Synthesis of New Optically Active 2-Pyrrolidinones

Laboratory of Organic Chemistry, Department of Chemistry, University of Athens, Panepistimiopolis, Athens 15771, Greece
*
Author to whom correspondence should be addressed.
Molecules 2013, 18(1), 50-73; https://doi.org/10.3390/molecules18010050
Submission received: 6 November 2012
/
Revised: 5 December 2012
/
Accepted: 12 December 2012
/
Published: 21 December 2012
(This article belongs to the Section Organic Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:A new class of optically active 2-pyrrolidinones was synthesized, starting from S-pyroglutamic acid, a well known natural chiral synthon. The synthetic design followed led to the insertion of various substituents at positions 1 and 5 of the 2-pyrrolidinone ring, including the imidazole moiety. Some of them possess two or three stereogenic centers, the configuration of which was retained under the mild conditions used. The new compounds also carry an imidazole moiety, which, along with the 2-pyrrolidinone template, may prove pivotal to several biological processes.
1. Introduction
Derivatives of 2-pyrrolidinone have shown significant biological and pharmacological activities. Some of them are well known medicines, e.g., 2-oxo-1-pyrrolidine acetamide (piracetam) for patients with seizures, Alzheimer’s and senile dementia, concussion and other neurological problems [1], 1-ethyl-4-(2-morpholin-4-ylethyl)-3,3-diphenyl-pyrrolidin-2-one (doxapram) for patients with respiratory failure [2], etc. Moreover, the 2-pyrrolidinone template, considered as an essential pharmacophore group, is also aggregation-inhibiting effects [4], or properties related to the treatment of a variety of diseases incorporated in more complicated chemical structures with anticonvulsant activity [3], pharmacological properties like modulated by H3 receptors, including those associated with the central nervous system [5] and many others.
In recent years, we have designed and synthesized 2-pyrrolidinones, starting from the naturally derived S-pyroglutamic acid (2-oxotetrahydropyrrol-5S-carboxylic acid), which is considered as a unique chiral synthon. The asymmetric use of S-pyroglutamic acid is based on its two differentiated carbonyls, the properties of which allow an extended derivatization on the 5-membered ring of the starting compound leading to a plethora of natural products, e.g., (−) domoic acid [6], the neurotoxin anatoxin-a [7], biologically interesting compounds, e.g., an inhibitor of angiotensin-converting enzyme (ACE) [8] for the treatment of hypertension as well as to chemical auxiliaries in asymmetric synthesis [9]. The properties and applications of pyroglutamic acid as a versatile building block in asymmetric synthesis has extensively been reviewed in the literature [10,11,12]. As it was mentioned above, in recent years, we have synthesized optically active pyrrolidinones based on the S-pyroglutamic acid, in which an imidazole ring has also been inserted. Some of them exhibited antihypertensive and anti-inflammatory activity [13,14,15,16]. In this paper we present the synthesis of seven new compounds (Figure 1), containing up to three stereogenic centers, with predetermined absolute configuration derived from the natural amino acid S-pyroglutamic acid, S-histidine and S-serine.
2. Results and Discussion
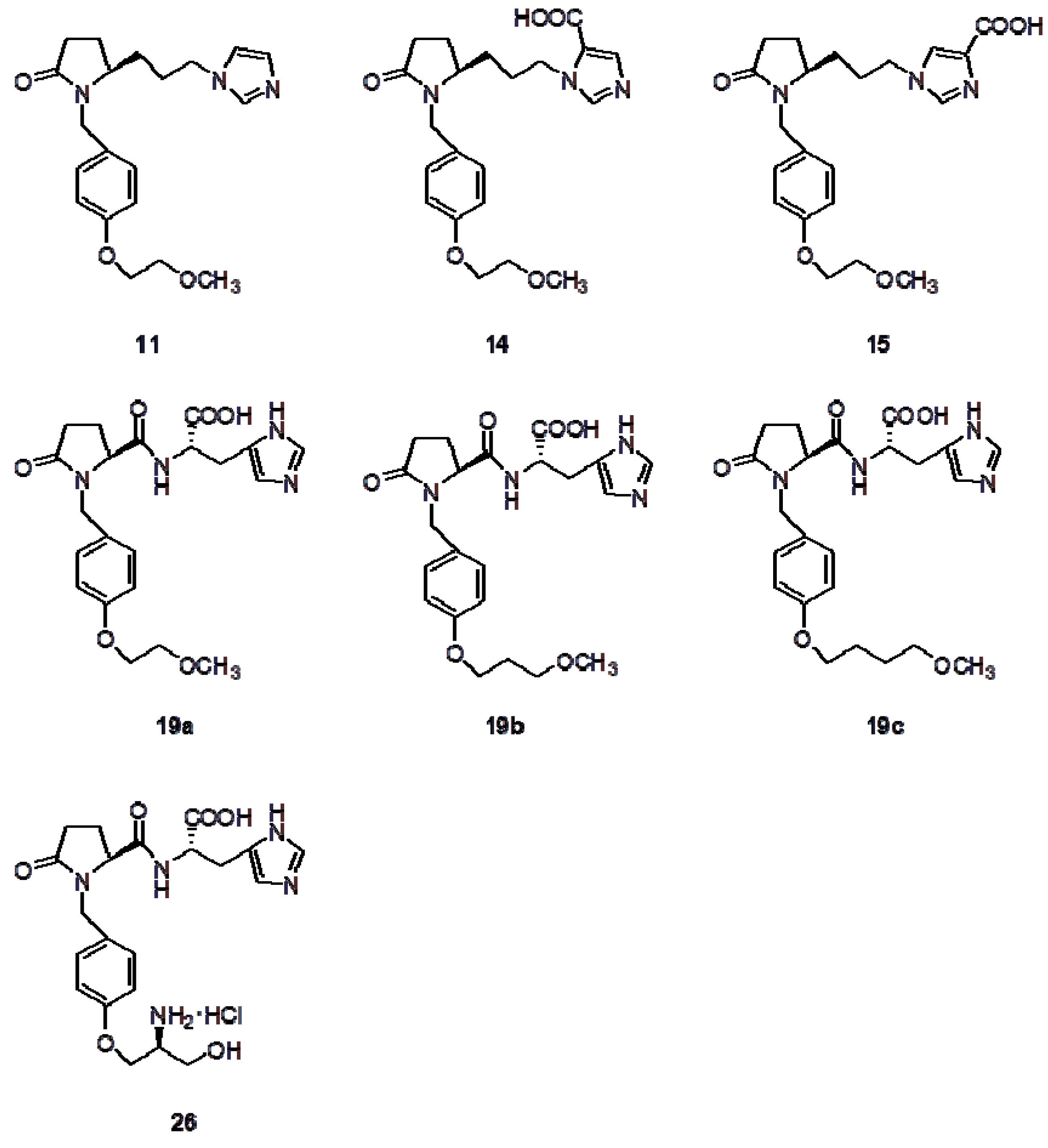
The 2-pyrrolidinones shown in Figure 1 possess an N-benzyl- type substituent, the 4-position of which is substituted by a methoxyalkyloxy group. In the case of compound 26 the alkoxy substituent is a chiral α-amino alcohol moiety.
The 5-position of the 2-pyrrolidinone template has retained the absolute configuration of the initial S-pyroglutamic acid, regardless of whether the carbon chain is elongated or not. At the other end of this chain, an imidazole ring (with or without substitution) has been inserted (compounds 11, 14, 15). In the case of compounds 19a–c and 26, the carboxyl group of the starting acid has been coupled to the amino acid S-histidine. Our approach for the synthesis of these 2-pyrrolidinones is depicted in Scheme 1, Scheme 2 and Scheme 3.
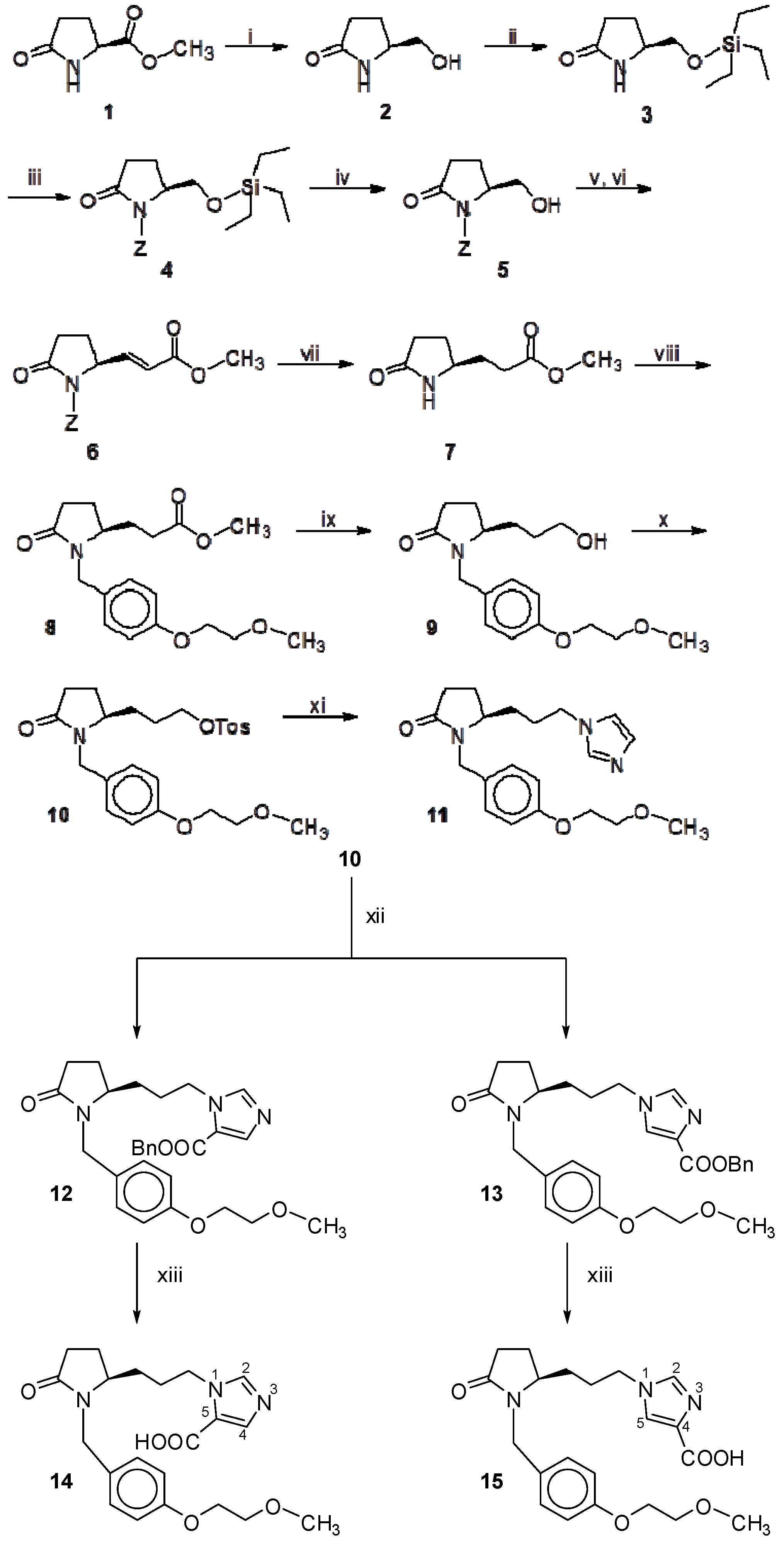
As shown in Scheme 1, the S-pyroglutaminol (2), derived from methyl S-pyroglutamate (1) using NaBH4 in absolute alcohol [17], was protected as the TES ether using TESCl, Et3N and DMAP [18] to yield compound 3. N-benzyloxy-carbonylation of 3 with benzyloxycarbonylchloride using NaH in dry THF [19], afforded compound 4. After deprotection of the TES group using TFA in CH2Cl2 [20], the resulting alcohol 5 was subjected to Moffatt oxidation [21] yielding the corresponding aldehyde, using DMSO, water soluble carbodiimide (EDC.HCl), pyridine and a trace of TFA. The aldehyde, without being isolated was immediately reacted with the stabilized ylide, phosphoranylidene methyl acetate [22], under the conditions of the Wittig reaction to give alkene 6. After catalytic hydrogenation, the N-unprotected, saturated methylester 7 was obtained, and its NH group was alkylated by benzylbromide 30 (Scheme 4) using NaH in dry DMF [19], to afford compound 8. The reduction of the ester group of 8 to the corresponding alcohol 9 was accomplished by LiBH4 in dry THF [23] and the alcohol 9 was converted to the activated tosyl ester 10. The desired products 11–13 were provided in higher yields (~60%) using imidazole or 1H-imidazole-4(5)-carboxylic acid phenylmethyl ester [16] together with Cs2CO3 in dry DMF at 50 °C. It is known that the nucleophilic strength of nitrogen is enhanced via complexes with Cs+, the so called “cesium effect” [24]. After catalytic hydrogenation the final products 14 and 15 were obtained. As shown in Scheme 1, compounds 14 and 15 as well as their precursors 12 and 13 are constitutional isomers in proportion 2:1, depending on which position (4 or 5) on the imidazole ring the substituent is located. In this case the separation of the two isomers was achieved by column chromatography, but we had to identify which isomer corresponds to each isolated compound. In a previous work on similar constitutional isomers [16], we had used 2D NOESY NMR spectroscopy, in order to distinguish whether the carboxyl group is attached on the 4- or 5- carbon of the imidazole ring. According to the 1H-NMR spectra of isomers 14 and 15 and taking into account the aforementioned studies, the H-4 of isomer 14 with the carboxylic group at the 5-position of the imidazole ring, resonates at lower field (ca. 7.80 ppm) in comparison to H-5 (ca. 7.68 ppm) of isomer 15 with the carboxylic group at the 4-position. In addition, the diastereotopic proton of the methylene group attached to nitrogen that points towards the carboxylate group in the isomer with substitution at 5-position resonates at lower field (ca. 4.71 ppm) in comparison to the corresponding diastereotopic proton that is not deshielded by the carboxylic group in the isomer with substitution at 4-position and resonates at ca. 4.60 ppm.
