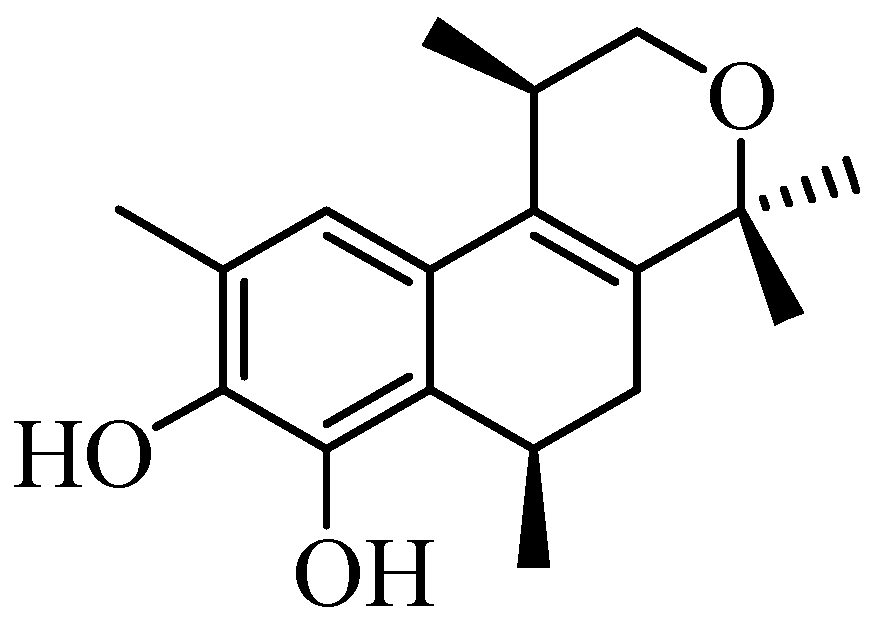
Populene D Analogues: Design, Concise Synthesis and Antiproliferative Activity

Abstract
:1. Introduction


2. Results and Discussion
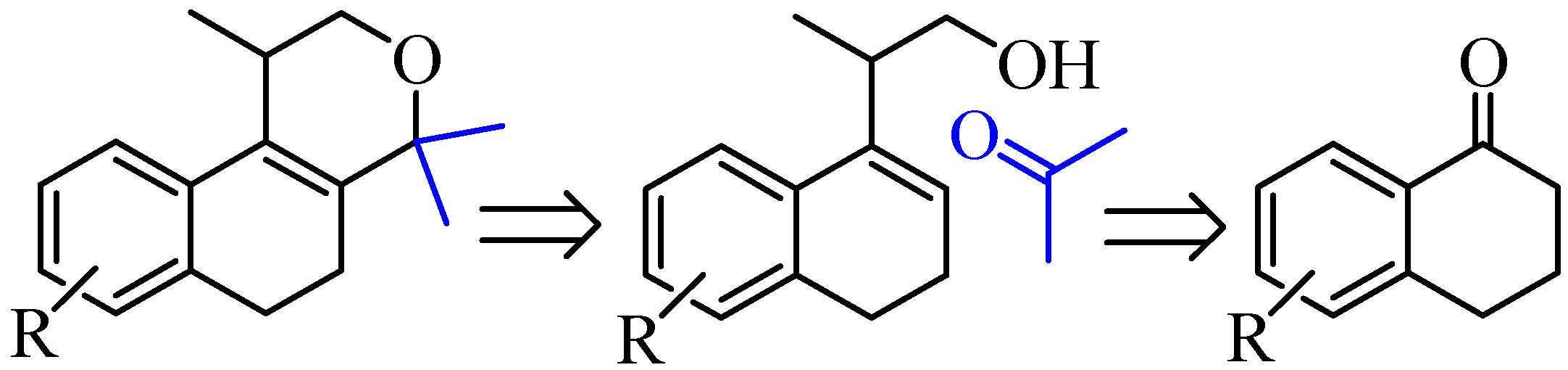
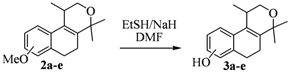
2.1. Synthesis of Populene D Analogues

{kind=link}
{kind=link}
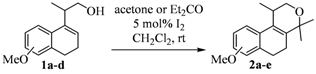 | ||
|---|---|---|
| Entry | Homoallylic alcohols 1a–d | Products 2a–e (yield) |
| 1 | 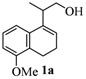 | 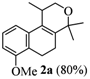 |
| 2 | 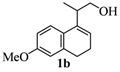 |  |
| 3 | 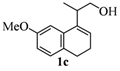 | 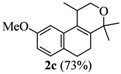 |
| 4 | 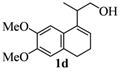 |  |
 | ||
|---|---|---|
| Entry | Substrates 2a–d | Products 3a–e (yield) |
| 1 | 2a | 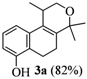 |
| 2 | 2b |  |
| 3 | 2c |  |
| 4 | 2d | 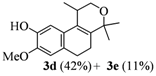 |
| 5 | 2d | 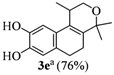 |
2.2. Antiproliferative Activity of Populene D Analogues
| Cell lines a | Doxorubicine b | 2a | 2b | 2c | 2d | 3a | 3b | 3c | 3d | 3e |
|---|---|---|---|---|---|---|---|---|---|---|
| U251 | 0.062 | 25.0 | 24.6 | 25.0 | >250 | >250 | 46.3 | 27.8 | 23.1 | 19.6 |
| MCF-7 | 0.21 | 25.0 | 23.2 | 25.0 | 38.8 | >250 | 107.8 | 24.7 | 24.6 | 5.5 |
| NCI-ADR/RES | 1.3 | 25.0 | 26.8 | 25.0 | 100.7 | >250 | >250 | 25.0 | 5.2 | 5.7 |
| 786-0 | 0.04 | 25.0 | 25.5 | 25.0 | 45.8 | 37.3 | 36.8 | 24.6 | 24.1 | 5.6 |
| NCI-H460 | <0.025 | N.T. | N.T. | N.T. | N.T. | >250 | 41.4 | 24.2 | 45.1 | 17.4 |
| PC-3 | 0.27 | 25.0 | 27.0 | 25.0 | 71.4 | >250 | >250 | 24.5 | N.T. | N.T. |
| OVCAR-3 | 0.26 | 25.0 | 24.5 | 25.0 | 193.4 | >250 | >250 | 1.8 | 24.3 | 3.9 |
| HT-29 | 0.22 | 25.0 | 6.6 | 25.0 | 23.1 | >250 | 78.4 | 22.9 | 6.0 | 19.5 |
| K-562 | 0.4 | 3.1 | 9.6 | 25.0 | 84.6 | >250 | 41.2 | 8.7 | N.T. | N.T. |
| HaCat | 0.23 | 25.0 | 26.1 | 25.0 | >250 | >250 | >250 | 3.6 | 4.2 | 4.2 |
| Mean logGI50c, d | −0.78 P | 1.17 W | 1.18 W | 1.21 W | >1.70 I | >2.31 I | >1.95 I | 1.22 W | 1.24 W | 0.95 M |
| Cell lines a | Doxorubicine b | 2a | 2b | 2c | 2d | 3a | 3b | 3c | 3d | 3e |
|---|---|---|---|---|---|---|---|---|---|---|
| U251 | 1.1 | 52.0 | 53.5 | 40.0 | >250 | >250 | >250 | 67.9 | 45.9 | 28.4 |
| MCF-7 | 8.8 | 51.3 | 46.9 | 45.3 | >250 | >250 | >250 | 45.0 | 58.2 | 41.6 |
