A Highly Sensitive Telomerase Activity Assay that Eliminates False-Negative Results Caused by PCR Inhibitors

Abstract
:1. Introduction
2. Results and Discussion
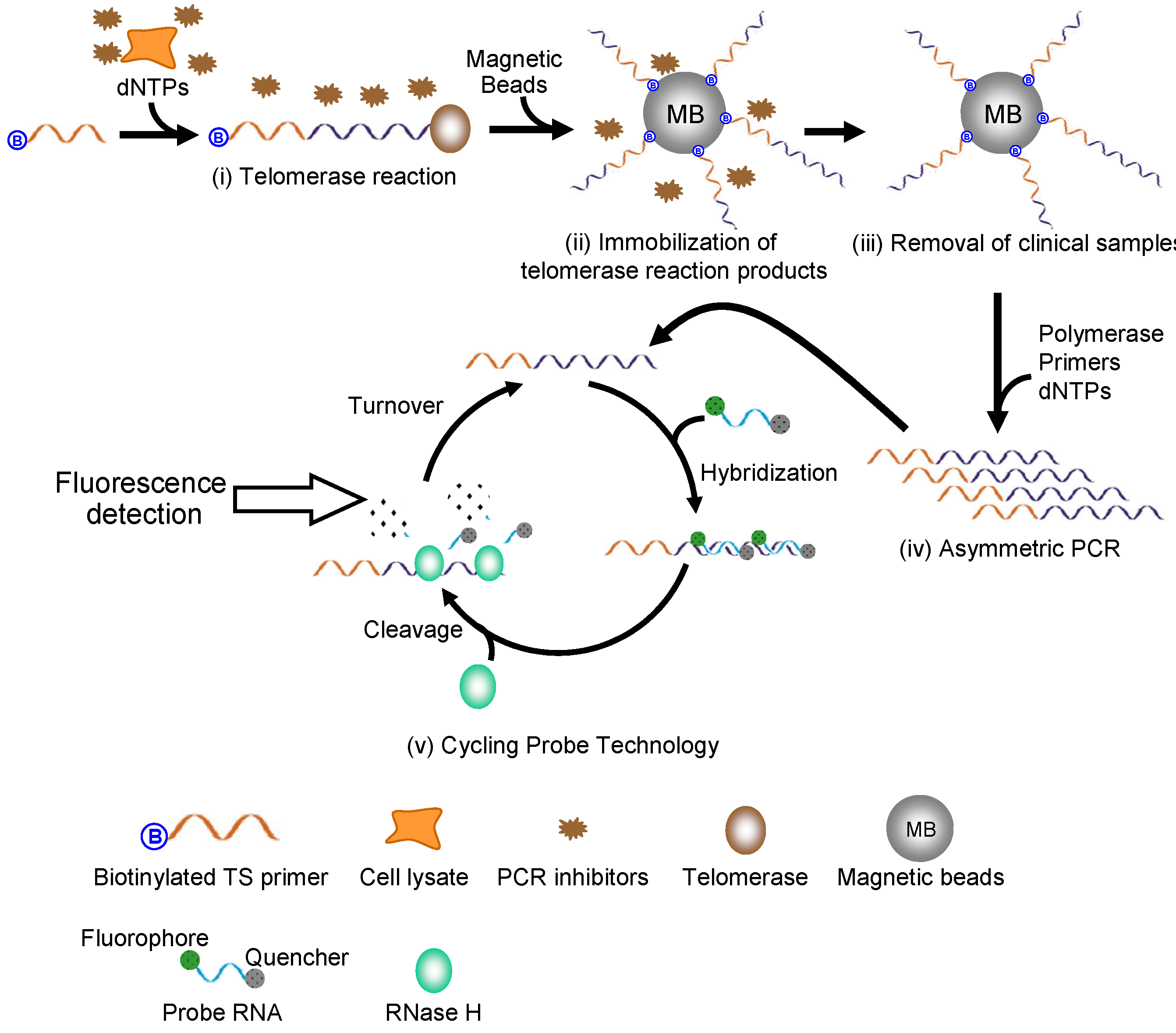
2.1. Detection Principle
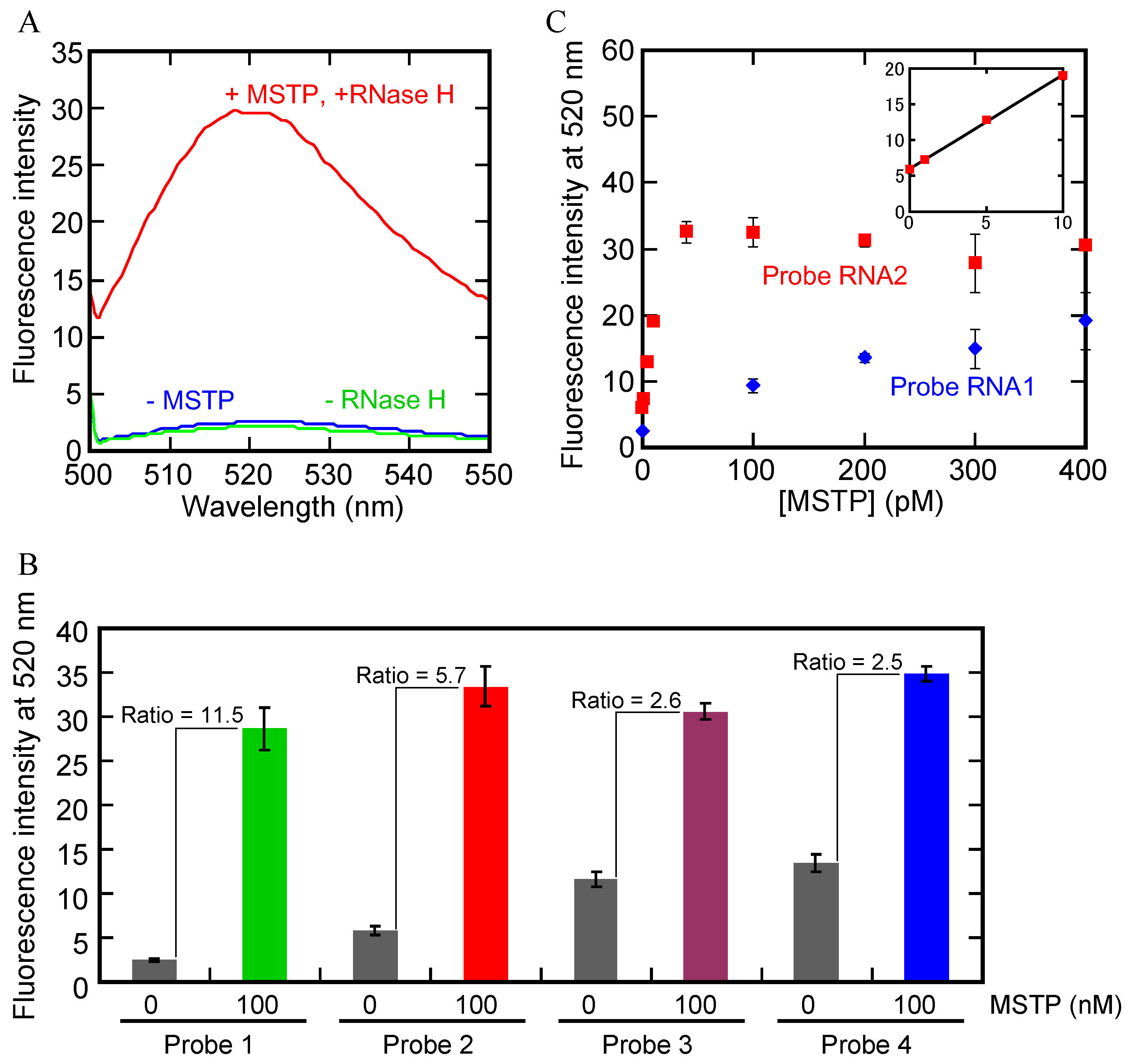
2.2. Design of the Probe RNA


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Modification |
|---|---|---|
| Probe 1 a | 5'-CCCUAA-3' | 5'-FITC/3'-Dabcyl |
| Probe 2 a | 5'-CCCUAACCC-3' | 5'-FITC/3'-Dabcyl |
| Probe 3 a | 5'-CUAACCCUAAC-3' | 5'-FITC/3'-Dabcyl |
| Probe 4 a | 5'-CCCUAACCCUAACCC-3' | 5'-FITC/3'-Dabcyl |
| MSTP b | 5'-(GGGTTA)16-3' | none |
| TS primer c | 5'-AATCCGTCGAGCAGAGTT-3' | none |
| Biotinylated TS primer d | 5'-AATCCGTCGAGCAGAGTT-3' | 5'-biotinylation |
| CX-ext e | 5'-GTGCCCTTACCCTTACCCTTACCCTAA-3' | none |


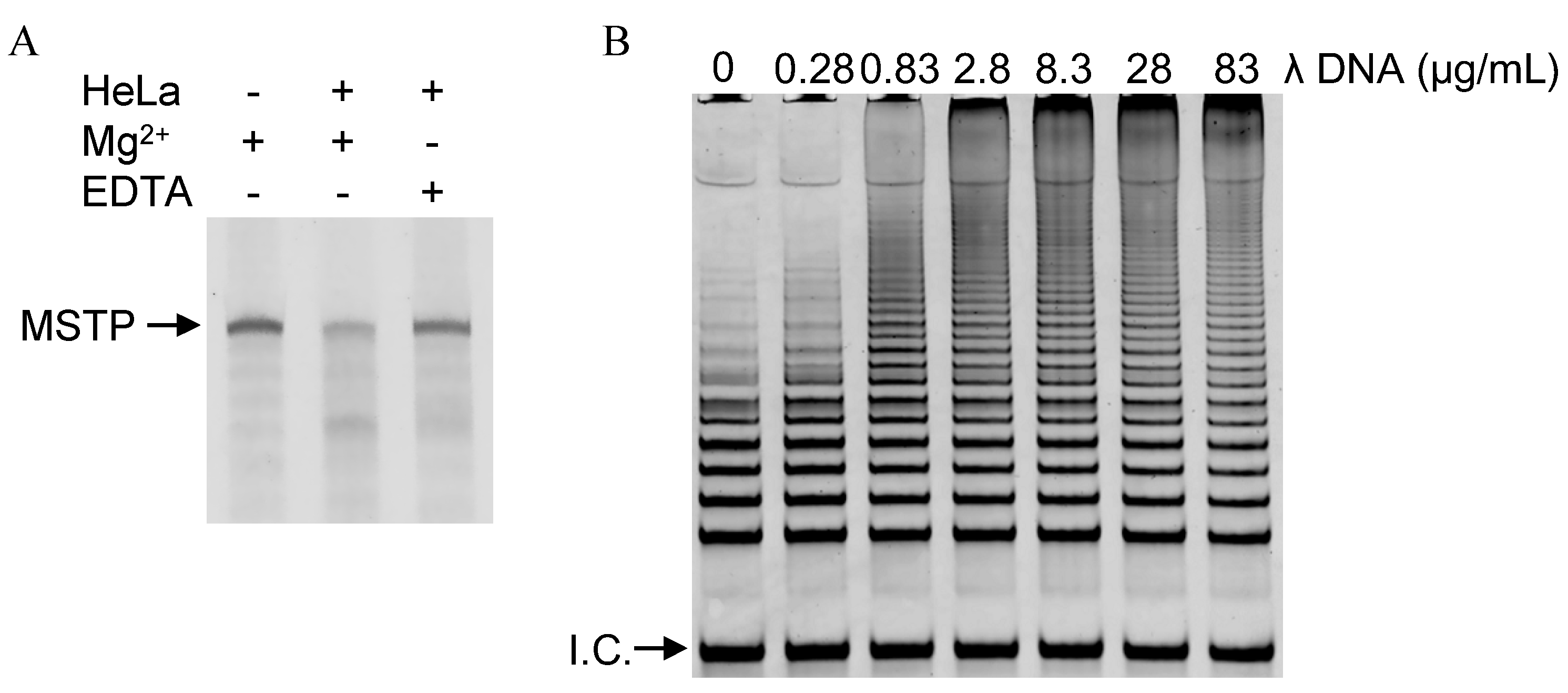
2.3. Inhibition of Degradation of Telomerase Reaction Products by Decoy DNA

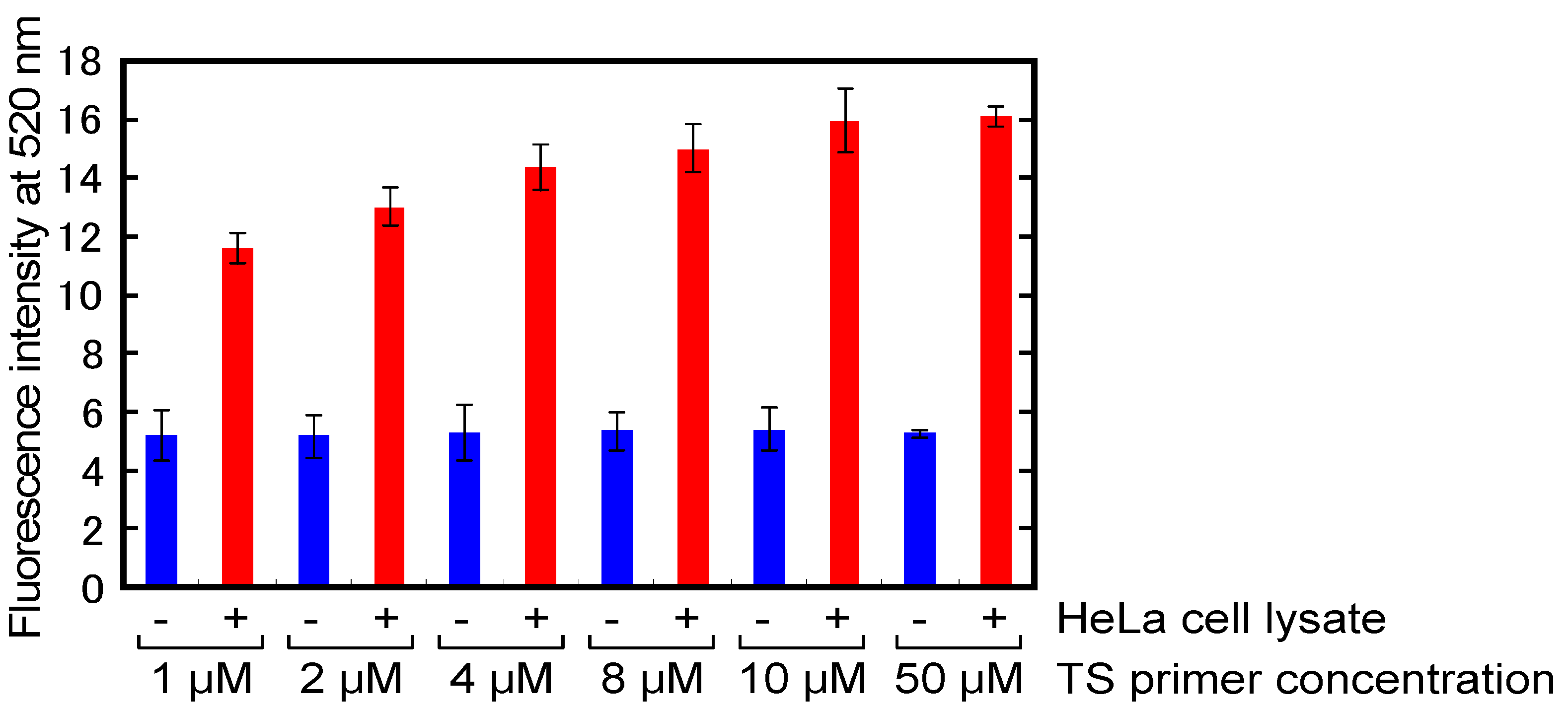
2.4. Optimization of Primers Concentrations for A-PCR

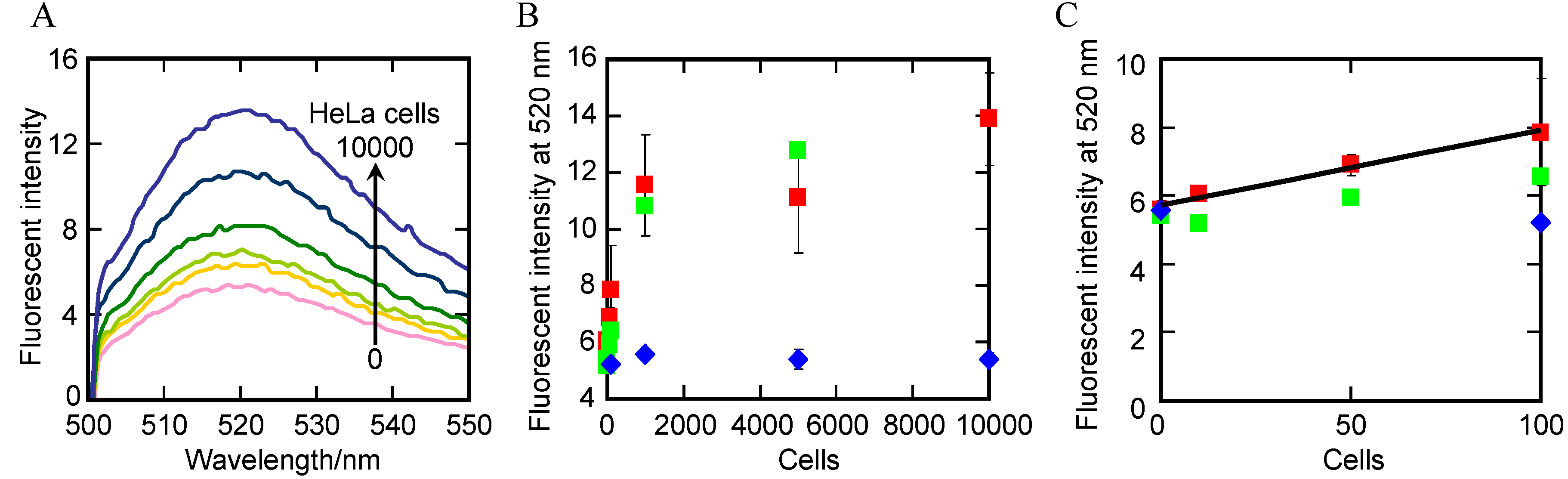
2.5. Detection of Telomerase Activity in Cells Lysate

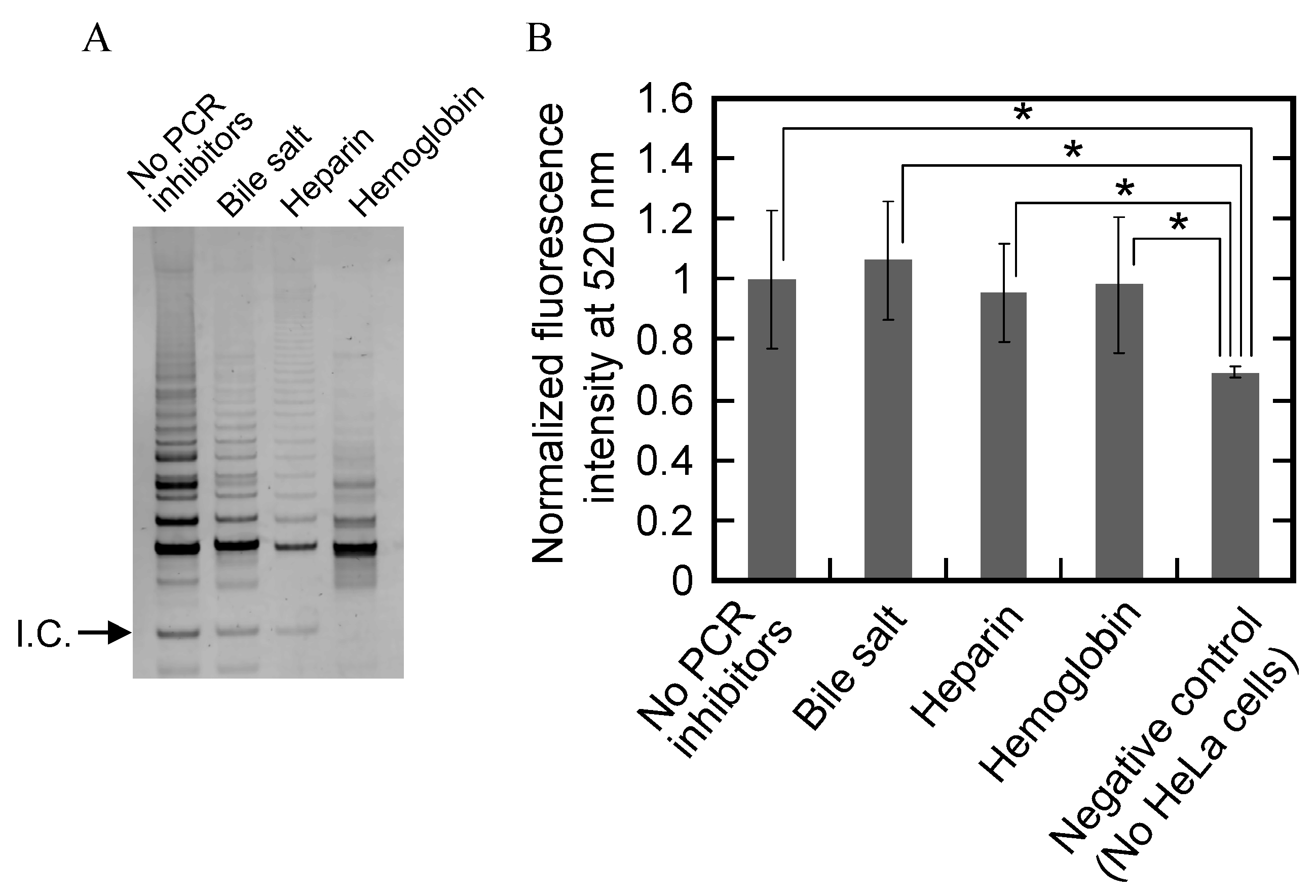
2.6. Eliminating False-Negative Results Caused by PCR Inhibitors

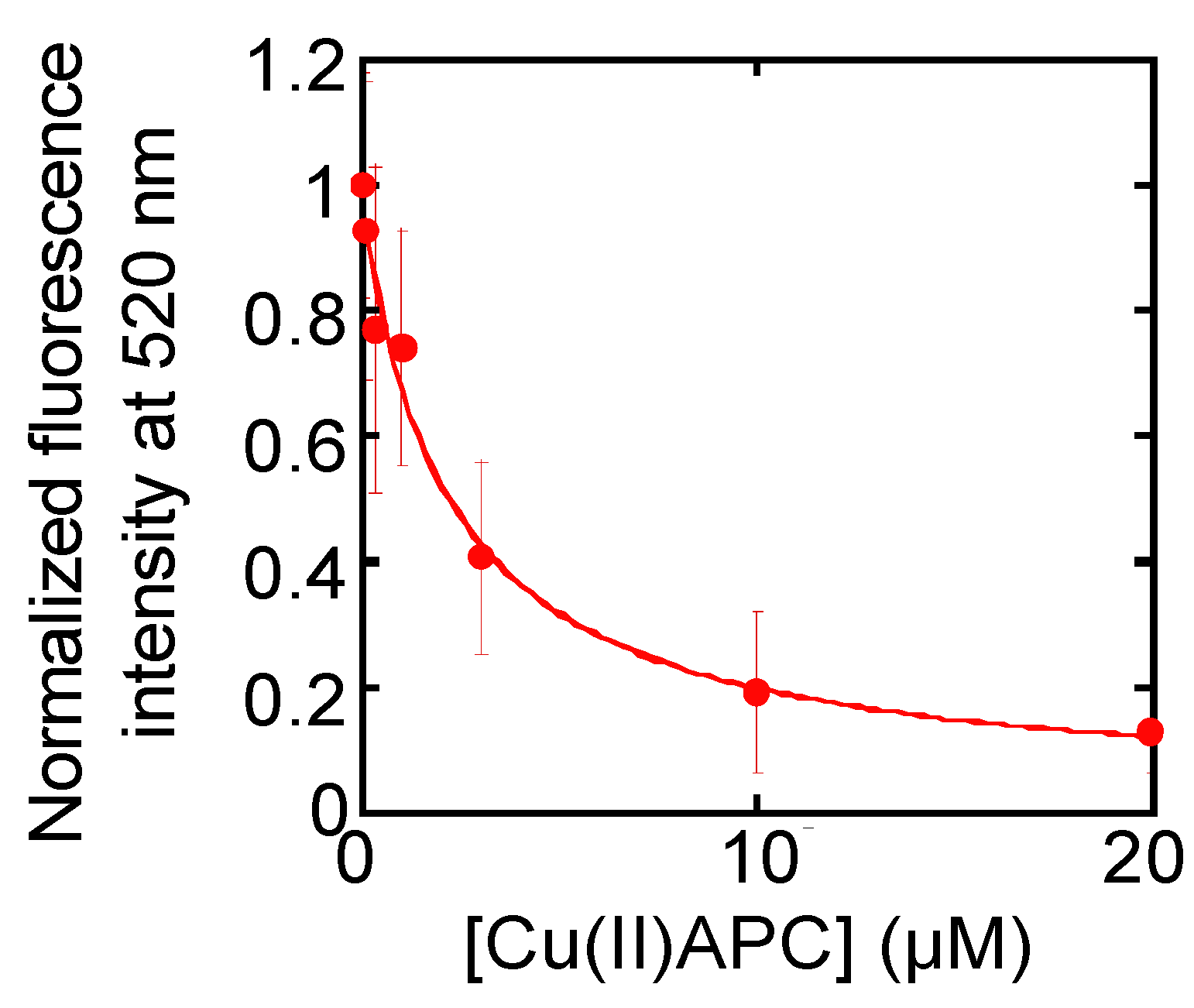
2.7. Evaluation of Telomerase Inhibitory Effects of Anionic Phthalocyanine

3. Experimental
3.1. Materials and Reagents
3.2. Preparation of Cell Lysate
3.3. CPT for MSTP Detection
3.4. Electrophoresis Analysis of MSTP
3.5. MSTP Digestion Assay
3.6. Telomerase Reaction
3.7. Immobilization of Telomerase Reaction Products on MBs