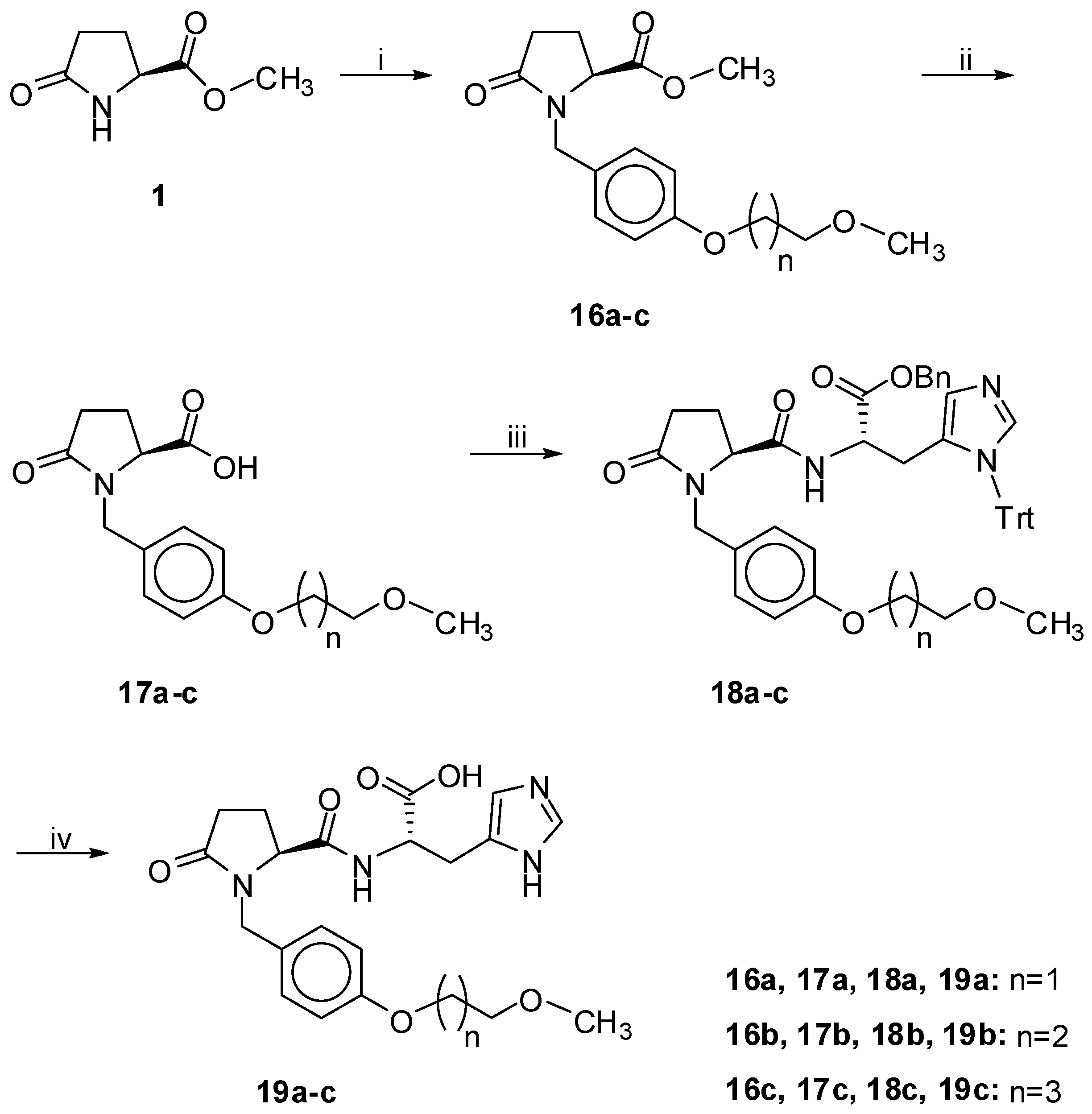
As shown in Scheme 2, methyl S-pyroglutamate was N-benzylated using bromides 30, 35 and 39 (Scheme 4, Scheme 5 and Scheme 6). After alkaline hydrolysis of the methyl ester of compounds 16a–c, the resulting carboxylic group was coupled with properly protected S-histidine (42, Scheme 7) using the water soluble carbodiimide method [25]. The benzylester group and the N-trityl group of the protected amino acid were removed in the end under catalytic hydrogenation.
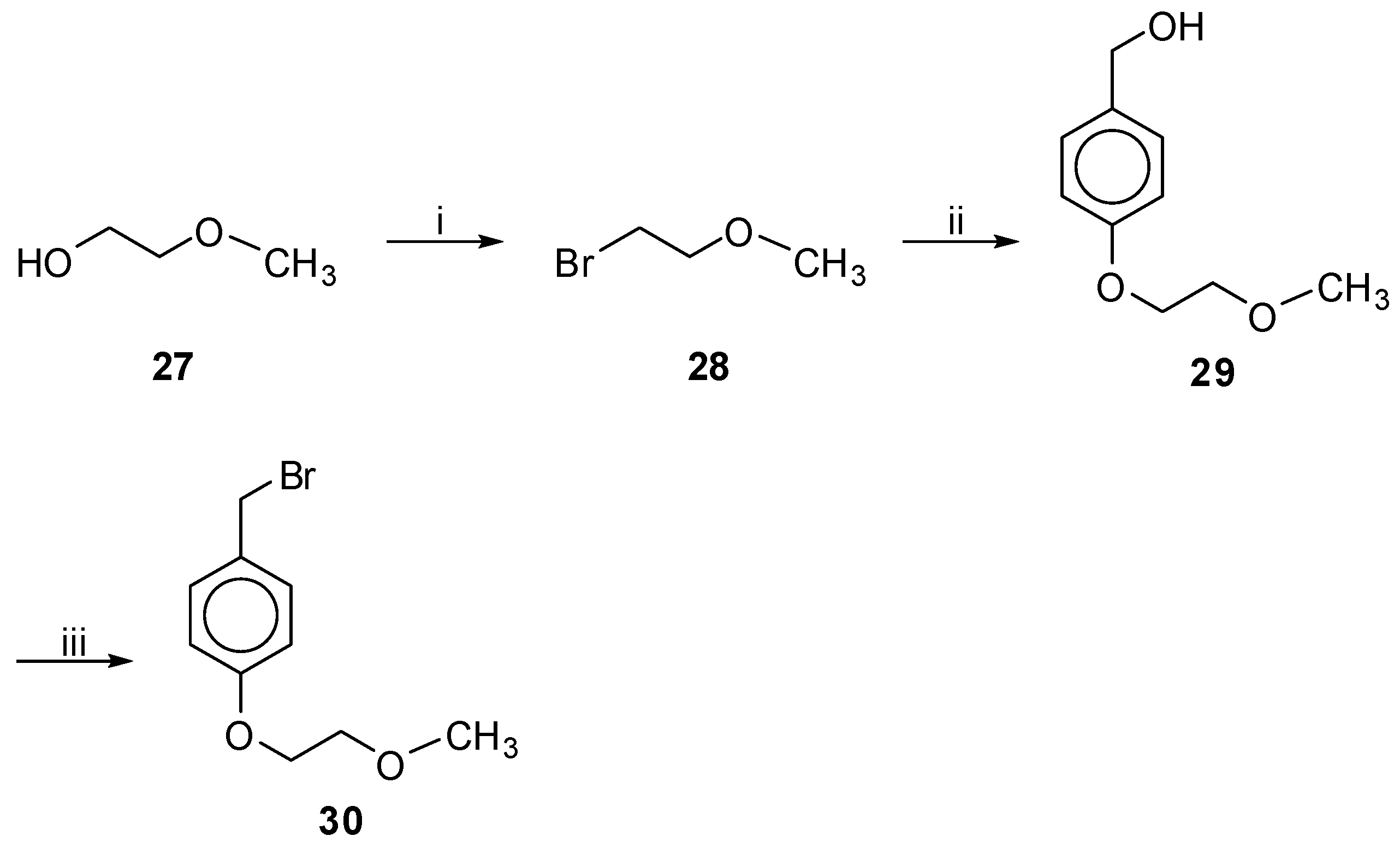
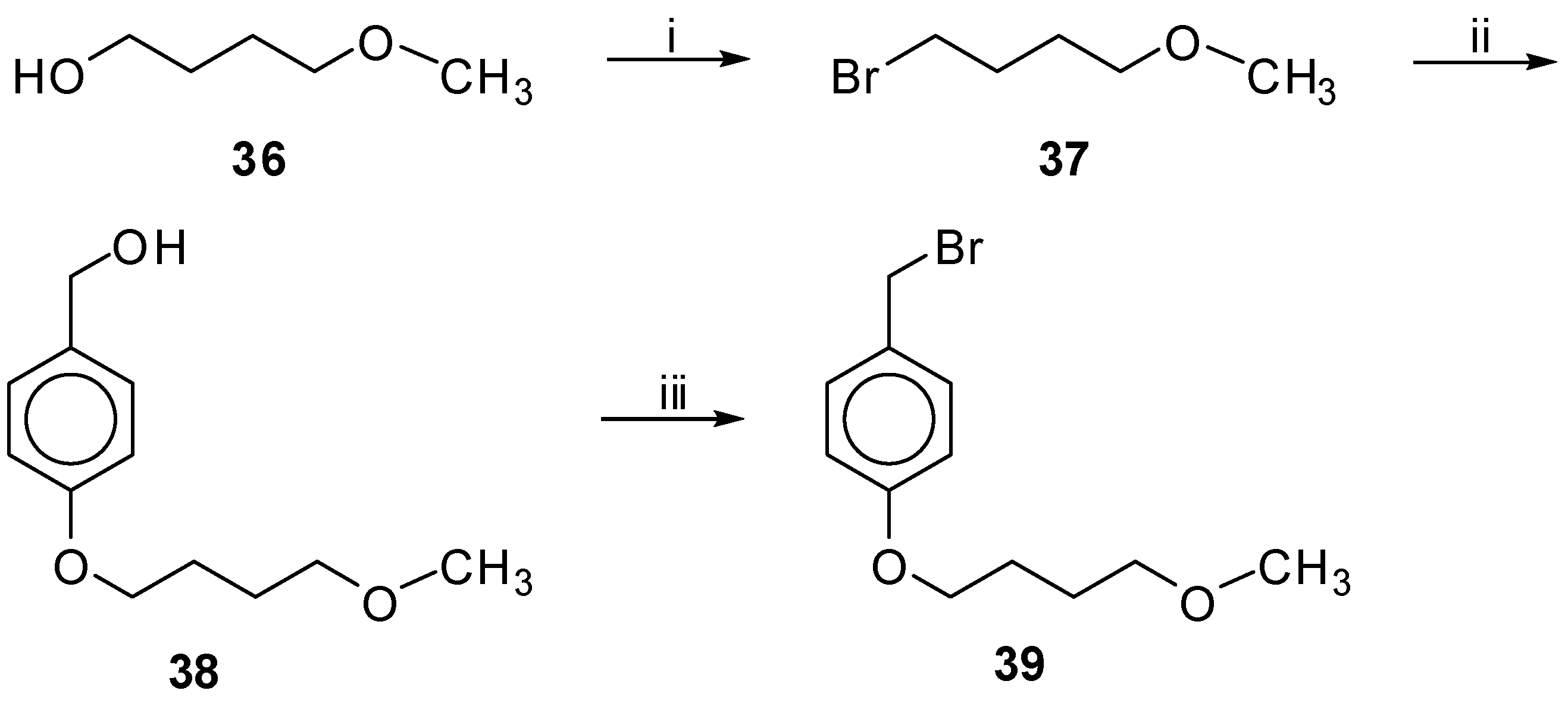
Bromides 30, 35 and 39 were prepared under the conditions depicted in Scheme 4, Scheme 5 and Scheme 6 respectively. Bromides 28 and 37 (Scheme 4 and Scheme 6), prepared from the commercially available alcohols 27 and 36 using PBr3 in Et2O, respectively [26], were used to convert 4-hydroxybenzyl alcohol to phenol ethers 29 and 38 using K2CO3 and 18-crown-6 in acetone [27]. Finally, using PBr3 in Et2O the bromide 30 was obtained in 60% yield, whereas bromide 39 was obtained in 42% yield only by the method of TMS-Cl, NaBr in CH3CN [28]. We tried to prepare the desired bromide 39 using many other methods like PBr3 in Et2O, Tos-Cl/Et3N and NaBr in acetone or DMF [29], MeSO2Cl/Et3N and NaBr in DMF or LiBr in THF [30], without any success. In the case of bromide 35, we followed a different approach, depicted in Scheme 5, since 3-bromo-propanol was not commercially available. Compound 32 was obtained by the reaction of 4-hydroxy benzaldehyde (31) and 1,3-dibromo propane with K2CO3 in acetone [31]. After conversion of the bromide group of 32 to methyl ether by MeONa [31], the aldehyde 33 was readily subjected to reduction by NaBH4 in dry THF to afford alcohol 34. The relatively low yield of the preparation of compound 32 can be explained by the fact that a parallel Cannizzaro reaction had occurred, under the alkaline conditions of that synthetic step, as revealed from the characterization of the isolated by-products. Both benzaldehydes 32 and 33 were unstable even if they were stored at −4 °C for more than 2–3 h.
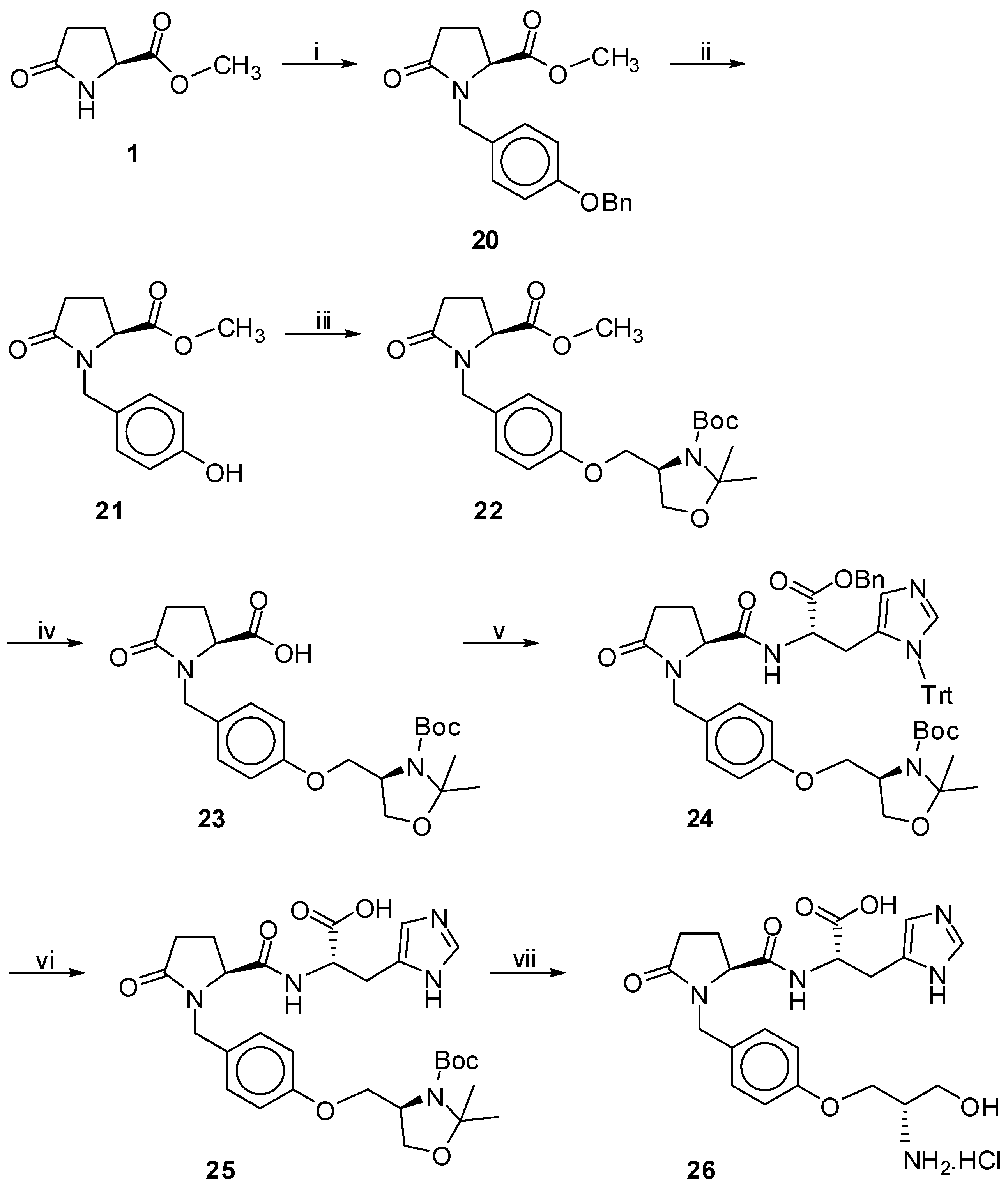
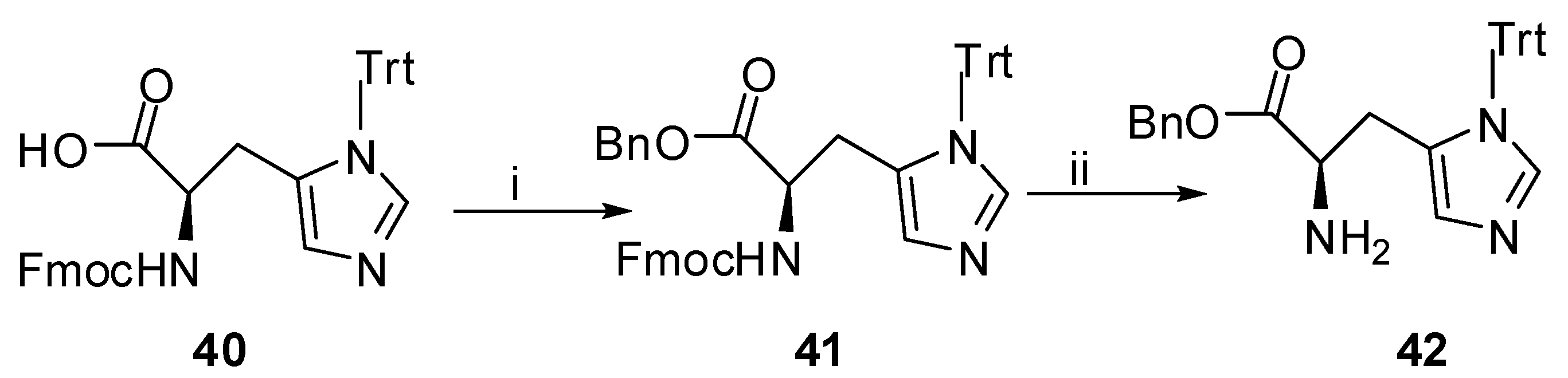
As depicted in Scheme 3, a different approach was followed for the synthesis of product 26. Methyl S-pyroglutamate was benzylated at the N-position by 1-(benzyloxy)-4-(bromomethyl) benzene [32] to afford product 20. After deprotection of the benzylether by catalytic hydrogenation, the phenol group of compound 21 reacted with the cyclic alcohol (tert-butyl (4R)-4-(hydroxymethyl)-2,2-dimethyl-1,3-oxazolidine-3-carboxylate), a precursor of Garner aldehyde and derived from the methylester of N-Boc-S-serine [33], under Mitsunobu reaction conditions [34]. The resulting product 22, was hydrolyzed and coupled with a properly protected derivative of S-histidine (42, Scheme 7). Catalytic hydrogenation, followed by acidic hydrolysis with 4 N HCl in dioxane, afforded the desired product 26 (Figure 1). Finally the synthesis of H-His(Trt)-OBn (42) is depicted in Scheme 7. The commercially available Fmoc-His(Trt)-OH (40) was converted to the corresponding benzyl ester (41) [35]. The desired, properly protected histidine (42) was obtained after Fmoc removal [36].
