Synthesis of Highly Substituted Oxazoles through Iodine(III)-Mediated Reactions of Ketones with Nitriles

Abstract
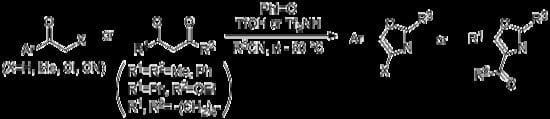
:
1. Introduction
2. Results and Discussion
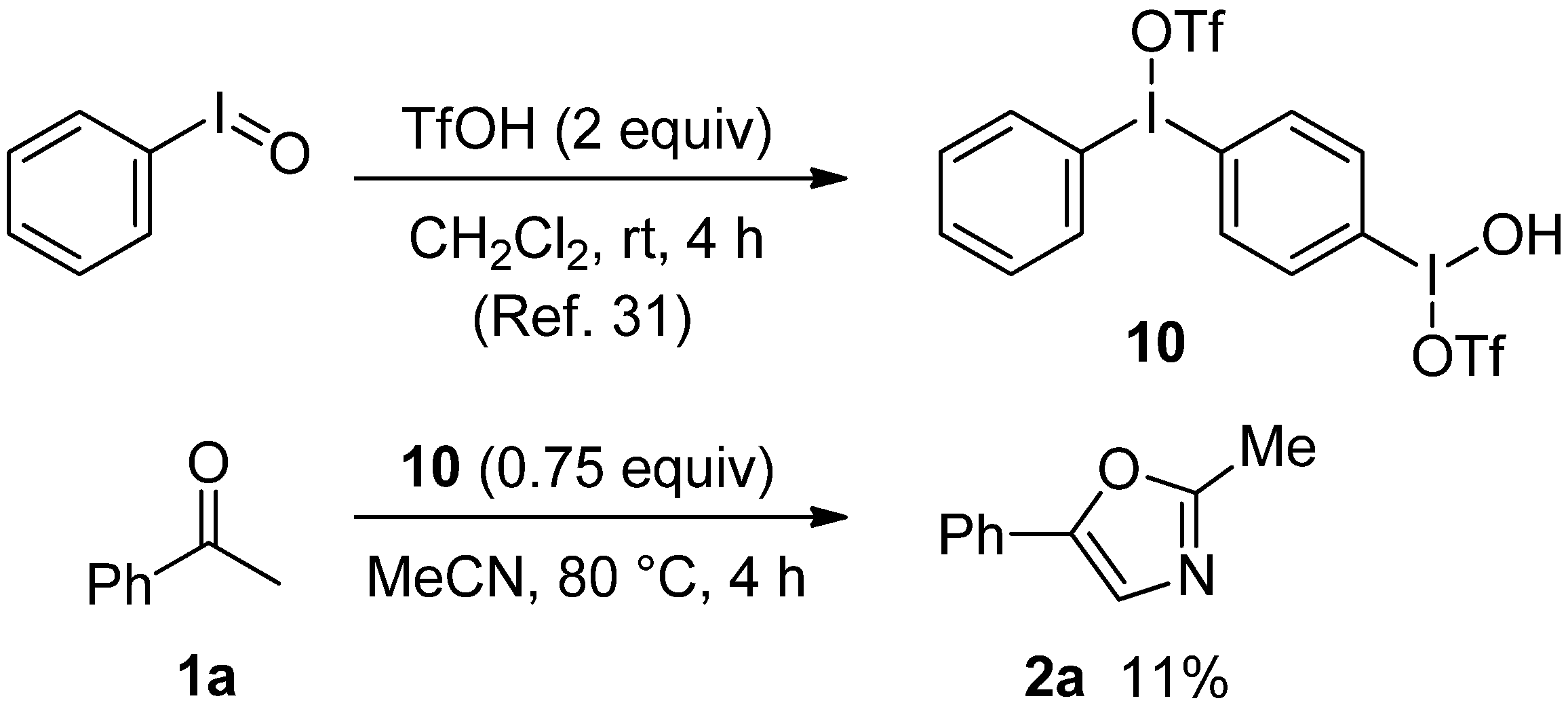
2.1. Evaluation of Oxidants for the Direct Synthesis of Oxazole

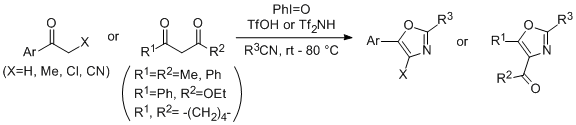{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Oxidant (equiv) | Acid (equiv.) | Temp. (°C) | Time (h) | 2a (%) [a] | |
|---|---|---|---|---|---|---|
| 1 [b] | PIDA (1.2) | TfOH (4.5) | 80 | 2 | 94 | |
| 2 | PIDA (1.2) | TfOH (4.5) | rt | 3 | 22 | |
| 3 | PhI=O (1.5) | TsOH (1.5) | 80 | 18 | - | ( 3 69) |
| 4 | PhI=O (1.5) | HBF4/Et2O (1.5) | 80 | 18 | 54 | |
| 5 | PhI=O (1.5) | TfOH (1.5) | 80 | 18 | 69 | |
| 6 | PhI=O (1.5) | Tf2O (1.5) | 80 | 18 | 21 [c] | |
| 7 | PhI=O (1.5) | TfOH (3.0) | 80 | 3 | 94 | |
| 8 | PhI=O (1.5) | Tf2NH (1.5) | 80 | 3 | 40 | ( 1a 16) |
| 9 | PhI=O (1.5) | Tf2NH (3.0) | rt | 3 | 86 |  |
| Entry | Oxidant | Acid | Time (h) | 5a (%) [a] | |
|---|---|---|---|---|---|
| 1 | PIDA | TfOH [b] | 24 | 4 | |
| 2 | PhI=O | TfOH | 25 | 7 | |
| 3 | PhI=O | TfOH | 115 | 41 | |
| 4 | PhI=O | Tf2NH | 16 | 51 | |
| 5 | PhI=O | Tf2NH | 72 | 79 | |
| 6 | PhI=O | Tf2NH [c] | 3 | 81  |
2.2. Scope of the Direct Synthesis of Oxazoles Using PhI=O with TfOH or Tf2NH
| Entry | 1 or 4 | R1 | R2 | Procedure | (°C) | (h) | 2 or 5 | Yield (%) [b] |
|---|---|---|---|---|---|---|---|---|
| 1 | 1a | Ph | H | A | 80 | 3 | 2a | 88 |
| 2 | 1a | B | rt | 3 | 2a | 86 | ||
| 3 | 1b | m-Me-C6H4 | H | A | 80 | 3 | 2b | 75 |
| 4 | 1c | p-Cl-C6H4 | H | A | 80 | 3 | 2c | 86 |
| 5 | 1d | p-NO2-C6H4 | H | A | 80 | 3 | 2d | 73 |
| 6 | 1e | Ph | Me | A | 80 | 3 | 2e | 94 |
| 7 | 1e | B | rt | 2 | 2e | 91 | ||
| 8 | 1f | Ph | Cl | A | 80 | 49 | 2f | 68 |
| 9 | 1g | Ph | CN | A | 80 | 20 | 2g | 53 |
| 10 | 1g | B | 80 | 20 | 2g | 38 | ||
| 11 | 1g | C | rt | 24 | 2g | 69 | ||
| 12 | 4a | Ph | OEt | B | 80 | 72 | 5a | 78 |
| 13 | 4a | C | 80 | 3 | 5a | 81 | ||
| 14 | 4b | Ph | Ph | B | 80 | 139 | 5b | 89 |