3.8. A-PCR Amplification of Telomerase Reaction Products
3.9. CPT for Detection of A-PCR Products
3.10. Normal PCR Amplification of Telomerase Reaction Products
3.11. One-Step TRAP Assay (Conventional TRAP Assay)
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A highly conserved repetitive DNA-sequence, (TTAGGG)n, present at the telomeres of human-chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Harley, C.B.; Vaziri, H.; Counter, C.M.; Allsopp, R.C. The telomere hypothesis of cellular aging. Exp. Gerontol. 1992, 27, 375–382. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase-activity in tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Morin, G.B. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 1989, 59, 521–529. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.C.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- De Cian, A.; Lacroix, L.; Douarre, C.; Temime-Smaali, N.; Trentesaux, C.; Riou, J.F.; Mergny, J.L. Targeting telomeres and telomerase. Biochimie 2008, 90, 131–155. [Google Scholar]
- Vidaurreta, M.; Maestro, M.; Rafael, S.; Veganzones, S.; Sanz-Casla, M.; Cerdan, J.; Arroyo, M. Telomerase activity in colorectal cancer, prognostic factor and implications in the microsatellite instability pathway. World J. Gastroenterol. 2007, 13, 3868–3872. [Google Scholar]
- Uen, Y.H.; Lin, S.R.; Wu, D.C.; Su, Y.C.; Wu, J.Y.; Cheng, T.L.; Chi, C.W.; Wang, J.Y. Prognostic significance of multiple molecular markers for patients with stage II colorectal cancer undergoing curative resection. Ann. Surg. 2007, 246, 1040–1046. [Google Scholar] [CrossRef]