3. Experimental
3.1. General
All chemicals and solvents were reagent grade and used without purification. Dry THF and extra dry DMF (99.8%) over molecular sieves were purchased from Acros. Melting points were determined on a Büchi 530 apparatus and are uncorrected. Specific rotations were measured at 25 °C on a Perkin–Elmer polarimeter using a 10 cm cell. Nuclear magnetic resonance spectra were obtained on a Varian Mercury spectrometer (1H-NMR recorded at 200 MHz and 13C-NMR recorded at 50 MHz and they are referenced in ppm relative to TMS as an internal standard). Mass spectra were recorded on a Finnigan Surveyor MSQ Plus with only molecular ions and major peaks being reported with intensities quoted as percentages of the base peak. TLC plates (Silica Gel 60 F254) were purchased from Merck. Visualization of spots was effected with UV light and/or phosphomolybdic acid in EtOH stain. All target compounds possessed 95% purity as determined by combustion analysis.
3.2. General Procedure for the Preparation of Compounds 28, 30, 35, 37
To an ice cooled solution of appropriate alcohol 27, 29, 34, 36 (10 mmol) in Et2O (25 mL), PBr3 (1.41 mL, 15 mmol) was added dropwise under argon. The mixture was stirred at room temperature for 3–7 h and the reaction was quenched by the addition of H2O (15 mL) in small portions at 0 °C. The aqueous phase was removed and the organic layer was washed with H2O, dried over Na2SO4 and evaporated in vacuo. In the case of products 30 and 35, purification was achieved by column chromatography using the appropriate solvent systems as it will be defined in each case below. In the case of bromides 28, 37, the oily products were not visible either in UV-light or in iodine stain. They were characterized by NMR and used in the next step without further purification. For this reason the yields of their preparation must be considered as crude yields.
1-Bromo-2-Methoxyethane (28). Prepared from the commercially available alcohol 27; Yield: 58% (colorless oil). 1H-NMR (CDCl3): δ 3.69 (t, J = 6.0 Hz, 2H, OCH2), 3.45 (t, J = 6.0 Hz, 2H, CH2Br), 3.38 (s, 3H, CH3); 13C-NMR: δ 72.3 (OCH2), 58.8 (CH3), 30.3 (CH2Br). Anal. calcd for C3H7BrO: C, 25.92; H, 5.08; Found: C, 25.68; H, 5.12.
1-(Bromomethyl)-4-(2-Methoxyethoxy)benzene (30). Prepared from alcohol 18. Eluent EtOAc–petroleum ether (bp. 40–60 °C), 3:7; white solid; yield 60%; mp. 47–49 °C; Rf 0.50 in EtOAc–petroleum ether (bp. 40–60 °C), 3:7. 1H-NMR (CDCl3): δ 7.30 (d, J = 8.7 Hz, 2H, Ph), 6.87 (d, J = 8.7 Hz, 2H, Ph), 4.48 (s, 2H, CH2Br), 4.09 (t, J = 4.5 Hz, 2H, PhOCH2), 3.73 (t, J = 4.8 Hz, 2H, CH2OCH3), 3.43 (s, 3H, CH3). 13C-NMR: δ 158.7, 130.3, 114.7, 70.8 (CH2OCH3), 67.2 (PhOCH2), 59.1 (CH3), 33.9 (CH2Br). Anal. calcd for C10H13BrO2: C, 49.00; H, 5.35; Found: C, 48.95; H, 5.25.
1-(Bromomethyl)-4-(3-Methoxypropoxy)benzene (35). Prepared from alcohol 34. Eluent EtOAc–petroleum ether (bp. 40–60 °C), 3:7; yield: 70% (colorless oil); Rf 0.35 in EtOAc–petroleum ether (bp. 40–60 °C), 3:7. 1H-NMR (CDCl3): δ 7.31 (d, J = 8.7 Hz, 2H, Ph), 6.86 (d, J = 8.7 Hz, 2H, Ph), 4.50 (s, 2H, CH2Br), 4.05 (t, J = 6.3 Hz, 2H, PhOCH2), 3.55 (t, J = 6.2 Hz, 2H, CH2ΟCH3), 3.35 (s, 3H, CH3), 2.11–1.98 (m, 2H, CH2CH2CH2). 13C-NMR: δ 159.1, 130.4, 129.8, 114.7, 69.1 (CH2OCH3), 64.8 (PhOCH2), 58.7 (CH3), 34.1 (CH2Br), 29.5 (CH2CH2CH2). Anal. calcd for C11H15BrO2: C, 50.98; H, 5.83; Found: C, 50.82; H, 5.93.
1-Bromo-4-Methoxybutane (37). Prepared from the commercially available alcohol 36; Yield: 60% (colorless oil). 1H-NMR (CDCl3): δ 3.47–3.37 (m, 4H, OCH2, CH2Br), 3.33 (s, 3H, CH3), 2.02–1.88 (m, 2H, CH2CH2Br), 1.78–1.64 (m, 2H, CH2CH2O). 13C-NMR: δ 71.7 (OCH2), 58.6 (CH3), 33.7 (CH2Br), 29.6 (CH2CH2Br), 28.2 (CH2CH2O). Anal. calcd for C5H11BrO: C, 35.95; H, 6.64; Found: C, 35.82; H, 6.59.
1-(Bromomethyl)-4-(4-Methoxybutoxy)benzene (39). To a stirred solution containing alcohol 38 (35 mg, 0.166 mmol) and NaBr (17 mg, 0.166 mmol) in CH3CN (0.5 mL), TMS-Cl (21 μL, 0.166 mmol) was added under argon. After stirring for 1.5h at rt, the solvent was evaporated in vacuo and the residue dissolved in Et2O was washed with brine and H2O. The organic layer was dried over Na2SO4 and evaporated under reduced pressure. After purification by column chromatography using EtOAc–petroleum ether (bp. 40–60 °C) 1:9 as eluent, the product was obtained as a colorless oil in 42% yield (19 mg). Rf 0.35 in EtOAc-petroleum ether (bp. 40–60 °C) 1: 9. 1H-NMR (CDCl3): δ 7.27 (d, J = 8.6 Hz, 2H, Ph), 6.84 (d, J = 8.6 Hz, 2H, Ph), 4.50 (d, J = 12.6 Hz, 2H, CH2Br), 3.95 (t, J = 5.9 Hz, 2H, PhOCH2), 3.42 (t, J = 6.2 Hz, 2H, CH2OCH3), 3.33 (s, 3H, CH3), 1.87–1.68 (m, 4H, CH2CH2CH2CH2). 13C-NMR: δ 159.0, 130.3, 114.5, 72.2 (CH2OCH3), 67.5 (PhOCH2), 58.5 (CH3), 34.0 (CH2Br), 26.1, 25.9. Anal. calcd for C12H17BrO2: C, 52.76; H, 6.27; Found: C, 52.82; H, 6.35.
3.3. General Procedure for the Preparation of Compounds 29, 38
To a stirred solution of 4-hydroxybenzylalcohol (1.24 g, 10 mmol) in acetone (45 mL), bromides 28 or 37 (10.5 mmol), K2CO3 (4.14 g, 30 mmol) and 18-crown-6 (0.020 g) were added consecutively. The mixture was refluxed for 3 days. After evaporation of the solvent, the residue was dissolved in EtOAc and washed with brine and H2O. The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure. Purification was achieved by column chromatography, eluting with the appropriate solvents as defined for each case below.
[4-(2-Methoxyethoxy)-phenyl]methanol (29). Prepared from bromide 28. Eluent CHCl3–MeOH, 9.5:0.5; Yield: 76% (colorless oil); Rf 0.48 in CHCl3–MeOH, 9.5:0.5. 1H-NMR (CDCl3): δ 7.27 (d, J = 8.7 Hz, 2H, Ph), 6.90 (d, J = 8.7 Hz, 2H, Ph), 4.61 (s, 2H, CH2OH), 4.11 (t, J = 4.6 Hz, 2H, PhOCH2), 3.75 (t, J = 4.9 Hz, 2H, CH2OCH3), 3.44 (s, 3H, CH3); 13C-NMR: δ 158.2, 133.3, 128.5, 114.5, 70.9 (CH2OCH3), 67.2 (PhOCH2), 64.8 (CH2OH), 59.2 (CH3). Anal. calcd for C10H14O3: C, 65.91; H, 7.74; Found: C, 65.79; H, 7.68.
[4-(4-Methoxybutoxy) Phenyl]methanol (38). Prepared from bromide 37. Eluent CHCl3–MeOH, 9:1; Yield: 67% (colorless oil); Rf 0.35 in CHCl3–MeOH, 9:1. 1H-NMR (CDCl3): δ 7.26 (d, J = 8.7 Hz, 2H, Ph), 6.86 (d, J = 8.7 Hz, 2H, Ph), 4.60 (s, 2H, CH2OH), 3.97 (t, J = 6.1 Hz, 2H, PhOCH2), 3.43 (t, J = 6.2 Hz, 2H, CH2OCH3), 3.34 (s, 3H, CH3), 1.88–1.69 (m, 4H, CH2CH2CH2CH2). 13C-NMR: δ 158.6, 132.9, 128.6, 114.5, 72.3 (CH2OCH3), 67.6 (PhOCH2), 65.0 (CH2OH), 58.5 (CH3), 26.2, 26.0. Anal. calcd for C12H18O3: C, 68.54; H, 8.63; Found: C, 68.62; H, 8.59.
4-(3-Bromopropoxy) Benzaldehyde (32). To a stirred solution of 4-hydroxy-benzaldehyde (1 g, 8.2 mmol) in CH3CN (15 mL), K2CO3 (1.70 g, 12.3 mmol) and 1,3-dibromopropane (8.3 mL, 81.9 mmol) were added consecutively. A tube filled with CaCl2 was fixed on the condenser and the mixture was refluxed overnight. After evaporation of the solvent, the residue was dissolved in EtOAc and washed with brine and H2O. The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure. Purification was achieved by column chromatography using EtOAc–petroleum ether (bp. 40–60 °C) 1:4 as eluent. The product was obtained as colorless oil in 40% yield (0.8 g) and stored at −4 °C, only for a short time. Rf 0.36 in EtOAc–petroleum ether (bp. 40–60 °C) 1:4. 1H-NMR (CDCl3): δ 9.88 (s, 1H, CHO), 7.83 (d, J = 8.9 Hz, 2H, Ph), 7.00 (d, J = 8.9 Hz, 2H, Ph), 4.19 (t, J = 5.8 Hz, 2H, PhOCH2), 3.60 (t, J = 6.3 Hz, 2H, CH2Br), 2.41–2.29 (m, 2H, CH2CH2CH2). 13C-NMR: δ 190.7 (CHO), 163.6, 131.9, 130.1, 114.7, 65.6 (PhOCH2), 32.0 (CH2Br), 29.6 (CH2CH2CH2). Anal. calcd for C10H11BrO3: C, 49.41; H, 4.56; Found: C, 49.56; H, 4.39.
4-(3-Methoxypropoxy)benzaldehyde (33). To a stirred solution of bromide 32 (46 mg, 0.19 mmol) in MeOH (2 mL), a freshly prepared solution of 1N CH3ONa/MeOH (200 μL) was added and the mixture was left stirring at rt for 2 days. The reaction was quenched by the addition of H2O and the mixture was acidified with aq. 1 N HCl. After evaporation of the solvent, the residue was dissolved in EtOAc and washed with brine and H2O. The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure. Purification was achieved by column chromatography using EtOAc–petroleum ether (bp. 40–60 °C) 2:3 as eluent. The product was obtained as a colorless oil in 38% yield (14 mg) and was immediately subjected to the following reaction. Rf 0.38 in EtOAc–petroleum ether (bp. 40–60 °C) 2:3. 1H-NMR (CDCl3): δ 9.87 (s, 1H, CHO), 7.82 (d, J = 8.8 Hz, 2H, Ph), 7.00 (d, J = 8.8 Hz, 2H, Ph), 4.14 (t, J = 6.3 Hz, 2H, PhOCH2), 3.56 (t, J = 6.0 Hz, 2H, CH2Ο), 3.35 (s, 3H, CH3), 2.14–2.01 (m, 2H, CH2CH2CH2). 13C-NMR: δ 190.8 (CHO), 164.0, 131.9, 129.8, 114.7, 68.8 (CH2OCH3), 65.2 (PhOCH2), 58.7 (CH3), 29.4 (CH2CH2CH2). MS (ESI): m/z = 195 (100) (M+H+). Anal. calcd for C11H14O3: C, 68.02; H, 7.27; Found: C, 68.18; H, 7.15.
[4-(3-Methoxypropoxy) Phenyl]methanol (34). To a stirred solution of aldehyde 33 (63 mg, 0.324 mmol) in dry THF (2 mL) at 0 °C NaBH4 (18 mg, 0.487 mmol) was added under argon. The reaction mixture was left stirring at rt overnight. The reaction was quenched by the addition of a 20% solution of AcOH at 0 °C, until the production of the gas was ceased. The organic solvent was evaporated under reduced pressure and the residue was dissolved in EtOAc. The organic phase was washed with 5% aq. NaHCO3, brine and H2O, dried over Na2SO4 and evaporated under reduced pressure. After purification with column chromatography using EtOAc–petroleum ether (bp. 40–60 °C) 2:3 as eluent, the product was obtained as a colorless oil in 75% yield (47 mg). Rf 0.25 in EtOAc-petroleum ether (bp. 40–60 °C) 2:3. 1H-NMR (CDCl3): δ 7.28 (d, J = 8.6 Hz, 2H, Ph), 6.89 (d, J = 8.6 Hz, 2H, Ph), 4.61 (s, 2H, CH2OH), 4.05 (t, J = 6.3 Hz, 2H, PhOCH2), 3.55 (t, J = 6.2 Hz, 2H, CH2ΟCH3), 3.35 (s, 3H, CH3), 2.11–1.98 (m, 2H, CH2CH2CH2). 13C-NMR: δ 158.6, 133.0, 114.5, 69.2 (CH2OCH3), 65.0 (CH2OH), 64.8 (PhOCH2), 58.7 (CH3), 29.5 (CH2CH2CH2). MS (ESI): m/z = 197 (100) (M+H+). Anal. calcd for C11H16O3: C, 67.32; H, 8.22; Found: C, 67.53; H, 8.09.