| NCI-ADR/RES | 25.0 | 83.5 | 104.4 | 66.3 | >250 | >250 | >250 | 199.5 | 37.6 | >250 |
| 786-0 | 0.62 | 50.6 | 44.0 | 36.1 | >250 | >250 | >250 | 43.0 | 47.8 | 19.4 |
| NCI-H460 | 0.025 | N.T. | N.T. | N.T. | N.T. | >250 | >250 | 45.9 | >250 | >250 |
| PC-3 | 4.4 | 64.6 | 41.4 | 69.2 | >250 | >250 | >250 | 51.8 | N.T. | N.T. |
| OVCAR-3 | 3.9 | 52.3 | 56.2 | 67.0 | >250 | >250 | >250 | 31.5 | 69.9 | 34.4 |
| HT-29 | 25.0 | 39.1 | 33.0 | 41.8 | >250 | >250 | >250 | 53.3 | 32.3 | 39.0 |
| K-562 | 25.0 | >250 | >250 | 128.1 | >250 | >250 | >250 | 39.2 | N.T. | N.T. |
| HaCat | 0.67 | 66.9 | 54.4 | 32.0 | >250 | >250 | >250 | >250 | 160.7 | >250 |
3. Experimental
3.1. General
3.2. General Procedure for Prins Cyclizations
3.3. General Procedure for the Deprotections of 3a–d
3.4. Antiproliferative Assays
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. [Google Scholar] [CrossRef]
- Wang, B.; Deng, J.; Gao, Y.; Zhu, L.; He, R.; Xuet, Y. The screening toolbox of bioactive substances from natural products: A review. Fitoterapia 2011, 82, 1141–1151. [Google Scholar] [CrossRef]
- Miller, J.S. The Discovery of Medicines from Plants: A Current Biological Perspective. Econ. Bot. 2011, 65, 396–407. [Google Scholar] [CrossRef]
- Kingston, D.G.I. Modern Natural Products Drug Discovery and Its Relevance to Biodiversity Conservation. J. Nat. Prod. 2011, 74, 496–511. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Carter, G.T. Natural products and Pharma 2011: Strategic changes spur new opportunities. Nat. Prod. Rep. 2011, 28, 1783–1789. [Google Scholar] [CrossRef]
- Wilson, R.M.; Danishefsky, S.J. Applications of total synthesis toward the discovery of clinically useful anticancer agents. Chem. Soc. Rev. 2007, 36, 1207–1226. [Google Scholar] [CrossRef]
- Kumar, K.; Waldmann, H. Synthesis of Natural Product Inspired Compound Collections. Angew. Chem. Int. Ed. 2009, 48, 3224–3242. [Google Scholar] [CrossRef]
- Goel, A.; Ram, V.J. Natural and synthetic 2H-pyran-2-ones and their versatility in organic synthesis. Tetrahedron 2009, 65, 7865–7913. [Google Scholar] [CrossRef]
- Ferreira, V.F.; Ferreira, S.B.; Silva, F.C. Strategies for the synthesis of bioactive pyran naphthoquinones. Org. Biomol. Chem. 2010, 8, 4793–4802. [Google Scholar] [CrossRef]