- Saleh, S.; Lam, A.K.Y.; Ho, Y.H. Real-time PCR quantification of human telomerase reverse transcriptase (hTERT) in colorectal cancer. Pathology 2008, 40, 25–30. [Google Scholar] [CrossRef]
- Lam, A.K.Y.; Ong, K.; Ho, Y.H. Aurora kinase expression in colorectal adenocarcinoma: Correlations with clinicopathological features, p16 expression, and telomerase activity. Hum. Pathol. 2008, 39, 599–604. [Google Scholar] [CrossRef]
- Lam, A.K.Y.; Saleh, S.; Smith, R.A.; Ho, Y.H. Quantitative analysis of survivin in colorectal adenocarcinoma: Increased expression and correlation with telomerase activity. Hum. Pathol. 2008, 39, 1229–1233. [Google Scholar] [CrossRef]
- Soreide, K.; Gudlaugsson, E.; Skaland, I.; Janssen, E.A.M.; van Diermen, B.; Korner, H.; Baak, J.P.A. Metachronous cancer development in patients with sporadic colorectal adenomas–multivariate risk model with independent and combined value of hTERT and survivin. Int. J. Colorectal Dis. 2008, 23, 389–400. [Google Scholar] [CrossRef]
- Lam, A.K.Y.; Ong, K.; Ho, Y.H. hTERT expression in colorectal adenocarcinoma: correlations with p21, p53 expressions and clinicopathological features. Int. J. Colorectal Dis. 2008, 23, 587–594. [Google Scholar] [CrossRef]
- Kim, N.W.; Wu, F. Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP). Nucleic Acids Res. 1997, 25, 2595–2597. [Google Scholar] [CrossRef]
- Schmidt, P.M.; Lehmann, C.; Matthes, E.; Bier, F.F. Detection of activity of telomerase in tumor cells using fiber optical biosensors. Biosens. Bioelectron. 2002, 17, 1081–1087. [Google Scholar] [CrossRef]
