USA, 2001. [Google Scholar]
- Nascimento, V.A.; Melnikov, P.; Zanoni, L.Z. Comparative structural modeling of cysteine and selenocysteine. J. Solids Struct. 2011, 5, 153–161. [Google Scholar]
- Kennard, O.; Isaacs, N.W.; Motherwell, W.D.S.; Copolla, J.C.; Wampler, D.L.; Larson, A.C.; Watson, D.G. The crystal and molecular structure of adenosine triphosphate. Proc. R. Soc. Lond. A 1971, 325, 401–436. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nascimento, V.A.; Melnikov, P.; Consolo, L.Z.Z. Computerized Modeling of Adenosine Triphosphate, Adenosine Triarsenate and Adenosine Trivanadate. Molecules 2012, 17, 9489-9495. https://doi.org/10.3390/molecules17089489
Nascimento VA, Melnikov P, Consolo LZZ. Computerized Modeling of Adenosine Triphosphate, Adenosine Triarsenate and Adenosine Trivanadate. Molecules. 2012; 17(8):9489-9495. https://doi.org/10.3390/molecules17089489
Chicago/Turabian StyleNascimento, Valter A., Petr Melnikov, and Lourdes Z. Z. Consolo. 2012. "Computerized Modeling of Adenosine Triphosphate, Adenosine Triarsenate and Adenosine Trivanadate" Molecules 17, no. 8: 9489-9495. https://doi.org/10.3390/molecules17089489