(5S)-(Hydroxymethyl)-2-Pyrrolidinone (2). See reference [17].
5-[[(Triethylsilyl)oxy] Methyl]-2-Pyrrolidinone (3). See reference [18].
3.4. General Procedure of for the Preparation of Compounds 4, 8, 16a–c, 20
To a cooled solution of methyl (S)-pyroglutamate (1), O-protected-S-pyroglutaminol (3), or methylester 7 (1 mmol) in dry THF (5 mL) NaH (60% in paraffin oil, 1.5 mmol) was added in small portions, followed by the addition of the appropriate benzyl bromide (or benzyloxycarbonylchloride in case of compound 4) (1.1 mmol). The stirring was continued for 15 min at 0 °C, and for 24 h at rt under argon. The reaction was quenched by the addition of a saturated solution of NH4Cl at 0 °C, the organic solvent was evaporated and the residue was dissolved in ethyl acetate. The organic layer was washed with brine, dried over Na2SO4 and evaporated under reduced pressure. The product was purified by column chromatography (Silica Gel 60) using the appropriate solvent systems as defined in each case below.
Benzyl (5S)-2-oxo-5-{[(triethylsilyl)oxy]methyl}pyrrolidine-1-carboxylate (4). Prepared from compound 3 and benzyloxycarbonyl chloride. Eluent EtOAc–petroleum ether (bp. 40–60 °C), 4:6. The product was obtained as colorless oil in 50% yield. Rf 0.53 in EtOAc–petroleum ether (bp. 40–60 °C), 4:6; [α]D −68.2 (c 1.1, CHCl3). 1H-NMR (CDCl3): δ 7.46–7.32 (m, 5H, Ph), 5.28 (dd, J1 = 12.4 Hz, J2 = 18.4 Hz, 2H, COOCH2), 4.28–4.20 (m, 1H, NCH), 3.88 (dd, J1 = 4.0 Hz, J2 = 10.6 Hz, 1H, CHHOSi), 3.67 (dd, J1 = 2.5 Hz, J2 = 10.7 Hz, 1H, CHHOSi), 2.84–2.03 (m, 4H, 2xCH2), 0.88 (t, J = 7.6 Hz, 9H, 3 × CH3), 0.51 (q, J = 7.6 Hz, 6H, 3 × (CH2CH3)) 13C-NMR: δ 174.7 (CON), 151.3 (COO), 135.2, 128.5, 128.3, 128.2, 67.8 (CH2OSi), 63.7 (OCH2Ph), 58.8 (NCH), 32.1, 21.2, 6.6 (CH2CH3), 4.1 (CH2CH3). MS (ESI): m/z = 364 (100) (M+H+). Anal. calcd for C19H29NO4Si: C, 62.78; H, 8.04; N, 3.85 Found: C, 62.83; H, 7.95; N, 3.92.
Methyl 3-{(2S)-1-[4-(2-Methoxyethoxy)benzyl]-5-oxopyrrolidin-2-yl} propanoate (8). Prepared from compound 7 and bromide 30. Eluent EtOAc–petroleum ether (bp. 40–60 °C), 9:1. The product was obtained as colorless oil in 40% yield. Rf 0.23 in EtOAc–petroleum ether (bp. 40–60 °C), 1:9; [α]D +5.2 (c 0.92, CHCl3). 1H-NMR (CDCl3): δ 7.13 (d, J = 8.6 Hz, 2H, Ph), 6.83 (d, J = 8.6 Hz, 2H, Ph), 4.90 (d, J = 14.8 Hz, 1H, NCHHPh), 4.09–4.04 (m, 2H, PhOCH2), 3.85 (d, J = 14.8 Hz, 1H, NCHHPh), 3.73–3.68 (m, 2H, CH2OCH3), 3.62 (s, 3H, COOCH3), 3.42–3.38 (m, 1H, NCH), 3.41 (s, 3H, OCH3), 2.45–1.54 (m, 8H, 4 × CH2). 13C-NMR: δ 174.8 (NCO), 173.0 (COO), 158.1, 129.2, 128.7, 114.6, 70.7 (CH2OCH3), 67.1 (PhOCH2), 59.1 (OCH3), 55.7 (NCH), 51.7 (COOCH3), 43.3 (NCH2Ph), 30.0, 29.0, 27.6, 23.3. MS (ESI): m/z = 358 (100) (M+Na+). Anal. calcd for C18H25NO5: C, 64.46; H, 7.51; N, 4.18 Found: C, 64.51; H, 7.62; N, 4.05.
Methyl 1-[4-(2-Methoxyethoxy)benzyl]-5-oxo-L-prolinate (16a). Prepared from methyl-S-pyroglutamate (1) and bromide 30. Eluent EtOAc. The product was obtained as a colorless oil in 73% yield. Rf 0.42 in EtOAc; [α]D +20.2 (c 1.0, CHCl3). 1H-NMR (CDCl3): δ 7.07 (d, J = 8.5 Hz, 2H, Ph), 6.82 (d, J = 8.5 Hz, 2H, Ph), 4.87 (d, J = 14.7 Hz, 1H, CHHPh), 4.07–4.02 (m, 2H, PhOCH2), 3.93–3.86 (m, 2H, CHHPh, NCHCOO), 3.71–3.66 (m, 2H, CH2OCH3), 3.62 (s, 3H, COOCH3), 3.39 (s, 3H, OCH3), 2.61–1.92 (m, 4H, 2 × CH2). 13C-NMR: δ 174.8 (NCO), 172.1 (COO), 158.2, 129.7, 127.8, 114.6, 70.8 (CH2OCH3), 67.1 (OCH2), 59.0 (NCH), 58.4 (OCH3), 52.2 (COOCH3), 44.8 (CH2Ph), 29.5, 22.6. MS (ESI): m/z = 308 (100) (M+Η+). Anal. calcd for C16H21NO5: C, 62.53; H, 6.89; N, 4.56 Found: C, 62.58; H, 6.75; N, 4.61.
Methyl 1-[4-(3-Methoxypropoxy)benzyl]-5-oxo-L-prolinate (16b). Prepared from methyl-S-pyroglutamate (1) and bromide 35. Eluent EtOAc. The product was obtained as a colorless oil in 72% yield. Rf 0.44 in EtOAc; [α]D +13.5 (c 1.0, CHCl3). 1H-NMR (CDCl3): δ 7.08 (d, J = 8.6 Hz, 2H, Ph), 6.80 (d, J = 8.6 Hz, 2H, Ph), 4.90 (d, J = 14.6 Hz, 1H, NCHHPh), 3.99 (t, J = 6.3 Hz, 2H, PhOCH2), 3.95–3.90 (m, 1H, NCHCOO), 3.89 (d, J = 4.6 Hz, 1H, NCHHPh), 3.65 (s, 3H, COOCH3), 3.51 (t, J = 6.2Hz, 2H, CH2OCH3), 3.31 (s, 3H, OCH3), 2.57–1.93 (m, 6H, 3 × CH2). 13C-NMR: δ 174.8 (NCO), 172.2 (COO), 158.5, 129.7, 127.5, 114.5, 69.0 (CH2OCH3), 64.7 (PhOCH2), 58.6 (NCH), 58.4 (OCH3), 44.8 (CH2Ph), 29.5, 29.4, 22.6. MS (ESI): m/z = 322 (100) (M+Η+). Anal. calcd for C17H23NO5: C, 63.54; H, 7.21; N, 4.36 Found: C, 63.48; H, 7.28; N, 4.18.
Methyl 1-[4-(4-Methoxybutoxy)benzyl]-5-oxo-L-prolinate (16c). Prepared from methyl-S-pyroglutamate (1) and bromide 39. Eluent EtOAc. The product was obtained as a colorless oil in 66% yield. Rf 0.47 in EtOAc; [α]D +11.7 (c 1.0, CHCl3). 1H-NMR (CDCl3): δ 7.08 (d, J = 8.6 Hz, 2H, Ph), 6.79 (d, J = 8.6 Hz, 2H, Ph), 4.90 (d, J = 14.7 Hz, 1H, NCHHPh), 3.97–3.89 (m, 1H, NCHCO), 3.92 (t, J = 6.30 Hz, 2H, PhOCH2), 3.90 (d, J = 14.7 Hz, 1H, NCHHPh), 3.65 (s, 3H, COOCH3), 3.40 (t, J = 6.2 Hz, 2H, CH2OCH3), 3.31 (s, 3H, OCH3), 2.58–1.96 (m, 4H, 2 × CH2), 1.84-1.67 (m, 4H, (OCH2CH2)2). 13C-NMR: δ 174.8 (NCO), 172.2 (COO), 158.6, 129.7, 127.5, 114.5, 72.2 (CH2OCH3), 67.5 (PhOCH2), 58.5 (NCH), 58.4 (OCH3), 52.2 (COOCH3), 44.9 (NCH2Ph), 29.5, 26.1, 25.9, 22.7. MS (ESI): m/z = 336 (100) (M+Η+). Anal. calcd for C18H25NO5: C, 64.46; H, 7.51; N, 4.18 Found: C, 64.38; H, 7.48; N, 4.27.
Methyl 1-[4-(Benzyloxy)benzyl]-5-oxo-L-prolinate (20). Prepared from methyl-S-pyroglutamate (1) and 1-(benzyloxy)-4-(bromomethyl) benzene [32]. Eluent EtOAc -petroleum ether (bp. 40–60 °C), 7:3. The product was obtained as a colorless oil in 47% yield. Rf 0.35 in EtOAc–petroleum ether (bp. 40–60 °C), 7:3; [α]D +22.5 (c 1, CHCl3). 1H-NMR (CDCl3): δ 7.44–7.28 (m, 5H, Ph), 7.14 (d, J = 8.5 Hz, 2H, Ph), 6.92 (d, J = 8.5 Hz, 2H, Ph), 5.05 (s, 2H, OCH2Ph), 4.93 (d, J = 14.7 Hz, 1H, NCHHPh), 4.00-3.92 (m, 2H, NCHHPh, NCH), 3.66 (s, 3H, CH3), 2.53–2.04 (m, 4H, 2 × CH2). 13C-NMR: δ 174.8 (NCO), 172.1 (COOCH3), 158.2, 136.7, 129.7, 128.4, 127.9, 127.8, 127.3, 114.8, 69.8 (OCH2Ph), 58.5 (NCH), 52.2 (CH3), 44.8 (NCHPh), 29.4, 22.6. MS (ESI): m/z = 340 (100) (M+Η+). Anal. calcd for C20H21NO4: C, 70.78; H, 6.24; N, 4.13 Found: C, 70.62; H, 6.27; N, 4.22.
Benzyl (2S)-2-(Hydroxymethyl)-5-oxopyrrolidine-1-carboxylate (5). To a stirred solution of compound 4 (1.20 g, 3.30 mmol) in DCM (20 mL), TFA was added (2.02 mL, 26.4 mmol) at room temperature. Once the reaction was finished (45 min), the solvent was evaporated to dryness. The residue was dissolved in toluene and the solvent was evaporated (twice) for the removal of the acid. Finally the residue was dissolved in ethyl acetate and the organic phase was washed with brine to neutral pH. After drying over Na2SO4 and evaporation of the solvent under reduced pressure, the product was obtained as a pure white solid. Yield: 0.710 g (86%); Rf 0.37 in EtOAc; mp. 83–86 °C; [α]D −54.2 (c 1.0, CHCl3). 1H-NMR (CDCl3): δ 7.37–7.33 (m, 5H, Ph), 5.23 (s, 2H, OCH2Ph), 4.29–4.23 (m, 1H, NCH), 3.95 (dd, J1 = 3.2 Hz, J2 = 11.8 Hz, 1H, CHHOH), 3.66 (dd, J1 = 3.2 Hz, J2 = 11.8 Hz, 1H, CHHOH), 3.09 (bs, 1H, OH), 3.86–1.95 (m, 4H, 2 × CH2). 13C-NMR: δ 175.5 (CON), 151.7 (COO), 135.0, 128.6, 128.4, 128.1, 68.0 (OCH2Ph), 64.1 (CH2OH), 59.4 (NCH), 32.1, 20.9. MS (ESI): m/z = 250 (100) (M+Η+). Anal. calcd for C13H15NO4: C, 62.64; H, 6.07; N, 5.62 Found: C, 62.59; H, 6.12; N, 5.59.
Benzyl (2S)-2-[(1E)-3-Methoxy-3-oxoprop-1-en-1-yl]-5-oxopyrrolidine-1-carboxylate (6). Alcohol 5 (0.22 g, 0.882 mmol) was dissolved in a mixture of dry toluene (5 mL) and dry DMSO (2.5 mL), followed by the addition of EDC·HCl (0.51 g, 2.64 mmol), the dropwise addition of dry pyridine (0.25 mL, 3.04 mmol) and finally TFA (33.2 μL, 0.441 mmol) under argon. After stirring at rt for 1.5 h the reaction was quenched by the addition of CHCl3 and the solution was washed with brine and H2O. The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure. The resulting crude aldehyde was subjected to Wittig reaction without further purification. More specifically, it was dissolved in dry THF (6 mL), methyl (triphenylphosphoranylidene)acetate (0.32 g, 0.95 mmol) was added and the mixture was refluxed under argon for 1 h. The reaction was quenched by the addition of saturated solution of NH4Cl. The solvent was evaporated under reduced pressure and the residue dissolved in EtOAc was successively washed with NH4Cl, H2O and brine. The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure. After purification by column chromatography using EtOAc as eluent, the product was obtained in 60% yield (0.16 g) as a colorless oil. Rf 0.63 in EtOAc; [α]D −71.1 (c 1.0, CHCl3). 1H-NMR (CDCl3): δ 7.38–7.31 (m, 5H, Ph), 6.89 (dd, J1 = 6.3 Hz, J2 = 6.2 Hz, 1H, CHCHCOOCH3), 5.25 (dd, J1 = 12.3 Hz, J2 = 12.2 Hz, 2H, CH2Ph), 4.87–4.81 (m, 1H, NCH), 3.73 (s, 3H, OCH3), 2.64–1.82 (m, 4H, 2 × CH2). 13C-NMR: δ 173.0 (NCOCH2), 166.0 (COOCH3), 150.8 (COOCH2), 145.1, 134.8, 128.5, 128.4, 128.2, 121.5 (CHCOOCH3), 68.2 (OCH2Ph), 57.8 (NCH), 51.7 (CH3), 30.7, 23.8. MS (ESI): m/z = 326 (100) (M+Na+). Anal. calcd for C17H17NO5: C, 63.36; H, 5.65; N, 4.62 Found: C, 63.29; H, 5.48; N, 4.75.
(5R)-5-(3-Hydroxypropyl)-1-[4-(2-methoxyethoxy)benzyl]-pyrrolidin-2-one (9). To a two necked flask, LiBH4 (14 mg, 0.656 mmol) was suspended in dry THF (0.5 mL) under argon at rt. A solution of compound 8 (0.110g, 0.656 mmol) in dry THF (1.5 mL) was added dropwise and the reaction mixture was stirred for 10 h. The reaction was quenched by the addition of a 20% aq. solution of AcOH at 0 °C until gas production ceased. The excess of acetic acid was neutralized by the addition of a small quantity of Na2CO3. The organic solvent was evaporated under reduced pressure and the residue was dissolved in EtOAc. The organic phase was then washed with brine, dried over Na2SO4 and evaporated under reduced pressure. After purification with column chromatography using EtOAc–MeOH 9:1 as eluent, the product was obtained as a colorless oil in 87% yield (87 mg). Rf 0.34 in EtOAc–MeOH 9:1; [α]D +7.6 (c 0.95, CHCl3). 1H-NMR (CDCl3): δ 7.13 (d, J = 8.6 Hz, 2H, Ph), 6.84 (d, J = 8.6 Hz, 2H, Ph), 4.89 (d, J = 14.8 Hz, 1H, NCHHPh), 4.10–4.05 (m, 2H, PhOCH2), 3.88 (d, J = 14.8 Hz, 1H, NCHHPh), 3.74–3.70 (m, 2H, CH2OCH3), 3.64–3.54 (m, 2H, CH2OH), 3.42 (s, 4H, NCH, CH3), 2.47–1.36 (m, 8H, 4 × CH2). 13C-NMR: δ 175.1 (NCO), 158.1, 129.2, 128.8, 114.6, 70.9 (CH2OCH3), 67.1 (PhOCH2), 62.2 (CH2OH), 59.2 (CH3), 56.6 (NCH), 43.4 (NCH2Ph), 30.2, 28.9, 27.3, 23.7. MS (ESI): m/z = 308 (90) (M+H+).Anal. calcd for C17H25NO4: C, 66.43; H, 8.20; N, 4.56 Found: C, 66.38; H, 8.26; N, 4.42.