- Wright, A.E.; Botelho, J.C.; Guzmán, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a macrolide from a lithistid sponge of the family neopeltidae. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef]
- Searle, P.A.; Molinski, T.F. Phorboxazoles A and B: Potent cytostatic macrolides from marine sponge Phorbas species. J. Am. Chem. Soc. 1995, 117, 8126–8131. [Google Scholar] [CrossRef]
- Boonsri, S.; Karalai, C.; Ponglimanont, C.; Chantrapromma, S.; Kanjana-opas, A. Cytotoxic and antibacterial sesquiterpenes from Thespesia populnea. J. Nat. Prod. 2008, 71, 1173–1177. [Google Scholar] [CrossRef]
- Silva, L.F., Jr.; Quintiliano, S.A.P. An expeditious synthesis of hexahydrobenzo f isochromenes and of hexahydrobenzo f isoquinoline via iodine-catalyzed Prins and aza-Prins cyclization. Tetrahedron Lett. 2009, 50, 2256–2260. [Google Scholar]
- Ferraz, H.M.C.; Silva, L.F., Jr. Construction of functionalized indans by thallium(III) promoted ring contraction of 3-alkenols. Tetrahedron 2001, 57, 9939–9949. [Google Scholar] [CrossRef]
- Schow, S.R.; Bloom, J.D.; Thompson, A.S.; Winzenberg, K.N.; Smith, A.B., III. Milbemycin avermectin studies. 5. Total Synthesis of Milbemmycin BETA-3 and its C(12) Epimer. J. Am. Chem. Soc. 1986, 108, 2662–2674. [Google Scholar]
- Ferraz, H.M.C.; Aguilar, A.M.; Silva, L.F., Jr. A diastereoselective total synthesis of the sesquiterpene (±)-mutisianthol. Tetrahedron 2003, 59, 5817–5821. [Google Scholar] [CrossRef]
- Fouche, G.; Cragg, G.M.; Pillay, P.; Kolesnikova, N.; Maharaj, V.J.; Senabe, J. In vitro anticancer screening of South African plants. J. Ethnopharmacol. 2008, 119, 455–461. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reddy, K.R.K.K.; Longato, G.B.; Carvalho, J.E.d.; Ruiz, A.L.T.G.; Silva, L.F., Jr. Populene D Analogues: Design, Concise Synthesis and Antiproliferative Activity. Molecules 2012, 17, 9621-9630. https://doi.org/10.3390/molecules17089621
Reddy KRKK, Longato GB, Carvalho JEd, Ruiz ALTG, Silva LF Jr. Populene D Analogues: Design, Concise Synthesis and Antiproliferative Activity. Molecules. 2012; 17(8):9621-9630. https://doi.org/10.3390/molecules17089621
Chicago/Turabian StyleReddy, Kachi R. Kishore Kumar, Giovanna B. Longato, João E. de Carvalho, Ana L. T. G. Ruiz, and Luiz F. Silva, Jr. 2012. "Populene D Analogues: Design, Concise Synthesis and Antiproliferative Activity" Molecules 17, no. 8: 9621-9630. https://doi.org/10.3390/molecules17089621