- Grimm, J.; Perez, J.M.; Josephson, L.; Weissleder, R. Novel nanosensors for rapid analysis of telomerase activity. Cancer Res. 2004, 64, 639–643. [Google Scholar] [CrossRef]
- Weizmann, Y.; Patolsky, F.; Lioubashevski, O.; Willner, I. Magneto-mechanical detection of nucleic acids and telomerase activity in cancer cells. J. Am. Chem. Soc. 2004, 126, 1073–1080. [Google Scholar] [CrossRef]
- Sharon, E.; Freeman, R.; Riskin, M.; Gil, N.; Tzfati, Y.; Willner, I. Optical, electrical and surface plasmon resonance methods for detecting telomerase activity. Anal. Chem. 2010, 82, 8390–8397. [Google Scholar] [CrossRef]
- Sato, S.; Kondo, H.; Nojima, T.; Takenaka, S. Electrochemical telomerase assay with ferrocenyl naphthalene diimide as a tetraplex DNA-specific binder. Anal. Chem. 2005, 77, 7304–7309. [Google Scholar] [CrossRef]
- Kim, K.W.; Shin, Y.; Perera, A.P.; Liu, Q.; Kee, J.S.; Han, K.; Yoon, Y.J.; Park, M.K. Label-free, PCR-free chip-based detection of telomerase activity in bladder cancer cells. Biosens. Bioelectron. 2013, 45, 152–157. [Google Scholar] [CrossRef]
- Maesawa, C.; Inaba, T.; Sato, H.; Iijima, S.; Ishida, K.; Terashima, M.; Sato, R.; Suzuki, M.; Yashima, A.; Ogasawara, S.; et al. A rapid biosensor chip assay for measuring of telomerase activity using surface plasmon resonance. Nucleic Acids Res. 2003, 31, e4. [Google Scholar] [CrossRef]
- Xu, S.Q.; He, M.; Yu, H.P.; Wang, X.Y.; Tan, X.L.; Lu, B.; Sun, X.; Zhou, Y.K.; Yao, Q.F.; Xu, Y.J.; et al. Bioluminescent method for detecting telomerase activity. Clin. Chem. 2002, 48, 1016–1020. [Google Scholar]