| 15 | 4b | C | 80 | 3 | 5b | 83 | ||
| 16 | 4c | Me | Me | B | 80 | 120 | 5c | 35 |
| 17 | 4c | C | 80 | 3 | 5c | 67 | ||
| 18 | 4d | -(CH2)4- | C | 80 | 167 | 5d | 40  | |

| Entry | 4 | R1 | R2 | R3 | (h) | 2 or 5 | Yield (%) [a] |
|---|---|---|---|---|---|---|---|
| 1 | 4a | Ph | OEt | Et | 2 | 8a | 83 |
| 2 | 4a | Ph | 3 | 9a | 72 | ||
| 3 | 4b | Ph | Ph | Et | 3 | 8b | 89 |
| 4 | 4b | Ph | 3 | 9b | 67 | ||
| 5 | 4c | Me | Me | Et | 20 | 8c | 56 |
| 6 | 4c | Ph | 20 | 9c | 60  |
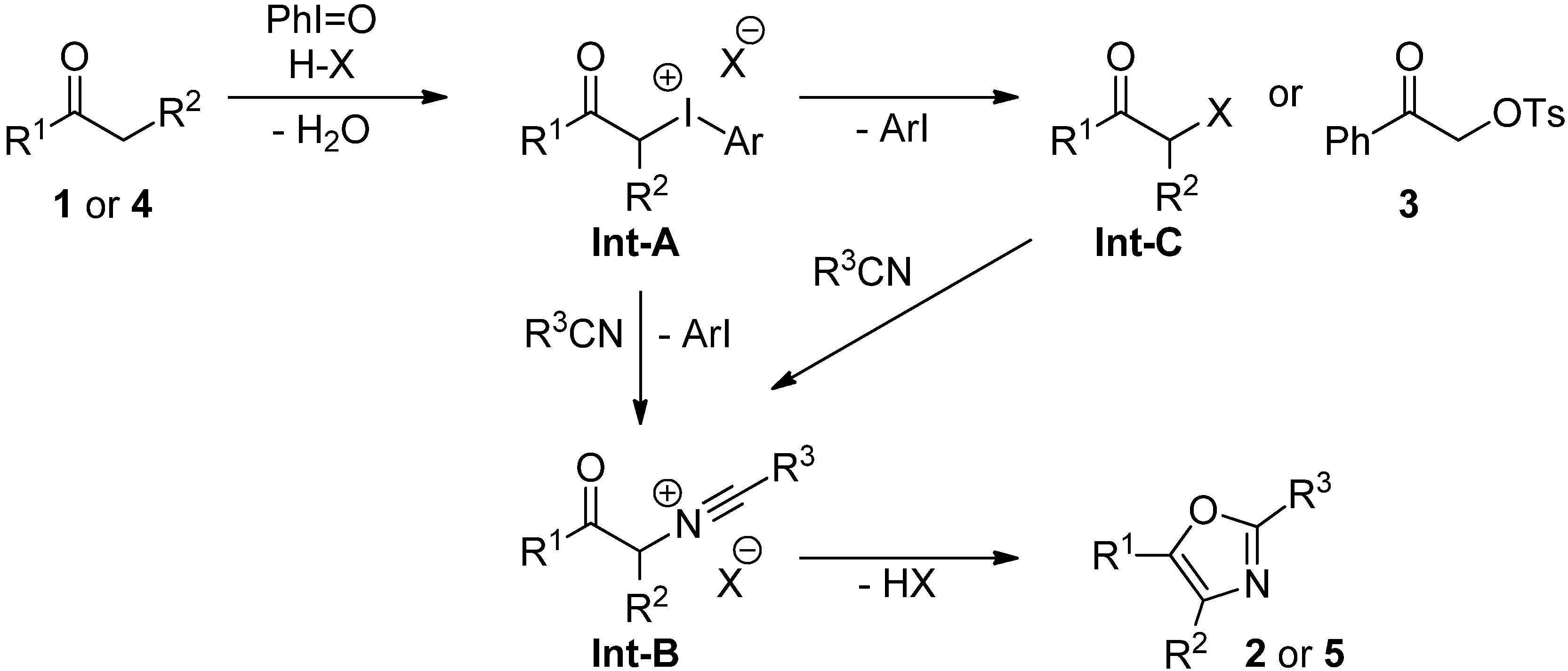
2.3. Mechanistic Considerations


3. Experimental
3.1. General
3.2. General Procedure for the Iodine(III)-Mediated Synthesis of Oxazoles
3.3. Formation of α-Tosyloxy Ketone 3 under the Iodine(III)-Mediated Conditions
4. Conclusions
Acknowledgments
References
- Wipf, P. Synthetic Studies of Biologically Active Marine Cyclopeptides. Chem. Rev. 1995, 95, 2115–2134. [Google Scholar] [CrossRef]
- Palmer, D.C.; Venkatraman, S.; Lowe, D.; Levin, J.L.; Laakso, L.M.; Gribble, G.W. The Chemistry of Heterocyclic Compounds, Oxazoles: Synthesis, Reactions, and Spectroscopy, Part A; Palmer, D.C., Ed.; Wiley & Sons: Hoboken, NJ, USA, 2003; Volume 60. [Google Scholar]
- Kunieda, T.; Matsunaga, H.; Palmer, D.C.; Cativiela, C.; Diaz-de-Villegas, M.D.; Wong, G.S.K.; Wu, W.; Ghosh, A.; Bilcer, G.; Fidanze, S. The Chemistry of Heterocyclic Compounds, Oxazoles: Synthesis, Reactions, and Spectroscopy, Part B; Palmer, D.C., Ed.; Wiley & Sons: Hoboken, NJ, USA, 2004; Volume 60. [Google Scholar]
- Robinson, R. A new synthesis of oxazole derivatives. J. Chem. Soc. 1909, 95, 2167–2174. [Google Scholar] [CrossRef]
- Gabriel, S. Synthese von Oxazolen und Thiazolen II. Ber. Dtsch. Chem. Soc. 1910, 43, 1283–1287. [Google Scholar] [CrossRef]
- Thalhammer, A.; Mecinović, J.; Schofield, C.J. Triflic anhydride-mediated synthesis of oxazoles. Tetrahedron Lett. 2009, 50, 1045–1047 and references cited therein. [Google Scholar]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem. Int. Ed. 1963, 2, 565–598. [Google Scholar]