3-{(2R)-1-[4-(2-Methoxyethoxy)benzyl]-5-oxopyrrolidin-2-yl}propyl 4-Methylbenzenesulfonate (10). To an ice cooled stirred solution of alcohol 9 (35 mg 0.114 mmol) in DCM (2 mL), p-toluenesulfonylchloride (43 mg, 0.228 mmol) was added, followed by the addition of Et3N (17.5 μL). The reaction mixture was stirred at 0 °C for 10 min and overnight at room temperature. The organic layer was subsequently washed with a 1N HCl aq. solution, brine, 5% aq. solution of NaHCO3 and brine. After drying over Na2SO4 and evaporation in vacuo, the residue was purified by column chromatography using EtOAc as eluent, and the product was obtained as a yellowish oil in 72% yield (37 mg). Rf 0.35 in EtOAc; [α]D +5.6 (c 1, CHCl3). 1H-NMR (CDCl3): δ 7.75 (d, J = 8.3 Hz, 2H, Ph), 7.34 (d, J = 8.0 Hz, 2H, Ph), 7.10 (d, J = 8.6 Hz, 2H, Ph), 6.85 (d, J = 8.6 Hz, 2H, Ph), 4.83 (d, J = 14.9 Hz, 1H, NCHHPh), 4.11–4.06 (m, 2H, PhOCH2), 3.95 (t, J = 5.9 Hz, 2H, CH2OSO2), 3.83 (d, J = 14.9Hz, 1H, NCHHPh), 3.75–3.71 (m, 2H, CH2OCH3), 3.43 (s, 3H, OCH3), 3.39-3.31 (m, 1H, NCH), 2.44 (s, 3H, CH3), 2.41–1.50 (m, 8H, 4 × CH2). 13C-NMR: δ 174.9 (NCO), 158.1, 144.9, 132.8, 129.9, 129.2, 128.7, 127.8, 114.7, 70.9 (CH2OCH3), 69.9 (CH2OSO2), 67.2 (PhOCH2), 59.2 (CH3), 56.1 (NCH), 43.5 (NCH2Ph), 30.1, 28.7, 23.9, 23.5, 21.6 (PhCH3). MS (ESI): m/z = 462 (100) (M+H+). Anal. calcd for C24H31NO6S: C, 62.45; H, 6.77; N, 3.03 Found: C, 62.32; H, 6.82; N, 3.18.
3.5. General Procedure of for the Preparation of Compounds 11–13
Imidazole or 1H-imidazole-4(5)-carboxylic acid phenylmethyl ester [16], (1.2 mmol) and cesium carbonate (0.39 g, 1.2 mmol) were dissolved in dry DMF (3 mL) under argon. After stirring of the mixture for 30 min at 50 °C, a solution of tosyl ester 10 (1 mmol) dissolved in dry DMF (3 mL) was added and the stirring was continued overnight at 50 °C under argon. After evaporation of DMF in high vacuo, the residue was dissolved in EtOAc and the organic phase was washed with brine to neutral pH, dried over Na2SO4 and evaporated under reduced pressure. The residual product was purified by column chromatography (silica gel) using the appropriate solvent systems as it will be defined, in each case, below.
(5R)-5-[3-(1H-Imidazol-1-yl)propyl]-1-[4-(2-methoxyethoxy)benzyl]-pyrrolidin-2-one (11). Prepared from tosyl ester 10 and imidazole as a colorless oil. Eluent EtOAc–MeOH, 7:3. Yield: 58%. Rf 0.29 in EtOAc–MeOH, 7:3. [α]D +4.1 (c 0.8, MeOH). 1H-NMR (CD3OD): δ 7.61 (s, 1H, NCHN), 7.11 (d, J = 8.6 Hz, 2H, Ph), 7.07 (s, 1H, CH2NCHCHN), 6.98 (s, 1H, CH = NCH = CHN), 6.89 (d, J = 8.6 Hz, 2H, Ph), 4.69 (d, J = 14.9 Hz, 1H, NCHHPh), 4.12–4.07 (m, 2H, PhOCH2), 4.03–3.93 (m, 3H, NCHHPh, CH2N), 3.75–3.71 (m, 2H, CH2OCH3), 3.52–3.42 (m, 1H, NCH), 3.41 (s, 3H, CH3), 2.47–1.30 (m, 8H, 4 × CH2). 13C-NMR: δ 177.8 (NCO), 159.8, 138.5, 130.4, 130.1, 129.1, 120.6, 115.8, 72.2 (CH2OCH3), 68.5 (PhOCH2), 59.3 (CH3), 58.7 (CONCH), 47.7 (CH2N), 44.7 (NCH2Ph), 31.2, 30.6, 26.9, 24.5. MS (ESI): m/z = 358 (100) (M+H+). Anal. calcd for C20H27N3O3: C, 67.20; H, 7.61; N, 11.76 Found: C, 67.28; H, 7.52; N, 11.82.
Benzyl 1-(3-{(2R)-1-[4-(2-Methoxyethoxy)benzyl]-5-oxopyrrolidin-2-ylpropyl)-1H-imidazole-5-carboxylate (12). Prepared from tosyl ester 10 and 1H-imidazole-4(5)-carboxylic acid phenylmethyl ester [16], as a mixture with its constitutional isomer 13. Eluent EtOAc-MeOH, 9:1. Yield: 55% (colorless oil). Rf 0.42 in EtOAc–MeOH, 7:3. [α]D +2.8 (c 1, CHCl3). 1H-NMR (CDCl3): δ 7.78 (s, 1H, NCHN), 7.58 (s, 1H, NCHCOO), 7.40–7.34 (m, 5H, Ph), 7.08 (d, J = 8.6 Hz, 2H, Ph), 6.85 (d, J = 8.6 Hz, 2H, Ph), 5.28 (s, 2H, OCH2Ph), 4.78 (d, J = 14.9 Hz, 1H, NCHHPh), 4.20 (t, J = 6.7 Hz, 2H, CH2N), 4.10–4.06 (m, 2H, PhOCH2), 3.87 (d, J = 14.9 Hz, 1H, NCHHPh), 3.74–3.70 (m, 2H, CH2OCH3), 3.42 (s, 3H, CH3), 3.41–3.33 (m, 1H, NCH), 2.43–1.24 (m, 8H, 4 × CH2). 13C-NMR: δ 174.9 (NCO), 159.8 (COO), 158.1, 141.8, 137.9, 135.5, 129.1, 128.8, 128.6, 128.5, 128.3, 128.0, 114.7, 70.9 (CH2OCH3), 67.2 (PhOCH2), 66.2 (COOCH2Ph), 59.1 (CH3), 56.3 (NCH), 46.6 (CH2NCCOO), 43.6 (NCH2Ph), 30.1, 29.6, 26.0, 23.6. MS (ESI): m/z = 492 (100) (M+H+). Anal. calcd for C28H33N3O5: C, 68.41; H, 6.77; N, 8.55 Found: C, 68.32; H, 6.60; N, 8.57.
Benzyl 1-(3-{(2R)-1-[4-(2-Methoxyethoxy)benzyl]-5-oxopyrrolidin-2-yl}propyl)-1H-imidazole-4-carboxylate (13). Prepared from tosyl ester 10 and 1H-imidazole-4(5)-carboxylic acid, phenylmethyl ester [16], as a mixture with its constitutional isomer 12. After the separation of isomer 12, the elution system (EtOAc–MeOH, 9:1) was gradually changed to the more polar EtOAc–MeOH, 3:2 in order to recover isomer 13. Yield: 30% (colorless oil). Rf 0.56 in EtOAc–MeOH, 3:2. [α]D +9.9 (c 1, CHCl3). 1H-NMR (CDCl3): δ 7.53 (s, 1H, NCHN), 7.51 (s, 1H, NCHCOO), 7.46–7.31 (m, 5H, Ph), 7.07 (d, J = 8.7 Hz, 2H, Ph), 6.84 (d, J = 8.7 Hz, 2H, Ph), 6.34 (s, 2H, COOCH2Ph), 4.73 (d, J = 14.9 Hz, 1H, NCHHPh), 4.09–4.05 (m, 2H, PhOCH2), 3.97–3.82 (m, 3H, NCHHPh, CH2N), 3.74–3.69 (m, 2H, CH2OCH3), 3.43 (s, 3H, CH3), 3.41–3.36 (m, 1H, NCH), 2.45–1.24 (m, 8H, 4 × CH2). 13C-NMR: δ 174.9 (NCO), 162.2 (COO), 158.2, 137.9, 136.0, 133.6, 129.1, 128.7, 128.5, 128.4, 128.2, 124.9, 114.7, 70.9 (CH2OCH3), 67.2 (PhOCH2), 66.2 (COOCH2Ph), 59.2 (CH3), 56.4 (NCH), 47.3 (CH2NCHN), 43.9 (NCH2Ph), 30.1, 29.7, 25.8, 23.6. MS (ESI): m/z = 492 (100) (M+H+). Anal. calcd for C28H33N3O5: C, 68.41; H, 6.77; N, 8.55 Found: C, 68.38; H, 6.82; N, 8.45.
3.6. General Procedure for the Preparation of Compounds 7, 14, 15, 19a–c, 21, 25
A mixture of a compound possessing a double bond and also the N- benzyloxycarbonyl group (compound 6), benzyl ester (compounds 12,13), benzyl ether (compound 20) or benzyl ester and N-trityl imidazole group (compounds 18a–c, 24) (1 mmol) in MeOH (10 mL) and 10% palladium on activated carbon (0.02 g or 0.04 g for compounds 6, 18a–c, 24) was hydrogenated for 1.5–3 h (overnight for compound 6) under atmospheric conditions. After filtration through a pad of Celite, the solvent was removed in vacuo to give the deprotected final compound in almost quantitative yield and high purity. In the case of compounds 18a–c and 24 the triphenylmethane produced from the hydrogenation of trityl protecting group was completely removed by adding a small quantity of CHCl3 and discounting the supernatant where the polar products (19a–c, 25) were insoluble.
Methyl 3-[(2S)-5-Oxopyrrolidin-2-yl] propanoate (7). Prepared from alkene 6. The product was obtained as a yellowish solid in 94% yield and high purity. Rf 0.14 in EtOAc; mp. 72–75 °C; [α]D +14.3 (c 0.95, CHCl3). 1H-NMR (CDCl3): δ 7.01 (bs, 1H, NH), 3.73–3.64 (m, 1H, NCH), 3.66 (s, 3H, CH3), 2.41–1.60 (m, 8H, 4 × CH2). 13C-NMR: δ 178.6 (NCO), 173.1 (COOCH3), 53.7 (NCH), 51.4 (CH3), 31.3, 30.1, 26.5. MS (ESI): m/z = 172 (100) (M+Η+). Anal. calcd for C8H13NO3: C, 56.13; H, 7.65; N, 8.18 Found: C, 56.22; H, 7.58; N, 8.22.
1-(3-{(2R)-1-[4-(2-Methoxyethoxy)benzyl]-5-oxopyrrolidin-2-yl}propyl)-1H-imidazole-5-carboxylic acid (14). Catalytic hydrogenation of compound 12 afforded product 14 as a pure colorless oil in 99% yield. Rf 0.18 in EtOAc–MeOH, 3:2. [α]D +2.1 (c 1, MeOH). 1H-NMR (CD3OD): δ 8.09 (s, 1H, NCHN), 7.80 (s, 1H, NCHCCOO), 7.12 (d, J = 8.5 Hz, 2H, Ph), 6.88 (d, J = 8.5 Hz, 2H, Ph), 4.71 (d, J = 15.0 Hz, 1H, NCHHPh), 4.41–4.34 (m, 2H, CH2NCHCOO), 4.11–3.98 (m, 3H, NCHHPh, PhOCH2), 3.75–3.71 (m, 2H, CH2OCH3), 3.55–3.48 (m, 1H, NCHCH2), 3.41 (s, 3H, OCH3), 2.46–1.28 (m, 8H, 4 × CH2). 13C-NMR: δ 177.7 (NCO), 162.8 (COOH), 159.8, 142.0, 134.2, 130.4, 130.0, 126.5, 115.8, 72.2 (CH2OCH3), 68.4 (PhOCH2), 59.3 (CH3), 58.6 (NCH), 48.1 (CH2NCCOOH), 44.5 (NCH2Ph), 31.2, 30.5, 27.0, 24.5. MS (ESI): m/z = 402 (100) (M+H+). Anal. calcd for C21H27N3O5: C, 62.83; H, 6.78; N, 10.47 Found: C, 62.88; H, 6.73; N, 10.38.
1-(3-{(2R)-1-[4-(2-Methoxyethoxy)benzyl]-5-oxopyrrolidin-2-yl}propyl)-1H-imidazole-4-carboxylic acid (15). Catalytic hydrogenation of compound 13 afforded product 15 as a pure colorless oil in 90% yield. Rf 0.15 in EtOAc–MeOH, 1:1. [α]D +0.9 (c 1, MeOH). 1H-NMR (CD3OD): δ 7.83 (s, 1H, NCHN), 7.68 (s, 1H, NCHCOO), 7.10 (d, J = 8.2 Hz, 2H, Ph), 6.88 (d, J = 8.2 Hz, 2H, Ph), 4.60 (d, J = 14.9 Hz, 1H, NCHHPh), 4.11–3.99 (m, 5H, PhOCH2, NCHHPh, CH2NCHCOO), 3.75–3.71 (m, 2H, CH2OCH3), 3.51–3.47 (m, 1H, NCH), 3.41 (s, 3H, OCH3), 2.47–1.21 (m, 8H, 4 × CH2). 13C-NMR: δ 177.8 (NCO), 165.6 (COOH), 159.8, 139.5, 136.0, 134.7, 130.4, 130.0, 126.4, 115.8, 72.3 (CH2OCH3), 68.4 (PhOCH2), 59.3 (CH3), 58.5 (NCH), 48.4 (CH2NCHN), 44.7 (NCH2Ph), 31.2, 30.6, 26.8, 24.6. MS (ESI): m/z = 402 (100) (M+H+). Anal. calcd for C21H27N3O5: C, 62.83; H, 6.78; N, 10.47 Found: C, 62.74; H, 6.84; N, 10.35.
1-[4-(2-Methoxyethoxy)benzyl]-5-oxo-L-prolyl-L-histidine (19a). Prepared from compound 18a as a colorless oil in 97% yield. Rf 0.1 in EtOAc–MeOH, 1:1. [α]D +3.0 (c 1, MeOH). 1H-NMR (CD3OD): δ 8.85 (s, 1H, NCHN), 7.34 (s, 1H, NCHNCH), 7.05 (d, J = 8.5 Hz, 2H, Ph), 6.88 (d, J = 8.5 Hz, 2H, Ph), 4.84–4.72 (m, 2H, NHCHCOO, NCHHPh), 4.10–4.06 (m, 2H, PhOCH2), 4.01–3.95 (m, 1H, NCHCON), 3.75–3.62 (m, 3H, CH2OCH3, NCHHPh), 3.42 (s, 3H, CH3), 3.15–3.02 (m, 2H, CH2CHN), 2.57–1.96 (m, 4H, 2 × CH2). 13C-NMR: δ 178.3 (NCO), 173.9, 173.2, 160.0, 135.3, 131.7, 130.8, 129.1, 118.3, 115.9, 72.2 (CH2OCH3), 68.5 (OCH2), 61.1 (NCHCON), 59.3 (OCH3), 52.7 (HNCHCO), 45.8 (CH2Ph), 30.8 (CH2CHN), 28.0, 24.2. MS (ESI): m/z = 431 (100) (M+Η+). Anal. calcd for C21H26N4O6: C, 58.59; H, 6.09; N, 13.02 Found: C, 58.65; H, 5.98; N, 13.14.
