2.2.2. Preparation of the Carotenoid Standards
The solvents used to dissolve the carotenoid standards were chosen considering either the previously reported carotenoid solubility or the availability of the absorption coefficient of each pigment.
Stock carotenoid solutions were prepared in EtOH, acetone and hexane [
31]. Carotenoid concentrations were determined spectrophotometrically.
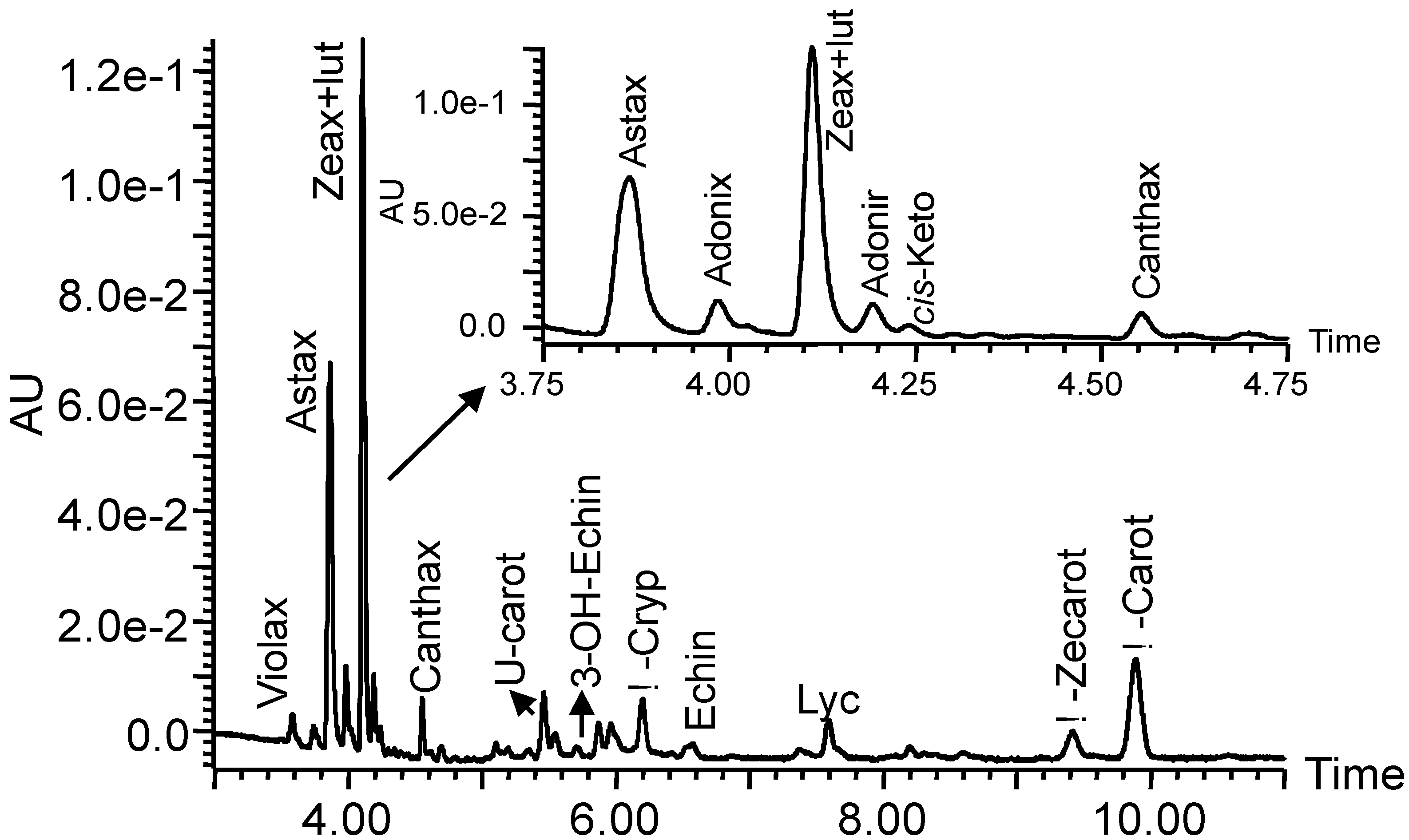
Table 4 shows the solvent and the value of A
![Molecules 17 11255 i004]()
used to quantify each pigment. Working solutions were prepared from stock solutions by sampling an aliquot and diluting it with injection solvent. Solution concentrations were assessed by UHPLC analysis. For those carotenoids dissolved in hexane (canthaxanthin,
β-cryptoxanthin,
β-carotene, lycopene and phytoene), working solutions were prepared from stock solutions by evaporating an aliquot under nitrogen and diluting it with injection solvent. Calibration curves were obtained from peak area by injecting mixtures of standards.
Table 5 shows the linear regression data for each carotenoid standard curve.
Table 4.
Concentrations of carotenoid stock solutions used to build calibration curves.
Table 4.