- Kha, H.; Zhou, W.; Chen, K.; Karan-Tamir, B.; Miguel, T.S.; Zeni, L.; Kearns, K.; Mladenovic, A.; Rasnow, B.; Robinson, M.; et al. A telomerase enzymatic assay that does not use polymerase chain reaction, radioactivity, or electrophoresis. Anal. Biochem. 2004, 331, 230–234. [Google Scholar] [CrossRef]
- Pavlov, V.; Willner, I.; Dishon, A.; Kotler, M. Amplified detection of telomerase activity using electrochemical and quartz crystal microbalance measurements. Biosens. Bioelectron. 2004, 20, 1011–1021. [Google Scholar] [CrossRef]
- Ding, C.F.; Li, X.L.; Ge, Y.; Zhang, S.S. Fluorescence detection of telomerase activity in cancer cells based on isothermal circular strand-displacement polymerization reaction. Anal. Chem. 2010, 82, 2850–2855. [Google Scholar] [CrossRef]
- Pavlov, V.; Xiao, Y.; Gill, R.; Dishon, A.; Kotler, M.; Willner, I. Amplified chemiluminescence surface detection of DNA and telomerase activity using catalytic nucleic acid labels. Anal. Chem. 2004, 76, 2152–2156. [Google Scholar] [CrossRef]
- Yi, X.; Pavlov, V.; Gill, R.; Bourenko, T.; Willner, I. Lighting up biochemiluminescence by the surface self-assembly of DNA-hemin complexes. ChemBioChem 2004, 5, 374–379. [Google Scholar] [CrossRef]
- Xiao, Y.; Pavlov, V.; Niazov, T.; Dishon, A.; Kotler, M.; Willner, I. Catalytic beacons for the detection of DNA and telomerase activity. J. Am. Chem. Soc. 2004, 126, 7430–7431. [Google Scholar]
- Patolsky, F.; Gill, R.; Weizmann, Y.; Mokari, T.; Banin, U.; Willner, I. Lighting-up the dynamics of telomerization and DNA replication by CdSe-ZnS quantum dots. J. Am. Chem. Soc. 2003, 125, 13918–13919. [Google Scholar] [CrossRef]