1-[4-(3-Methoxypropoxy)benzyl]-5-oxo-L-prolyl-L-histidine (19b). Prepared from compound 18b as a colorless oil in 99% yield. Rf 0.11 in EtOAc–MeOH, 1:1. [α]D +11.5 (c 1, MeOH). 1H-NMR (CD3OD): δ 8.75 (s, 1H, NCHN), 7.32 (s, 1H, NCHNC), 7.04 (d, J = 8.5 Hz, 2H, Ph), 6.85 (d, J = 8.5 Hz, 2H, Ph), 4.88 (d, J = 14.8 Hz, 1H, NCHHPh), 4.75–4.65 (m, 1H, NHCHCOO), 4.04–3.98 (m, 1H, NCHCO), 4.01 (t, J = 6.3 Hz, 2H, PhOCH2), 3.63 (d, J = 14.8 Hz, 1H, NCHHPh), 3.54 (t, J = 6.2 Hz, 2H, CH2OCH3), 3.33 (s, 3H, OCH3), 3.28–3.03 (m, 2H, CH2CN), 2.53–1.93 (m, 6H, 3 × CH2). 13C-NMR: δ 178.3 (NCO), 173.7, 160.2, 135.2, 132.1, 131.0, 130.7, 128.9, 118.2, 115.8, 70.3 (CH2OCH3), 65.9 (PhOCH2), 61.2 (NCH), 58.9 (OCH3), 53.4 (CHCOOH), 45.8 (NCH2Ph), 30.9, 30.6, 28.4, 24.2. MS (ESI): m/z = 445 (100) (M+Η+). Anal. calcd for C22H28N4O6: C, 59.45; H, 6.35; N, 12.61 Found: C, 59.52; H, 6.37; N, 12.50.
1-[4-(4-Methoxybutoxy)benzyl]-5-oxo-L-prolyl-L-histidine (19c). Prepared from compound 18c as a colorless oil in 92% yield. Rf 0.16 in EtOAc–MeOH, 1:1. [α]D +6.9 (c 1, MeOH). 1H-NMR (CD3OD): δ 8.84 (s, 1H, HNCHN), 7.36 (s, 1H, NCCHN), 7.04 (d, J = 8.5 Hz, 2H, Ph), 6.85 (d, J = 8.5 Hz, 2H, Ph), 4.89 (d, J = 14.7 Hz, 1H, NCHHPh), 4.80–4.73 (m, 1H, NHCHCOO), 4.02–3.96 (m, 1H, NCH), 3.95 (t, J = 5.4 Hz, 2H, PhOCH2), 3.63 (d, J = 14.7 Hz, 1H, NCHHPh), 3.45 (t, J = 6.1 Hz, 2H, CH2OCH3), 3.33 (s, 3H, CH3), 3.17–3.05 (m, 2H, CH2CNH), 2.49-1.96 (m, 4H, 2 × CH2), 1.84–1.65 (m, 4H, (OCH2CH2)2). 13C-NMR: δ 178.2 (NCO), 173.9, 160.3, 135.2, 131.8, 131.1, 130.7, 128.8, 118.3, 115.8, 73.5 (CH2OCH3), 68.9 (PhOCH2), 61.1 (NCH), 58.8 (OCH3), 52.9 (CHCOOH), 45.8 (NCH2Ph), 30.8, 28.0, 27.3, 27.2, 24.1. MS (ESI): m/z = 459 (100) (M+Η+). Anal. calcd for C23H30N4O6: C, 60.25; H, 6.59; N, 12.12 Found: C, 60.32; H, 6.47; N, 12.30.
Methyl 1-(4-Hydroxybenzyl)-5-oxo-L-prolinate (21). Prepared from compound 20 as a colorless oil in 99% yield. Rf 0.69 in EtOAc. [α]D +35.5 (c 1.1, CHCl3). 1H-NMR (CDCl3): δ 7.03 (d, J = 7.5 Hz, 2H, Ph), 6.80 (d, J = 7.5 Hz, 2H, Ph), 4.89 (d, J = 14.5 Hz, 1H, NCHHPh), 4.02–3.91 (m, 2H, NCHHPh, NCH), 3.67 (s, 3H, CH3), 2.55–2.04 (m, 4H, 2xCH2). 13C-NMR: δ 175.7 (NCO), 172.2 (COOCH3), 156.3, 130.0, 126.5, 115.7, 58.9 (NCH), 52.5 (COOCH3), 45.3 (NCHPh), 29.8, 22.7. MS (ESI): m/z = 250 (97) (M+Η+). Anal. calcd for C13H15NO4: C, 62.64; H, 6.07; N, 5.62 Found: C, 62.59; H, 6.17; N, 5.54.
1-(4-{[(4R)-3-(tert-Butoxycarbonyl)-2,2-dimethyl-1,3-oxazolidin-4-yl]methoxy}benzyl)-5-oxo-L-prolyl-L-histidine (25). Prepared from compound 24 as a colorless oil in 97% yield. Rf 0.18 in EtOAc–MeOH, 1:1. [α]D −24.9 (c 0.8, MeOH). 1H-NMR (CD3OD): δ 8.90 (s, 1H, HNCHN), 7.40 (s, 1H, HNCHNCH), 7.05 (d, J = 8.3 Hz, 2H, Ph), 6.93 (d, J = 8.3 Hz, 2H, Ph), 4.87–4.75 (m, 2H, CHCOOH, NCHHPh), 4.17–3,81 (m, 6H, NCHCO, PhOCH2CHCH2O), 3.64 (d, J = 15.0 Hz, 1H, NCHHPh), 3.41–3.06 (m, 2H,HOOCCHCH2), 2.58–1.94 (m, 4H, 2xCH2), 1.58–1.45 (m, 15H, 5 × CH3). 13C-NMR: δ 173.9 (NCO), 172.8, 159.8, 153.4 (NCOO), 135.3, 131.5, 130.8, 129.5, 129.3, 118.4, 115.9, 95.0 (NC(CH3)2O), 82.2 (C(CH3)3), 67.3, 66.2, 61.1 (NCHCO), 57.5 (PhOCH2), 52.5 (NHCHCOOH), 45.8 (NCH2Ph), 30.8, 28.8 (CH3), 27.9, 27.7, 24.5, 24.2. MS (ESI): m/z = 586 (100) (M+Η+). Anal. calcd for C29H39N5O8: C, 59.47; H, 6.71; N, 11.96 Found: C, 59.52; H, 6.68; N, 12.05.
3.7. General Procedure for the Preparation of Compounds 17a–c, 23
To a stirred solution of methylester (compounds 16a–c, 22) (1 mmol), in MeOH (3 mL), an aq. solution of 2N NaOH (0.5 ml, 1 mmol) was added and the reaction mixture was left stirring at rt for 2–3 h. Upon completion of the reaction, the MeOH was evaporated under reduced pressure and the residual product was diluted with H2O and extracted with Et2O (1 × 10 mL). The aqueous phase was acidified with 1 N HCl aq. solution at pH 2 and extracted with EtOAc (2 × 10 mL). The combined organic phases were neutralized by washing with brine and H2O, dried over Na2SO4 and evaporated under reduced pressure to afford the carboxylic products in quantitative yield and high purity.
1-[4-(2-Methoxyethoxy)benzyl]-5-oxo-L-proline (17a). Prepared from methylester 16a as a colorless oil in 94% yield. Rf 0.36 in EtOAc–MeOH, 3:2. [α]D +43.8 (c 1, CHCl3). 1H-NMR (CDCl3): δ 8.77 (bs, 1H, COOH), 7.13 (d, J = 8.6 Hz, 2H, Ph), 6.85 (d, J = 8.6 Hz, 2H, Ph), 5.02 (d, J = 14.7 Hz, 1H, CHHPh), 4.11–4.06 (m, 2H, PhOCH2), 3.99–3.93 (m, 1H, NCHCOO), 3.91 (d, J = 4.7 Hz, 1H, CHHPh), 3.77–3.72 (m, 2H, CH2OCH3), 3.44 (s, 3H, OCH3), 2.68–2.09 (m, 4H, 2 × CH2). 13C-NMR: δ 176.2 (NCO), 174.1(COOH), 158.4, 129.9, 127.6, 114.7, 70.9 (CH2OCH3), 67.1 (OCH2), 59.1 (NCH), 58.5 (OCH3), 45.0 (CH2Ph), 29.6, 22.8. MS (ESI): m/z = 292 (100) (M−Η+). Anal. calcd for C15H19NO5: C, 61.42; H, 6.53; N, 4.78 Found: C, 61.38; H, 6.57; N, 4.82.
1-[4-(3-Methoxypropoxy)benzyl]-5-oxo-L-proline (17b). Prepared from methylester 16b, as a colorless oil in 99% yield. Rf 0.24 in EtOAc–MeOH, 3:2. [α]D +17.2 (c 1, CHCl3). 1H-NMR (CDCl3): δ 9.62 (bs, 1H, COOH), 7.13 (d, J = 8.6 Hz, 2H, Ph), 6.83 (d, J = 8.6 Hz, 2H, Ph), 5.05 (d, J = 4.7 Hz, 1H, CHHPh), 4.04–3.94 (m, 3H, NCHCOO, PhOCH2), 3.91 (d, J = 14.7 Hz, 1H, CHHPh), 3.56 (t, J = 6.2 Hz, 2H, CH2OCH3), 3.35 (s, 3H, OCH3), 2.66–2.09 (m, 4H, 2 × CH2), 2.09–1.96 (m, 2H, CH2CH2CH2). 13C NMR: δ 176.2 (NCO), 174.2 (COOH), 158.6, 129.9, 127.2, 114.6, 69.2 (CH2OCH3), 64.7 (PhOCH2), 58.6 (NCH), 58.5 (OCH3), 45.0 (CH2Ph), 29.7, 29.4, 22.8. MS (ESI): m/z = 306 (100) (M−Η+). Anal. calcd for C16H21NO5: C, 62.53; H, 6.89; N, 4.56 Found: C, 62.58; H, 6.77; N, 4.53.
1-[4-(4-Methoxybutoxy)benzyl]-5-oxo-L-proline (17c). Prepared from methylester 16c, as a colorless oil in 96% yield. Rf 0.30 in EtOAc–MeOH, 3:2. [α]D +13.3 (c 1, CHCl3). 1H-NMR (CDCl3): δ 9.98 (bs, 1H, COOH), 7.12 (d, J = 8.6 Hz, 2H, Ph), 6.81 (d, J = 8.6 Hz, 2H, Ph), 5.03 (d, J = 14.7 Hz, 1H, NCHHPh), 4.00–3.88 (m, 4H, NCHHPh, NCH, PhOCH2), 3.44 (t, J = 6.2Hz, 2H, CH2OCH3), 3.34 (s, 3H, OCH3), 2.65–2.08 (m, 4H, 2xCH2), 1.85–1.68 (m, 4H, (OCH2CH2)2). 13C-NMR: δ 176.2 (NCO), 173.8 (COO), 158.6, 129.9, 127.1, 114.6, 72.3 (CH2OCH3), 67.5 (PhOCH2), 58.4 (NCH), 58.3 (OCH3), 45.0 (CH2Ph), 29.6, 26.0, 25.8, 22.8. MS (ESI): m/z = 320 (100) (M−Η+). Anal. calcd for C17H23NO5: C, 63.54; H, 7.21; N, 4.36 Found: C, 63.38; H, 7.19; N, 4.42.
1-(4-{[(4R)-3-(tert-Butoxycarbonyl)-2,2-dimethyl-1,3-oxazolidin-4-yl]methoxy}benzyl)-5-oxo-L-proline (23). Prepared from methylester 22, as a colorless oil in 94% yield. Rf 0.43 in EtOAc–MeOH, 1:1. [α]D −11.5 (c 1, CHCl3). 1H-NMR (CDCl3): δ 7.62 (bs, 1H, COOH), 7.13 (d, J = 7.4 Hz, 2H, Ph), 6.88 (d, J = 4 Hz, 2H, Ph), 5.03 (dd, J1 = 14.6 Hz, J2 = 3.1 Hz, 1H, NCHHPh), 4.28–3.80 (m, 7H, NCHHPh, NCHCOOH, OCH2CHCH2O), 4.68–2.15 (m, 4H, 2 × CH2), 1.64-1.52 (m, 15H, 5 × CH3). 13C-NMR: δ 176.1 (NCO), 173.9 (COOH), 158.1, 152.4 (NCOO), 129.9, 127.6, 114.8, 93.6 (OC(CH3)2N), 80.9 (C(CH3)3), 66.0, 65.1, 58.4 (NCHCOOH), 55.9 (PhOCH2), 45.0 (NCH2Ph), 29.6, 28.3 (CH3), 27.4, 24.2, 22.8. MS (ESI): m/z = 447 (100) (M-Η+). Anal. calcd for C23H32N2O7: C, 61.59; H, 7.19; N, 6.25 Found: C, 61.53; H, 7.24; N, 6.32.
3.8. General Procedure for the Preparation of Compounds 18a–c, 24
To a stirred solution of carboxylic acids (compounds 17a–c, 23) (1 mmol) in CH2Cl2 (10 mL) at 0 °C, 1-hydroxybenzotriazole (HOBt) (0.135 g, 1 mmol), freshly prepared benzyl 3-trityl-L-histidinate (42) (0.537 g, 1.1 mmol), Et3N (0.154 mL, 1.1 mmol) and N-ethyl-N-dimethylaminopropyl-carbodiimide hydrochloride (EDC.HCl) (0.210 g, 1.1 mmol) were added consecutively. The reaction mixture was left stirring at 0 °C for 1 h and then warmed to room temperature and left stirring for 18 h. The solvents were evaporated under reduced pressure and the crude product was dissolved in EtOAc (20 mL). The organic layer was washed with 5% aq. H2SO4, H2O, 5% aq. NaHCO3, and brine. After drying over Na2SO4 and evaporation of the solvent in vacuo, the crude ester was purified using column chromatography with the appropriate solvent systems as it will be defined in each case below.
Benzyl 1-[4-(2-Methoxyethoxy)benzyl]-5-oxo-L-prolyl-3-trityl-L-histidinate (18a). Prepared from compound 17a. Eluent EtOAc–MeOH 9:1. Yield: 55% (colorless oil); Rf 0.55 in EtOAc–MeOH 9:1; [α]D +3.8 (c 1, CHCl3). 1H-NMR (CDCl3): δ 8.22 (d, J = 7.8 Hz, 1H, NH), 7.39 (d, J = 1.3 Hz, 1H, NCHN), 7.33–7.03 (m, 22H, Ph), 6.80 (d, J = 8.6 Hz, 2H, Ph), 6.47 (s, 1H, NCHNCHC), 5.01 (d, J = 14.3 Hz, 1H, CHHPh), 5.00 (s, 2H, OCH2Ph), 4.86–4.77 (m, 1H, NHCHCOO), 4.12–4.04 (m, 2H, PhOCH2), 3.89–3.83 (m, 1H, NCHCON), 3.75 (d, J = 14.3 Hz, 1H, CHHPh), 3.74–3.69 (m, 2H, CH2OCH3), 3.43 (s, 3H, OCH3), 3.13–2.91 (m, 2H, COCHCH2CHN), 2.68–1.98 (m, 4H, 2 × CH2). 13C-NMR: δ 175.5 (NCO), 171.6, 170.6, 158.2, 142.0, 138.7, 135.9, 135.2, 129.9, 129.6, 128.5, 128.4, 128.3, 128.2, 128.1, 119.5, 119.4, 114.6, 75.4 (NCPh), 70.9 (CH2OCH3), 67.1 (OCH2), 67.0 (OCH2Ph), 60.0 (NCH), 59.2 (OCH3), 52.5 (HNCHCO), 44.5 (CH2Ph), 29.7, 28.8 (NHCHCH2C), 23.2. MS (ESI): m/z = 763 (100) (M+Η+). Anal. calcd for C47H46N4O6: C, 74.00; H, 6.08; N, 7.34 Found: C, 74.15; H, 6.16; N, 7.27.