- Ibata, T.; Fukushima, K. Formation and reaction of acyl-substituted nitrile ylide through the rhodium complex Rh2(OAc)4-catalyzed reaction of α-diazocarbonyl compounds with benzonitrile. Chem. Lett. 1992, 2197–2200. [Google Scholar]
- Austeri, M.; Rix, D.; Zeghida, W.; Lacour, J. CpRu-Catalyzed O-H Insertion and Condensation Reactions of α-Diazocarbonyl Compounds. Org. Lett. 2011, 13, 1394–1397. [Google Scholar]
- Lora-Tamayo, M.; Madroñero, R.; Leipprand, H. Die Anwendung Der Nitriliumsalze bei der Synthese heterocyclischer Verbindungen, V. Derivate des 4.5-Diphenyl-oxazols. Chem. Ber. 1964, 97, 2230–2233. [Google Scholar]
- Schmittel, M.; Roeck, M. Enol cation radicals in solution. 3. Reaction of enol cation radicals in the presence of nucleophiles. Chem. Ber. 1992, 125, 1611–1620. [Google Scholar] [CrossRef]
- Lai, P.-S.; Taylor, M.S. Preparation of substituted oxazoles by Ritter reactions of α-oxo tosylates. Synthesis 2010, 1449–1452. [Google Scholar]
- Gogonas, E.P.; Hadjiarapoglou, L.P. [3+2]-Cycloaddition reactions of 2-phenyliodonio-5,5-dimethyl-1,3-dioxacyclohexanemethylide. Tetrahedron Lett. 2000, 41, 9299–9303. [Google Scholar] [CrossRef]
- Lee, J.C.; Hong, T. A novel and direct synthesis of 2-alkyl-5-aryl disubstituted oxazoles. Tetrahedron Lett. 1997, 38, 8959–8960. [Google Scholar] [CrossRef]
- Lee, J.C.; Song, I.-G. Mercury(II) p-toluenesulfonate mediated synthesis of oxazoles under microwave irradiation. Tetrahedron Lett. 2000, 41, 5891–5894. [Google Scholar]
- Kotani, E.; Kobayashi, S.; Adachi, M.; Tsujioka, T.; Nakamura, K.; Tobinaga, S. Synthesis of oxazole by the reaction of ketones with Iron(III) solvates of nitriles. Chem. Pharm. Bull. 1989, 37, 606–609. [Google Scholar]
- Nagayoshi, K.; Sato, T. One-step Synthesis of oxazole from ketones and nitriles using copper(II) trifluoromethanesulfonate as a key reagent. Chem. Lett. 1983, 1355–1356. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Stang, P.J. Chemistry of polyvalent iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef]