- Zheng, G.F.; Daniel, W.L.; Mirkin, C.A. A new approach to amplified telomerase detection with polyvalent oligonucleotide nanoparticle conjugates. J. Am. Chem. Soc. 2008, 130, 9644–9645. [Google Scholar] [CrossRef]
- Zhou, X.M.; Xing, D.; Zhu, D.B.; Jia, L. Magnetic bead and nanoparticle based electrochemiluminescence amplification assay for direct and sensitive measuring of telomerase activity. Anal. Chem. 2009, 81, 255–261. [Google Scholar] [CrossRef]
- Li, Y.; Liu, B.W.; Li, X.; Wei, Q.L. Highly sensitive electrochemical detection of human telomerase activity based on bio-barcode method. Biosens. Bioelectron. 2010, 25, 2543–2547. [Google Scholar] [CrossRef]
- Innis, M.A.; Myambo, K.B.; Gelfand, D.H.; Brow, M.A.D. DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase chain reaction-amplified DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 9436–9440. [Google Scholar] [CrossRef]
- Williams, J.F. Optimization strategies for the polymerase chain reaction. Biotechniques 1989, 7, 762–769. [Google Scholar] [CrossRef]
- Bekkaoui, F.; Poisson, I.; Crosby, W.; Cloney, L.; Duck, P. Cycling probe technology with RNase H attached to an oligonucleotide. Biotechniques 1996, 20, 240–248. [Google Scholar]
- Saldanha, S.N.; Andrews, L.G.; Tollefsbol, T.O. Analysis of telomerase activity and detection of its catalytic subunit, hTERT. Anal. Biochem. 2003, 315, 1–21. [Google Scholar] [CrossRef]
- Krupp, G.; Kuhne, K.; Tamm, S.; Klapper, W.; Heidorn, K.; Rott, A.; Parwaresch, R. Molecular basis of artifacts in the detection of telomerase activity and a modified primer for a more robust ‘TRAP’ assay. Nucleic Acids Res. 1997, 25, 919–921. [Google Scholar] [CrossRef]