Concentrations of carotenoid stock solutions used to build calibration curves.
| Carotenoid | Solvent | ![Molecules 17 11255 i005]() | Stock carotenoid concentration (μg/mL) |
|---|
| cis-Neoxanthin | EtOH | 2,380 | 19.64 a |
| Violaxanthin | EtOH | 2,550 | 16.26 a |
| Antheraxanthin | EtOH | 2,350 | 17.53 a |
| Astaxanthin | EtOH | 2,100 | 1.55 a and 4.96 b |
| Astaxanthin | Injection solvent | - | 5.12 b |
| Zeaxanthin | Acetone | 2,340 | 32.31 a |
| Lutein | EtOH | 2,550 | 21.57 a |
| Canthaxanthin | Hexane | 2,200 | 0.27 a and 4.53 b |
| Canthaxanthin | Injection solvent | - | 5.70 b |
| β-Cryptoxanthin | Hexane | 2,400 | 35.00 a |
| β-Carotene | Hexane | 2,590 | 24.85 a |
| Lycopene | Hexane | 3,450 | 8.26 a |
| cis-Phytoene | Hexane | 915 | 16.16 a |
Table 5.
Linear regression data obtained for several carotenoid standard curves under UHPLC conditions.
Table 5.
Linear regression data obtained for several carotenoid standard curves under UHPLC conditions.
| Carotenoid | Linear range (μg/mL) | Slope | Intercept | R 2 |
|---|
| cis-Neoxanthin | 0.04−19.64 | 2,481 ± 7.57 | −136.62 ± 16.28 | 0.9999 |
| Violaxanthin | 0.03−16.26 | 2,516 ± 5.59 | −121.93 ± 12.97 | 0.9994 |
| Antheraxanthin | 0.03−15.53 | 2,509 ± 22.13 | −413.83 ± 4.13 | 0.9970 |
| Astaxanthin | 0.04−5.12 | 1,825 ± 6.43 | −50.37 ± 10.51 | 0.9999 |
| Astaxanthin a | − | 14,149 | 4414.1 | 0.7741 |
| Lutein | 0.02−17.25 | 2,475 ± 81.74 | −626.20 ± 35.78 | 0.9952 |
| Zeaxanthin | 0.03−17.23 | 2,578 ± 38.04 | −86.96 ± 25.8 | 0.9996 |
| Canthaxanthin | 0.02−5.70 | 1,787 ± 4.24 | −43.96 ± 16.13 | 0.9995 |
| Canthaxanthin b | − | 9,613 | 1583.1 | 0.9320 |
| β-Cryptoxanthin | 0.04−18.67 | 2,379 ± 0.35 | −444.17 ± 31.46 | 0.9988 |
| Lycopene | 0.3−3.11 | 1,398 ± 104.40 | −121.72 ± 24.88 | 0.9998 |
| β-Carotene | 0.1−24.85 | 1,484 ± 27.22 | −189.02 ± 29.80 | 0.9998 |
| cis-Phytoene | 0.08−16.16 | 1,990 ± 285.46 | −259.82 ± 37.32 | 0.9989 |
Solubilization problems were encountered for carotenes (when lycopene and
β-carotene were dissolved in hexane) and for ketocarotenoids (when astaxanthin and canthaxanthin were dissolved in EtOH and hexane respectively). Chloroform, dichloromethane, hexane, ethyl acetate and THF [
6,
24,
32,
33] are known to dissolve lycopene and
β-carotene. Thus, we chose hexane to solubilize these compounds. Initially, we attempted to prepare 100 μg/mL stock solutions of lycopene and
β-carotene in hexane, but a precipitate was observed in the bottom of the vessels. Consequently, to ensure that carotenes were completely dissolved, stock solutions of carotenes were prepared again in hexane but in lower concentrations (
Table 4). Thus, stock solutions of 24.85 μg/mL for
β-carotene and 8.26 μg/mL for lycopene were prepared. We did not encounter solubilization problems with these concentrations. In addition, the calibration curves of these pigments (
Table 5) indicated that the chromatographic peak areas of carotenes gave a linear plot throughout the concentration range studied.
Similarly, canthaxanthin and astaxanthin were not properly dissolved in hexane and EtOH respectively. Because of the problems of solubilization observed with these compounds, we determined and compared their concentrations using two different methods: dividing the mass of the carotenoid by the total volume of solution (theoretical concentration) and spectrophotometrically (experimental concentration).
The theoretical and experimental concentration obtained for canthaxanthin was 4.53 and 0.27 μg/mL, respectively whereas for astaxanthin it was 4.96 and 1.55 μg/mL, respectively. The lower concentrations of ketocarotenoids obtained experimentally indicated that hexane and EtOH were not appropriate solvents for canthaxanthin and astaxanthin, respectively. In addition,
Table 5 shows that the calibration curves of these two pigments were characterized by a poor r-squared (R
2 < 0.94). Therefore, we prepared stock ketocarotenoid solutions in the injection solvent of 5.12 μg/mL for astaxanthin and 5.70 μg/mL for canthaxanthin (
Table 4).