- Koser, G.F. Hydroxy(tosyloxy)iodo]benzene and closely related iodanes: The second stage of development. Aldrich. Acta 2001, 34, 89–102. [Google Scholar]
- Moroda, A.; Togo, H. Biphenyl- and terphenyl-based recyclable organic trivalent iodine reagents. Tetrahedron 2006, 62, 12408–12414. [Google Scholar] [CrossRef]
- Lee, J.C.; Choi, H.J.; Lee, Y.C. Efficient synthesis of multi-substituted oxazoles under solvent-free microwave irradiation. Tetrahedron Lett. 2003, 44, 123–125. [Google Scholar] [CrossRef]
- Varma, R.S.; Kumar, D. A facile one-pot synthesis of 2,5-disubstituted oxazoles using iodobenzene diacetate. J. Heterocyclic Chem. 1998, 35, 1533–1534. [Google Scholar] [CrossRef]
- Ishiwata, Y.; Togo, H. Iodoarene-mediated one-pot preparation of 2,5-disubstituted and 2,4,5-trisubstituted oxazoles from alkyl aryl ketones with oxone in nitriles. Tetrahedron 2009, 65, 10720–10724. [Google Scholar] [CrossRef]
- Lee, J.E.; Koh, H.Y.; Seo, S.H.; Baek, Y.Y.; Rhim, H.; Cho, Y.S.; Choo, H.; Pae, A.N. Synthesis and biological evaluation of oxazole derivatives as T-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2010, 20, 4219–4222. [Google Scholar] [CrossRef]
- Very recently, an elegant method using tetra-n-butylammonium iodide and tert-butyl hydroperoxide has been reported for the single-step synthesis of oxazole from dicarbonyl compounds and benzylamine derivatives. See, Xie, J.; Jiang, H.; Cheng, Y.; Zhu, C. Metal-free, organocatalytic cascade formation of C-N and C-O bonds through dual sp3 C-H activation: oxidative synthesis of oxazole derivatives. Chem. Commun. 2012, 48, 979–981. [Google Scholar] [CrossRef]
- Saito, A.; Matsumoto, A.; Hanzawa, Y. PIDA-mediated synthesis of oxazoles through oxidative cycloisomerization of propargylamides. Tetrahedron Lett. 2010, 51, 2247–2250. [Google Scholar]