Benzyl 1-[4-(3-Methoxypropoxy)benzyl]-5-oxo-L-prolyl-3-trityl-L-histidinate (18b). Prepared from compound 17b. Eluent EtOAc–MeOH 9:1. Yield: 68% (colorless oil); Rf 0.63 in EtOAc–MeOH 9:1; [α]D +8.5 (c 1, CHCl3). 1H-NMR (CDCl3): δ 8.19 (d, J = 7.8 Hz, 1H, NH), 7.40 (d, J = 1.3 Hz, 1H, NCHN), 7.34–7.04 (m, 22H, Ph), 6.78 (d, J = 8.7 Hz, 2H, Ph), 6.48 (d, J = 1.2 Hz, 1H, NCHCN), 5.02 (d, J = 14.5 Hz, 1H, NCHHPh), 5.01 (s, 2H, CH2Ph), 4.00 (t, J = 6.3 Hz, 2H, PhOCH2), 3.89–3.83 (m, 1H, NCHCO), 3.76 (d, J = 14.5 Hz, 1H, NCHHPh), 3.53 (t, J = 6.2 Hz, 2H, CH2OCH3), 3.34 (s, 3H, OCH3), 3.14–2.92 (m, 2H, HNCHCH2), 2.69–2.08 (m, 4H, 2 × CH2), 2.05–1.95 (m, 2H, CH2CH2CH2). 13C-NMR: δ 175.5 (NCO), 171.6, 170.6, 158.4, 142.1, 138.7, 136.0, 135.2, 130.0, 129.6, 128.5, 128.3, 128.2, 128.1, 128.0, 119.5, 114.5, 75.4 (CPh), 69.2 (CH2OCH3), 67.0 (OCH2Ph), 64.7 (PhOCH2), 60.0 (NCH), 58.7 (OCH3), 52.5 (HNCHCO), 44.5 (CH2Ph), 29.8, 29.5 (CH2CH2CH2), 28.9 (NHCHCH2C), 23.2. MS (ESI): m/z = 777 (100) (M+Η+). Anal. calcd for C48H48N4O6: C, 74.21; H, 6.23; N, 7.21 Found: C, 74.11; H, 6.32; N, 7.28.
Benzyl 1-[4-(4-Methoxybutoxy)benzyl]-5-oxo-L-prolyl-3-trityl-L-histidinate (18c). Prepared from compound 17c. Eluent EtOAc–MeOH 9:1. Yield: 63% (colorless oil); Rf 0.56 in EtOAc–MeOH 9:1; [α]D +3.8 (c 1, CHCl3). 1H-NMR (CDCl3): δ 8.21 (d, J = 7.9 Hz, 1H, NH), 7.40 (d, J = 1.3 Hz, 1H, NCHN), 7.35–7.04 (m, 22H, Ph), 6.76 (d, J = 8.6 Hz, 2H, Ph), 6.48 (d, J = 1.1 Hz, 1H, NCHCN), 5.02 (d, J = 14.5 Hz, 1H, NCHHPh), 5.01 (s, 2H, COOCH2Ph), 4.87–4.79 (m, 1H, NCHCOO), 3.92 (t, J = 5.9 Hz, 2H, PhOCH2), 3.88–3.83 (m, 1H, NCH), 3.76 (d, J = 14.5 Hz, 1H, NCHHPh), 3.43 (t, J = 6.1 Hz, 2H, CH2OCH3), 3.34 (s, 3H, OCH3), 3.13–2.92 (m, 2H, CHCH2CN), 2.65–1.98 (m, 4H, 2 × CH2), 1.86–1.65 (m, 4H, (OCH2CH2)2). 13C-NMR: δ 175.4 (NCO), 171.6, 170.5, 158.4, 142.0, 138.7, 135.9, 135.2, 129.9, 129.6, 128.4, 128.3, 128.1, 128.0, 127.9, 119.4, 114.5, 75.3 (CPh), 72.2 (CH2OCH3), 67.5 (OCH2Ph), 66.9 (PhOCH2), 59.9 (NCH), 58.5 (OCH3), 52.5 (HNCHCO), 44.5 (CH2Ph), 29.7, 28.8, 26.1, 25.9, 23.2. MS (ESI): m/z = 791 (100) (M+Η+). Anal. calcd for C49H50N4O6: C, 74.41; H, 6.37; N, 7.08 Found: C, 74.35; H, 6.42; N, 7.18.
Benzyl 1-(4-{[(4R)-3-(tert-Butoxycarbonyl)-2,2-dimethyl-1,3-oxazolidin-4-yl]methoxy}benzyl)-5-oxo-L-prolyl-3-trityl-L-histidinate (24). Prepared from compound 23. Eluent EtOAc-petroleum ether (bp. 40–60 °C) 9:1, followed by EtOAc. Yield: 60% (colorless oil); Rf 0.64 in EtOAc; [α]D −16.4 (c 1, CHCl3). 1H-NMR (CDCl3): δ 8.23 (t, J = 8.3 Hz, 1H, NH), 7.40–6.80 (m, 25H, Ph), 6.47 (s, 1H, Ph), 5.05–4.98 (m, 3H, COOCH2, NCHHPh), 4.86–4.77 (m, 1H, HNCHCOO), 4.26–3.69 (m, 7H, NCHHPh, NCHCONH, PhOCH2CHCH2O), 3.13–2.92 (m, 2H, HNCHCH2C), 2.68–2.03 (m, 4H, 2×CH2), 1.61–1.48 (m, 15H, 5 × CH3). 13C-NMR: δ 175.5 (NCO), 171.6, 170.5, 158.0, 152.2 (NCOO), 142.0, 138.6, 135.8, 135.2, 130.0, 129.9, 129.6, 128.6, 128.5, 128.3, 128.1, 119.6, 114.7, 93.5 (OC(CH3)2N), 80.2 (C(CH3)3), 75.4 (CPh3), 67.0 (COOCH2Ph), 66.1, 65.3, 60.0 (NCHCO), 55.7 (PhOCH2), 52.5 (NHCHCOO), 44.5 (NCH2Ph), 29.7, 28.5 (CH3), 27.5, 24.2, 23.3, 23.0. MS (ESI): m/z = 919 (100) (M+Η+). Anal. calcd for C55H59N5O8: C, 71.95; H, 6.48; N, 7.63 Found: C, 72.07; H, 6.24; N, 7.58.
Μethyl 1-(4-{[(4R)-3-(tert-Butoxycarbonyl)-2,2-dimethyl-1,3-oxazolidin-4-yl]methoxy}benzyl)-5-oxo-L-prolinate (22). To a stirred solution of alcohol (tert-butyl (4R)-4-(hydroxymethyl)-2,2-dimethyl-1,3-oxazolidine-3-carboxylate) [33], (0.231g, 1 mmol) in dry toluene (5 mL), compound 21 (0.249 g, 1 mmol), DEAD (0.19 mL, 1.2 mmol), and PPh3 (0.314 g, 1.2 mmol) were added consecutively and the reaction mixture was refluxed for 15 h under argon. After evaporation of the solvent under reduced pressure the product was purified using column chromatography and EtOAc–petroleum ether (bp. 40–60 °C) 7:3 as eluent and isolated as a colorless oil in 60% yield (0.278 g). Rf 0.45 in EtOAc–petroleum ether (bp. 40–60 °C) 7:3; [α]D −17.7 (c 1, CHCl3). 1H-NMR (CDCl3): δ 7.10 (d, J = 7.9 Hz, 2H, Ph), 6.70 (bs, 2H, Ph), 4.94 (d, J = 14.6 Hz, 1H, NCHHPh), 4.21–3.81 (m, 7H, NCHHPh, NCHCOOCH3, PhOCH2CHCH2O), 3.67 (s, 3H, OCH3), 2.64–2.02 (m, 4H, 2 × CH2), 1.60–1.48 (m, 15H, 5 × CH3). 13C-NMR: δ 174.8 (NCO), 172.2 (COOCH3), 158.1, 152.2 (NCOO), 129.8, 127.9, 114.7, 93.4 (NC(CH3)2O), 80.4 (C(CH3)3), 66.0, 65.1, 58.5 (NCHCOO), 55.6 (PhOCH2), 52.3 (COOCH3), 44.8 (NCHPh), 29.5, 28.3 (C(CH3)3), 27.4, 24.2, 22.6. MS (ESI): m/z = 480 (100) (M+NΗ4+). Anal. calcd for C24H34N2O7: C, 62.32; H, 7.41; N, 6.06 Found: C, 62.28; H, 7.39; N, 6.15.
1-(4-{[(2S)-2-Ammonio-3-Hydroxypropyl]oxy}benzyl)-5-oxo-L-prolyl-L-histidine hydrochloride (26). To a stirred solution of compound 25 (21 mg, 0.036 mmol) in dioxane (1 mL) at 0 °C, a 4 M solution of HCl in dioxane (0.108 mL, 0.432 mmol) was added and the reaction mixture was left stirring at room temperature for 1 h. After evaporation of the solvent under reduced pressure, the product was precipitated by the addition of Et2O and recrystallized from dioxane/Et2O twice. It was obtained as a white gummy solid in 90% yield (16 mg). [α]D +3.4 (c 0.7, H2O). 1H-NMR (D2O): δ 8.87 (s, 1H, HNCHN), 7.32 (s, 1H, HNCHNCH), 7.14 (d, J = 8.4 Hz, 2H, Ph), 6.99 (d, J = 8.4 Hz, 2H, Ph), 4.75–4.70 (m, 2H, CHCOOH, NCHHPh), 4.33–4.19 (m, 2H, PhOCH2), 4.16–4.12 (m, 1H, NCH), 3.98–3.80 (m, 4H, NCHHPh, CHNH2, CH2OH), 3.33 (dd, J1 = 5.4 Hz, J2 = 15.6 Hz, 1H, HNCHCHHC), 3.15 (dd, J1 = 9.0 Hz, J2 = 15.5 Hz, 1H, HNCHCHHC), 2.61-1.87 (m, 4H, 2 × CH2). 13C-NMR: δ 179.6 (NCO), 174.2, 173.6, 158.0, 134.2, 130.3, 129.5, 128.7, 117.7, 115.5, 65.6 (PhOCH2), 61.3 (NCHCO), 59.3 (CH2OH), 52.8, 52.3, 45.7 (NCH2Ph), 30.4, 26.6, 23.2. MS (ESI): m/z = 446 (100) (M+Η+). Anal. calcd for C21H28N5O6Cl: C, 52.34; H, 5.86; N, 14.53 Found: C, 52.28; H, 5.92; N, 14.47.
Benzyl N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-1-trityl-L-histidinate (41). To a solution of Fmoc-S-His(Trt)-OH (0.20 g, 0.33 mmol) in DMF (1 mL) Cs2CO3 (53 mg, 0.162 mmol) was added with some drops of H2O to dissolve the inorganic salt. The solvent was evaporated under reduced pressure to dryness and the residue, dissolved in DMF (3 mL) was stirred for 5 min at rt, followed by the addition of benzyl bromide (44 μL, 0.371 mmol). After stirring overnight at rt and evaporation of DMF under reduced pressure, the residue was dissolved in EtOAc and the organic phase was washed with H2O, 5% aq. solution of NaHCO3 and H2O. The organic layer was dried over Na2SO4, evaporated in vacuo and the residue was purified by column chromatography on silica gel, eluting with EtOAc-petroleum ether (bp. 40–60 °C) 1:1. The product was isolated as colorless oil in 87% yield (0.20 g). Rf 0.50 in EtOAc–petroleum ether (bp. 40–60 °C) 1:1; [α]D −2.7 (c 1, CHCl3). 1H-NMR (CDCl3): δ 7.77–7.06 (m, 29H, Ph, NCHN), 6.61 (d, J = 8.2 Hz, 1H, NH), 6.54 (s, 1H, NCHNCHC), 5.07 (dd, J1 = 12.5 Hz, J2 = 14.9 Hz, 2H, OCH2Ph), 4.73–4.64 (m, 1H, HNCHCO), 4.40–4.19 (m, 3H, CHCH2OCO), 3.12 (d, J = 5.0Hz, 2H, CH2CHN). 13C-NMR: δ 171.2 (HNCHCOO), 156.2 (COONH), 144.1, 143.9, 142.0, 141.2, 138.4, 135.8, 135.4, 129.7, 128.5, 128.2, 128.1, 127.6, 127.0, 125.4, 125.3, 119.8, 75.6 (CPh), 67.2 (CH2COONH), 66.9 (OCH2Ph), 54.3 (HNCHCOO), 47.1 (CHCH2COO), 29.7 (CH2CHN). MS (ESI): m/z = 710 (100) (M+Η+). Anal. calcd for C47H39N3O4: C, 79.53; H, 5.54; N, 5.92 Found: C, 79.60; H, 5.58; N, 5.85.
Benzyl-3-trityl-L-histidinate (42). Compound 41 (0.15 g, 0.21 mmol) was dissolved in EtOH (1.5 mL) and Et2NH (153 μL, 1.48 mmol) was added. After leaving overnight at rt the solvent was evaporated under reduced pressure. The crude product was redissolved in water, washed by EtOAc (3 × 5mL) and the combined organic layers were dried over Na2SO4 and evaporated in vacuo. After purification by column chromatography and using EtOAc–MeOH (9:1) as eluent the product was isolated as colorless oil in 82% yield (84 mg). Rf 0.37 in EtOAc–MeOH 9:1; [α]D −11.7 (c 1, CHCl3). 1H-NMR (CDCl3): δ 7.32–7.08 (m, 21H, 4 × Ph, NCHN), 6.54 (s, 1H, NCHNCHC), 5.04 (dd, J1 = 12.3 Hz, J2 = 20.8 Hz, 2H, OCH2Ph), 3.90–3.85 (m, 1H, H2NCH), 3.08–2.82 (m, 2H, CH2CHN). 13C-NMR: δ 174.4 (COO), 142.3, 138.6, 137.0, 135.6, 129.7, 128.5, 128.3, 128.2, 128.1, 128.0, 119.5, 75.2 (CPh), 66.6 (CH2Ph), 54.6 (H2NCH), 32.9 (CH2CHN). MS (ESI): m/z = 488 (100) (M+Η+). Anal. calcd for C32H29N3O2: C, 78.82; H, 5.99; N, 8.62 Found: C, 78.68; H, 5.79; N, 8.74.
4. Conclusions
In this paper the synthesis of new, optically active 2-pyrrolidinones starting from the natural chiral synthon of S-pyroglutamic acid is described.
Acknowledgments
Financial support from the EPEAEK Program “Organic Synthesis and Applications in Chemical Industry” as well as from the Special Research Account of the University of Athens is highly appreciated.