- Gang, Z.R.; Wang, X.W.; Yuan, J.H.; Guo, L.X.; Xie, H. Using a non-radioisotopic, quantitative TRAP-based method detecting telomerase activities in human hepatoma cells. Cell Res. 2000, 10, 71–77. [Google Scholar] [CrossRef]
- Tian, T.; Peng, S.; Xiao, H.; Zhang, X.E.; Guo, S.; Wang, S.R.; Zhou, X.; Liu, S.M. Highly sensitive detection of telomerase based on a DNAzyme strategy. Chem. Commun. 2013, 49, 2652–2654. [Google Scholar]
- Al-Soud, W.A.; Radstrom, P. Purification and characterization of PCR-inhibitory components in blood cells. J. Clin. Microbiol. 2001, 39, 485–493. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Klug, A. Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 1989, 342, 825–829. [Google Scholar] [CrossRef]
- Zahler, A.M.; Williamson, J.R.; Cech, T.R.; Prescott, D.M. Inhibition of telomerase by G-quartet DNA structures. Nature 1991, 350, 718–720. [Google Scholar]
- Sun, D.Y.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of human telomerase by a G-quadruplex-interactive compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef]
- De Cian, A.; Cristofari, G.; Reichenbach, P.; De Lemos, E.; Monchaud, D.; Teulade-Fichou, M.P.; Shin-Ya, K.; Lacroix, L.; Lingner, J.; Mergny, J.L. Reevaluation of telomerase inhibition by quadruplex ligands and their mechanisms of action. Proc. Natl. Acad. Sci. USA 2007, 104, 17347–17352. [Google Scholar] [CrossRef]
- Yaku, H.; Murashima, T.; Miyoshi, D.; Sugimoto, N. Anionic phthalocyanines targeting G-quadruplexes and inhibiting telomerase activity in the presence of excessive DNA duplexes. Chem. Commun. 2010, 46, 5740–5742. [Google Scholar]