- Saito, A.; Anzai, T.; Matsumoto, A.; Hanzawa, Y. PIFA-mediated oxidative cycloisomerization of 2-propargyl-1,3-dicarbonyl compounds: divergent synthesis of furfuryl alcohols and furfurals. Tetrahedron Lett. 2011, 52, 4658–4661. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Tykwinski, R.; Caple, R.; Berglund, B.; Koz'min, A.S.; Zefirov, N.S. Reaction of PhIO·HBF4/silyl enol ether adduct with olefins as general approach to carbon-carbon bond formation in AdE reactions using hypervalent iodine reagents. Tetrahedron Lett. 1988, 29, 3703–3704. [Google Scholar]
- Wenderski, T.A.; Hoarau, C.; Mejorado, L.; Pettus, T.R.R. Dearomatization applications of I(III) reagents and some unusual reactivity amongst resorcinol derived cyclohexadienones. Tetrahedron Lett. 2010, 66, 5873–5883. [Google Scholar]
- Kitamura, T.; Nagata, K.; Nakamura, T.; Furuki, R. Self-condensation of iodosylbenzene. Formation of a (p-phenylene) type of bisiodine(III) reagents. Tetrahedron 1995, 51, 6229–6236. [Google Scholar]
- Kuwano, Y.; Togo, H. Iodoarene-catalyzed one-pot preparation of 2,4,5-trisubstituted oxazoles from alkyl aryl ketones with mCPBA in nitriles. Tetrahedron 2006, 62, 6251–6256. [Google Scholar]
- Schuh (nee Müller), K.; Glorius, F. A domino copper-catalyzed C-N-/C-O-coupling process for the synthesis of oxazoles. Synthesis 2007, 2297–2306. [Google Scholar]
- Wan, C.; Zhang, J.; Wang, S.; Fan, J.; Wang, Z. Facile Synthesis of polysubstituted oxazoles via acopper-catalyzed tandem oxidative cyclization. Org. Lett. 2010, 12, 2338–2341. [Google Scholar]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Saito, A.; Hyodo, N.; Hanzawa, Y. Synthesis of Highly Substituted Oxazoles through Iodine(III)-Mediated Reactions of Ketones with Nitriles. Molecules 2012, 17, 11046-11055. https://doi.org/10.3390/molecules170911046
Saito A, Hyodo N, Hanzawa Y. Synthesis of Highly Substituted Oxazoles through Iodine(III)-Mediated Reactions of Ketones with Nitriles. Molecules. 2012; 17(9):11046-11055. https://doi.org/10.3390/molecules170911046
Chicago/Turabian StyleSaito, Akio, Nao Hyodo, and Yuji Hanzawa. 2012. "Synthesis of Highly Substituted Oxazoles through Iodine(III)-Mediated Reactions of Ketones with Nitriles" Molecules 17, no. 9: 11046-11055. https://doi.org/10.3390/molecules170911046