References
- Winbal, B. Piracetam: A review of pharmacological properties and clinical uses. CNS Drug Rev. 2005, 11, 169–182. [Google Scholar]
- Singh, P.; Dimitriou, V.; Malajan, R.P.; Crossley, A.W. Double-blind comparison between doxapram and pethidine in the treatment of postanaesthetic shivering. Br. J. Anaesth. 1993, 71, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Ahsan, W.; Alam, M.S.; Ali, R.; Srivastava, K. Design, synthesis and evaluation of anticonvulsant activity of pyridinyl-pyrrolidones: A pharmacophore hybrid approach. Arch. Pharm. Chem. Life Sci. 2012, 345, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Volkhard, A.; Eisert, W.; Himmelsbach, F.; Linz, G.; Mueller, T.; Pieper, H.; Weisenberger, J. 2-Pyrrolidinones, Pharmaceutical composition containing these compounds and processes for preparing them. U.S. Patent 5455348, 3 November 1995. [Google Scholar]
- Zhongli, G.; Ryan, H.; David, S. Substituted tetrahydropyran spiro pyrrolidinone and piperidinone, preparation and therapeutic use thereof. U.S. Patent 20120238757, 20 September 2012. [Google Scholar]
- Ohfune, Y.; Tomita, M. Total synthesis of (-) Domoic Acid. A revision of the original structure. J. Am. Chem. Soc. 1982, 104, 3511–3513. [Google Scholar] [CrossRef]
- Petersen, J.S.; Rapoport, H. Chirospecific syntheses of (+)- and (-) Anatoxin a. J. Am. Chem. Soc. 1984, 106, 4539–4547. [Google Scholar] [CrossRef]
- Fang, F.; Danishefsky, J.S. Total synthesis of the angiotensin-converting enzyme inhibitor A58365: On the use of pyroglutamate as a chiral educt. Tetrahedron Lett. 1989, 30, 3621–3624. [Google Scholar] [CrossRef]
- Davies, G.S.; Dixon, J.D.; Doisneau, J.-M.D.; Prodger, C.J.; Sanganee, J.H. Synthesis and utility of the 3,3-dimethyl-5-substituted-2-pyrrolidinone “Quat” chiral auxiliary. Tetrahedron: Asymmetry 2002, 13, 647–658. [Google Scholar] [CrossRef]
- Coppola, G.M.; Schuster, H.F. Asymmetric Synthesis. Construction of Chiral Molecules Using Amino Acids; John Wiley: New York, NY, USA, 1987. [Google Scholar]
- Najera, C.; Yus, M. Pyroglutamic acid: A versatile block in asymmetric synthesis. Tetrahedron: Asymmetry 1999, 10, 2245–2303. [Google Scholar] [CrossRef]
- Panday, S.K.; Prasad, J.; Dikshit, K.D. Pyroglutamic acid: A unique chiral synthon. Tetrahedron: Asymmetry 2009, 20, 1581–1632. [Google Scholar] [CrossRef]
- Moutevelis-Minakakis, P.; Gianni, M.; Stougiannou, H.; Zoumpoulakis, P.; Zoga, A.; Vlahakos, E.; Iliodromitis, E.; Mavromoustakos, T. Design and synthesis of novel antihypertensive drugs. Bioorg. Med. Chem. Lett. 2003, 13, 1737–1740. [Google Scholar] [CrossRef]
- Mavromoustakos, T.; Moutevelis-Minakakis, P.; Kokotos, C.G.; Kontogianni, P.; Politi, A.; Zoumpoulakis, P.; Findlay, A.; Cox, A.; Balmforth, A.; Zoga, A.; et al. Synthesis, binding studies and in vivo biological evaluation of novel non-peptide antihypertensive analogues. Bioorg. Med. Chem. 2006, 14, 4353–4360. [Google Scholar] [CrossRef] [PubMed]
- Mavromoustakos, T.; Fotakis, C.; Siapi, E.; Potamitis, C.; Viras, K.; Moutevelis-Minakakis, P.; Kokotos, G.; Durgasi, S.; Grdadolnik, S.; Sartori, B.; et al. Interactions at the bilayer interface and receptor site induced by the novel synthetic pyrrolidinone analog MMK3. BBA-Biomembranes 2010, 1798, 422–432. [Google Scholar]
- Moutevelis-Minakakis, P.; Papavassilopoulou, E.; Michas, G.; Georgikopoulou, K.; Ragoussi, M.E.; Neophytou, N.; Zoumpoulakis, P.; Mavromoustakos, T.; Hadjipavlou-Litina, D. Synthesis, in silico docking experiments of new 2-pyrrolidinone derivatives and study of their anti-inflammatory activity. Bioorg. Med. Chem. 2011, 19, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Masuda, T.; Haupt, A.; Ohno, M.; Shiraki, T.; Sugiura, Y.; Maeda, K. Synthetic studies on antitumor, antibiotic bleomycin. 27. Man-designed bleomycin with altered sequence specificity in DNA cleavage. J. Am. Chem. Soc. 1990, 112, 838–845. [Google Scholar] [CrossRef]
- Roush, W.R.; Russo-Rodriguez, S. Trichothecene Degradation Studies. 2. Synthesis of [13-14C] Anguidine. J. Org. Chem. 1987, 52, 598–603. [Google Scholar] [CrossRef]
- Lee, Y.S.; Cho, D.J.; Kim, S.N.; Choi, J.H.; Park, H. Chiral synthesis of trans-1-aminoindolo[2,3-a]quinolizidine and trans-1-aminobenzo[a]quinolizidine derivatives from L-pyroglutamic acid. J. Org. Chem. 1999, 64, 9727–9730. [Google Scholar] [CrossRef]
- Bauer, J.; Brandenburg, K.; Zähringer, U.; Rademann, J. Chemical synthesis of a glycolipid library by a solid-phase strategy allows elucidation of the structural specificity of immunostimulation by rhamnolipids. Chem. Eur. J. 2006, 12, 7116–7124. [Google Scholar] [CrossRef] [PubMed]
- Chida, N.; Takeoka, J.; Ando, K.; Tsutsumi, N.; Ogawa, S. Stereoselective total synthesis of (+)-lactacystin from D-glucose. Tetrahedron 1997, 53, 16287–16298. [Google Scholar] [CrossRef]
- Elemes, Y.; Foote, C.S. Stepwise mechanisms in the ene reaction of α, β-unsaturated esters with N-phenyl-1,2,4-triazoline-3,5-dione and singlet oxygen. Intermolecular primary and secondary hydrogen isotope effects. J. Am. Chem. Soc. 1992, 114, 6044–6050. [Google Scholar] [CrossRef]
- Breña-Valle, L.J.; Sánchez, C.R.; Cruz-Almanza, R. Diastereoselective alkylation of 1-benzyl-(5S)-substituted 2-pyrrolidinones. Tetrahedron: Asymmetry 1996, 7, 1019–1026. [Google Scholar] [CrossRef]
- Salvatore, R.N.; Nagle, A.S.; Jung, K.W. Cesium effect: High chemoselectivity in direct N-alkylation of amines. J. Org. Chem. 2002, 67, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.C.; Cruickshank, P.A.; Boshart, G.L. A convenient synthesis of water-soluble carbodiimides. J. Org. Chem. 1961, 26, 2525–2528. [Google Scholar] [CrossRef]
- Oeveren, V.A.; Jansen, J.F.G.; Feringa, B.L. Enantioselective synthesis of natural dibenzylbutyrolactone lignans (−)-enterolactone, (−)-hinokinin, (−)-pluviatolide, (−)-enterodiol, and furofuran lignan (−) eudesmin via tandem conjugate addition to γ-alkoxybutenolides. J. Org. Chem. 1994, 59, 5999–6007. [Google Scholar] [CrossRef]
- Mandoli, A.; Calamante, M.; Feringa, B.L.; Salvadori, P. An insoluble polymer-bound phosphoramidite for the copper-catalysed enantioselective 1,4-additio of ZnEt2 to 2-cyclohexenone. Tetrahedron: Asymmetry 2003, 14, 3647–3650. [Google Scholar] [CrossRef]
- Zinieris, N.; Kokinaki, S.; Leontiadis, L.; Ferderigos, N. Synthesis of 4-(Fmoc-aminoacyloxymethyl)phenoxyacetic acids for use in solid-phase peptide synthesis. Synthesis 2006, 16, 1789–2793. [Google Scholar]
- Chong, J.M.; Sokoll, K.K. Enantiotopic group differentiation and kinetic resolution: Asymmetric reduction of meso-1, 3- dihalides. Tetrahedron Lett. 1992, 33, 879–882. [Google Scholar] [CrossRef]
- Ziegler, E.F.; Klein, S.I.; Pati, U.K.; Wang, T.F. Acyclic diastereoselection as a synthetic route to quassinoids: A Claisen rearrangement based strategy for bruceantin. J. Am. Chem. Soc. 1985, 107, 2730–2737. [Google Scholar] [CrossRef]
- Güschke, R.; Stutz, S.; Heinzelmann, W.; Maibaum, J. The nonchiral bislactim diethoxy ether as a highly stereo-inducing synthon for sterically hindered, γ-branched α-amino acids: A practical, large-scale route to an intermediate of the novel rennin inhibitor Aliskiren. Helv. Chim. Acta 2003, 86, 2848–2870. [Google Scholar] [CrossRef]
- Thakkar, K.; Geahlen, R.L.; Cushman, M. Synthesis and protein-tyrosine kinase inhibitory activity of polyhydroxylated stilbene analogues of piceatannol. J. Med. Chem. 1993, 36, 2950–2955. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.; Zhang, Z.; Shao, F.; Carroll, P.J.; Joullié, M.M. Grignard reactions to chiral oxazolidine aldehydes. Tetrahedron 1996, 52, 11673–11694. [Google Scholar] [CrossRef]
- Pavé, G.; Usse-Versluys, S.; Viaud-Massuard, M.C.; Guillaumet, G. Synthesis of 3-aminochroman derivatives by radical cyclization. Org. Lett. 2003, 5, 4253–4256. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gisin, B.F.; Winter, D.P.; Makofske, R.; Kulesha, I.D.; Tzougraki, C.; Meienhofer, J. Facile synthesis of amino acid and peptide esters under mild conditions via cesium salts. J. Org. Chem. 1977, 42, 1286–1290. [Google Scholar] [CrossRef]
- Zvilichovsky, G.; Gurvich, V. Products of ozonolysis of L-1,4-cyclohexadienylalanine. Intramolecular cyclization and cyclization with hydroxylamine. The synthesis of two isomers of L-isoxazolylalanine. Tetrahedron 1997, 53, 4457–4468. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
Figure 1.
The structure of the new synthesized 2-pyrrolidinones.

Scheme 1.
Synthesis of products 11, 14, 15.

Reagents and Conditions: (i) NaBH4, dry EtOH; 94%; (ii) TES-Cl, Et3N, DMAP in CH2Cl2; 83%; (iii) NaH, Z-Cl in dry THF; 50%; (iv) TFA in CH2Cl2; 86%; (v) Moffatt oxidation: EDC.HCl, Pyr, TFA drops in dry Tol and dry DMSO, under Ar; (vi) Ph3P=CHCOOMe in dry THF, 1h reflux under Ar; 60%; (vii) H2, 10% Pd/C in MeOH, overnight; 97%; (viii) 30, NaH in dry DMF; 40%; (ix) LiBH4, dry THF; 87%; (x) TosCl, Et3N in CH2Cl2; 72%; (xi) ![Molecules 18 00050 i001]() , Cs2CO3 in dry DMF; 58%; (xii)
, Cs2CO3 in dry DMF; 58%; (xii) ![Molecules 18 00050 i002]() Cs2CO3 in dry DMF; 55% for 12; 30% for 13; (xiii) H2, 10% Pd/C in MeOH; 99% for 14; 90% for 15.
Cs2CO3 in dry DMF; 55% for 12; 30% for 13; (xiii) H2, 10% Pd/C in MeOH; 99% for 14; 90% for 15.
 , Cs2CO3 in dry DMF; 58%; (xii)
, Cs2CO3 in dry DMF; 58%; (xii)  Cs2CO3 in dry DMF; 55% for 12; 30% for 13; (xiii) H2, 10% Pd/C in MeOH; 99% for 14; 90% for 15.
Cs2CO3 in dry DMF; 55% for 12; 30% for 13; (xiii) H2, 10% Pd/C in MeOH; 99% for 14; 90% for 15. Scheme 2.
Synthesis of products 19a–c.

Reagents and Conditions: (i) 30, 35, 39, NaH in dry DMF; 66–73%; (ii) 2N NaOH in MeOH; 94–99%; (iii) 42, Et3N, HOBt, EDC.HCl in CH2Cl2; 55–68%; (iv) H2, 10% Pd/C; 92–99%.
Scheme 3.
Synthesis of product 26.

Reagents and Conditions: (i) ![Molecules 18 00050 i003]() , NaH in dry THF; 47%; (ii) H2, 10% Pd/C in MeOH; 99%; (iii)
, NaH in dry THF; 47%; (ii) H2, 10% Pd/C in MeOH; 99%; (iii) ![Molecules 18 00050 i004]() , PPh3, DEAD in dry Tol; 60%; (iv) 2N NaOH in MeOH; 94%;(v) 42, Et3N, HOBt, EDC.HCl in CH2Cl2; 48%; (vi) H2, 10% Pd/C in MeOH; 97%; (vii) 4M HCl/dioxane; 90%.
, PPh3, DEAD in dry Tol; 60%; (iv) 2N NaOH in MeOH; 94%;(v) 42, Et3N, HOBt, EDC.HCl in CH2Cl2; 48%; (vi) H2, 10% Pd/C in MeOH; 97%; (vii) 4M HCl/dioxane; 90%.
 , NaH in dry THF; 47%; (ii) H2, 10% Pd/C in MeOH; 99%; (iii)
, NaH in dry THF; 47%; (ii) H2, 10% Pd/C in MeOH; 99%; (iii)  , PPh3, DEAD in dry Tol; 60%; (iv) 2N NaOH in MeOH; 94%;(v) 42, Et3N, HOBt, EDC.HCl in CH2Cl2; 48%; (vi) H2, 10% Pd/C in MeOH; 97%; (vii) 4M HCl/dioxane; 90%.
, PPh3, DEAD in dry Tol; 60%; (iv) 2N NaOH in MeOH; 94%;(v) 42, Et3N, HOBt, EDC.HCl in CH2Cl2; 48%; (vi) H2, 10% Pd/C in MeOH; 97%; (vii) 4M HCl/dioxane; 90%. Scheme 4.
Synthesis of bromide 30.

Reagents and Conditions: (i) PBr3 in Et2O; 58%; (ii) ![Molecules 18 00050 i005]() , K2CO3, 18-Crown-6 in acetone; 76%; (iii) PBr3 in Et2O; 60%.
, K2CO3, 18-Crown-6 in acetone; 76%; (iii) PBr3 in Et2O; 60%.
 , K2CO3, 18-Crown-6 in acetone; 76%; (iii) PBr3 in Et2O; 60%.
, K2CO3, 18-Crown-6 in acetone; 76%; (iii) PBr3 in Et2O; 60%. Scheme 5.
Synthesis of bromide 35.

Reagents and Conditions: (i) ![Molecules 18 00050 i006]() , K2CO3 in acetone; 40%; (ii) 1N MeONa/MeOH; 38%; (iii) NaBH4 in dry THF; 80%; (iv) PBr3 in Et2O; 70%.
, K2CO3 in acetone; 40%; (ii) 1N MeONa/MeOH; 38%; (iii) NaBH4 in dry THF; 80%; (iv) PBr3 in Et2O; 70%.
 , K2CO3 in acetone; 40%; (ii) 1N MeONa/MeOH; 38%; (iii) NaBH4 in dry THF; 80%; (iv) PBr3 in Et2O; 70%.
, K2CO3 in acetone; 40%; (ii) 1N MeONa/MeOH; 38%; (iii) NaBH4 in dry THF; 80%; (iv) PBr3 in Et2O; 70%. Scheme 6.
Synthesis of bromide 39.

Reagents and Conditions: (i) PBr3 in Et2O; 60%; (ii) ![Molecules 18 00050 i007]() K2CO3, 18-Crown-6 in acetone; 67%; (iii) TMS-Cl, NaBr in CH3CN; 42%.
K2CO3, 18-Crown-6 in acetone; 67%; (iii) TMS-Cl, NaBr in CH3CN; 42%.
 K2CO3, 18-Crown-6 in acetone; 67%; (iii) TMS-Cl, NaBr in CH3CN; 42%.
K2CO3, 18-Crown-6 in acetone; 67%; (iii) TMS-Cl, NaBr in CH3CN; 42%. Scheme 7.
Synthesis of compound 42.

Reagents and Conditions: (i) Bn-Br, Cs2CO3 in DMF; 87%; (ii) Et2NH in abs. EtOH, 82%.
© 2013 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Moutevelis-Minakakis, P.; Papavassilopoulou, E.; Mavromoustakos, T. Synthesis of New Optically Active 2-Pyrrolidinones. Molecules 2013, 18, 50-73. https://doi.org/10.3390/molecules18010050
AMA Style
Moutevelis-Minakakis P, Papavassilopoulou E, Mavromoustakos T. Synthesis of New Optically Active 2-Pyrrolidinones. Molecules. 2013; 18(1):50-73. https://doi.org/10.3390/molecules18010050
Chicago/Turabian StyleMoutevelis-Minakakis, Panagiota, Eleni Papavassilopoulou, and Thomas Mavromoustakos. 2013. "Synthesis of New Optically Active 2-Pyrrolidinones" Molecules 18, no. 1: 50-73. https://doi.org/10.3390/molecules18010050