- Yaku, H.; Murashima, T.; Miyoshi, D.; Sugimoto, N. Specific binding of anionic porphyrin and phthalocyanine to the G-quadruplex with a variety of in vitro and in vivo applications. Molecules 2012, 17, 10586–10613. [Google Scholar] [CrossRef]
- Yaku, H.; Fujimoto, T.; Murashima, T.; Miyoshi, D.; Sugimoto, N. Phthalocyanines: A new class of G-quadruplex-ligands with many potential applications. Chem. Commun. 2012, 48, 6203–6216. [Google Scholar]
- Yaku, H.; Murashima, T.; Miyoshi, D.; Sugimoto, N. Study on effects of molecular crowding on g-quadruplex-ligand binding and ligand-mediated telomerase inhibition. Methods 2013, in press. [Google Scholar]
- Sample Availability: Commercially available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yaku, H.; Murashima, T.; Miyoshi, D.; Sugimoto, N. A Highly Sensitive Telomerase Activity Assay that Eliminates False-Negative Results Caused by PCR Inhibitors. Molecules 2013, 18, 11751-11767. https://doi.org/10.3390/molecules181011751
Yaku H, Murashima T, Miyoshi D, Sugimoto N. A Highly Sensitive Telomerase Activity Assay that Eliminates False-Negative Results Caused by PCR Inhibitors. Molecules. 2013; 18(10):11751-11767. https://doi.org/10.3390/molecules181011751
Chicago/Turabian StyleYaku, Hidenobu, Takashi Murashima, Daisuke Miyoshi, and Naoki Sugimoto. 2013. "A Highly Sensitive Telomerase Activity Assay that Eliminates False-Negative Results Caused by PCR Inhibitors" Molecules 18, no. 10: 11751-11767. https://doi.org/10.3390/molecules181011751