Table 5 shows that the calibration curves of these pigments dissolved in the injection solvent gave a linear plot throughout the concentration range studied. The lack of information about carotenoid absorption coefficients in a variety of organic solvents hampers the use of more appropriate solvents for ketocarotenoids.
We did not encounter any solubilization problems with the concentration range used for: (a) violaxanthin, antheraxanthin, neoxanthin and lutein, dissolved in EtOH; or (b) zeaxathin, dissolved in acetone and (c)
β-cryptoxanthin, dissolved in hexane. As reported previously [
26,
27,
34,
35], oxygen-functionalized carotenoids showed satisfactory solubility in MeOH, EtOH and acetone.
Given the concentrations of carotenoids expected in maize samples, we did not prepare standard concentrations above 40 μg/mL. However, in our experience, higher concentrations of oxygen-functionalized carotenoids can be prepared with the injection solvent used here when needed. For example, concentrations of 100 μg/mL can be prepared for violaxanthin and neoxanthin. If higher carotene concentrations were required, changes in the injection solvent should be made to increase their solubility. For example, when lycopene is the target analyte, the following injection solvents have been used: chloroform 100%, to analyze extracts of tomato fruit pericarps [
36]; ethyl acetate 100% [
37] and
n-butanol–ACN–dichloromethane (3:7:0.1, v/v/v) [
38] to analyze extracts of tomato fruit.
Konings
et al. [
35] prepared stock solutions of lutein, zeaxanthin,
β-carotene and lycopene with the same solvents used in this study. However, they used a mixture of MeOH–THF (7.5:2.5, v/v) as injection solvent. Under the chromatographic conditions applied, they observed a higher linear range for lutein, zeaxanthin and
β-carotene than for lycopene. The smaller linearity range of lycopene (from 0 to 3.5 μg/mL) was explained by the lower solubility of this compound in the injection solvent. Nevertheless, the choice of the injection solvent was a compromise between satisfactory solubility of carotenoids, compatibility with the mobile phase, and the absence of peak distortions.
When carotenoid standard solutions are used several times and stored under N2 or Ar, their concentrations should be evaluated since the inert gas introduced various times into the vial evaporates the solvent, thereby changing the original carotenoid concentration. Thus, it is advisable to either divide the volume of carotenoid standard solutions into vials, putting only the volume required for each analysis into single vials, or to dry the standard solutions and redissolve these in each analysis. In addition, attention should be paid when many carotenoid standards at high concentrations are solubilized in the same solvent as some might precipitate. Thus, it is preferable to prepare various mixtures of carotenoids to ensure the complete solubilization of all analytes.



 = absorption coefficient, which is defined as the theoretical absorbance of a solution of 1% (w/v) concentration (i.e., g in 100 mL) in a cuvette with a path length of 1 cm. Lutein and zeaxanthin are the major carotenoids in maize. Therefore, an average value for A
= absorption coefficient, which is defined as the theoretical absorbance of a solution of 1% (w/v) concentration (i.e., g in 100 mL) in a cuvette with a path length of 1 cm. Lutein and zeaxanthin are the major carotenoids in maize. Therefore, an average value for A  of 2,332 was used [15]. C = total carotenoid content (μg/g) in a given sample on dry weight basis. Abs = absorbance measured at 450 nm. V = volume (mL). W = weight of sample (g). 104 = conversion factor to obtain the concentration in units of μg/g.
of 2,332 was used [15]. C = total carotenoid content (μg/g) in a given sample on dry weight basis. Abs = absorbance measured at 450 nm. V = volume (mL). W = weight of sample (g). 104 = conversion factor to obtain the concentration in units of μg/g.
{kind=link}
{kind=link}
{kind=link}

 used to quantify each pigment. Working solutions were prepared from stock solutions by sampling an aliquot and diluting it with injection solvent. Solution concentrations were assessed by UHPLC analysis. For those carotenoids dissolved in hexane (canthaxanthin, β-cryptoxanthin, β-carotene, lycopene and phytoene), working solutions were prepared from stock solutions by evaporating an aliquot under nitrogen and diluting it with injection solvent. Calibration curves were obtained from peak area by injecting mixtures of standards. Table 5 shows the linear regression data for each carotenoid standard curve.
used to quantify each pigment. Working solutions were prepared from stock solutions by sampling an aliquot and diluting it with injection solvent. Solution concentrations were assessed by UHPLC analysis. For those carotenoids dissolved in hexane (canthaxanthin, β-cryptoxanthin, β-carotene, lycopene and phytoene), working solutions were prepared from stock solutions by evaporating an aliquot under nitrogen and diluting it with injection solvent. Calibration curves were obtained from peak area by injecting mixtures of standards. Table 5 shows the linear regression data for each carotenoid standard curve.