3.1. General
All reactions were conducted under an atmosphere of dry nitrogen in oven-dried glassware unless otherwise noted. THF was dried by distillation from sodium-benzophenone. Toluene, CH
2Cl
2, 2,6-lutidine, Et
3N, pyridine, DBU, and CH
3CN were freshly distilled from CaH
2. Anhydrous DMF was obtained by stirring overnight over anhydrous CuSO
4 followed by distillation under reduced pressure. All starting materials and reagents were commercially available and were used as received with the exception of
5,
7, and
29 which were prepared as described previously [
18,
21,
27]. Flash chromatography was conducted using 230–400 mesh silica gel obtained from Dynamic Adsorbents, Inc. Melting points were obtained on a Thomas-Hoover apparatus and are uncorrected. Proton (400 MHz) and carbon (125 MHz) NMR spectra were recorded on a Bruker Avance III spectrometer in CDCl
3 unless otherwise noted. Chemical shifts are reported as δ units relative to internal tetramethylsilane. IR spectra were recorded on a Thermo-Nicolet Avatar 360 FT instrument as thin films on NaCl unless otherwise noted. High resolution mass spectra were provided by the Washington University Mass Spectrometry Resource, St. Louis, MO.
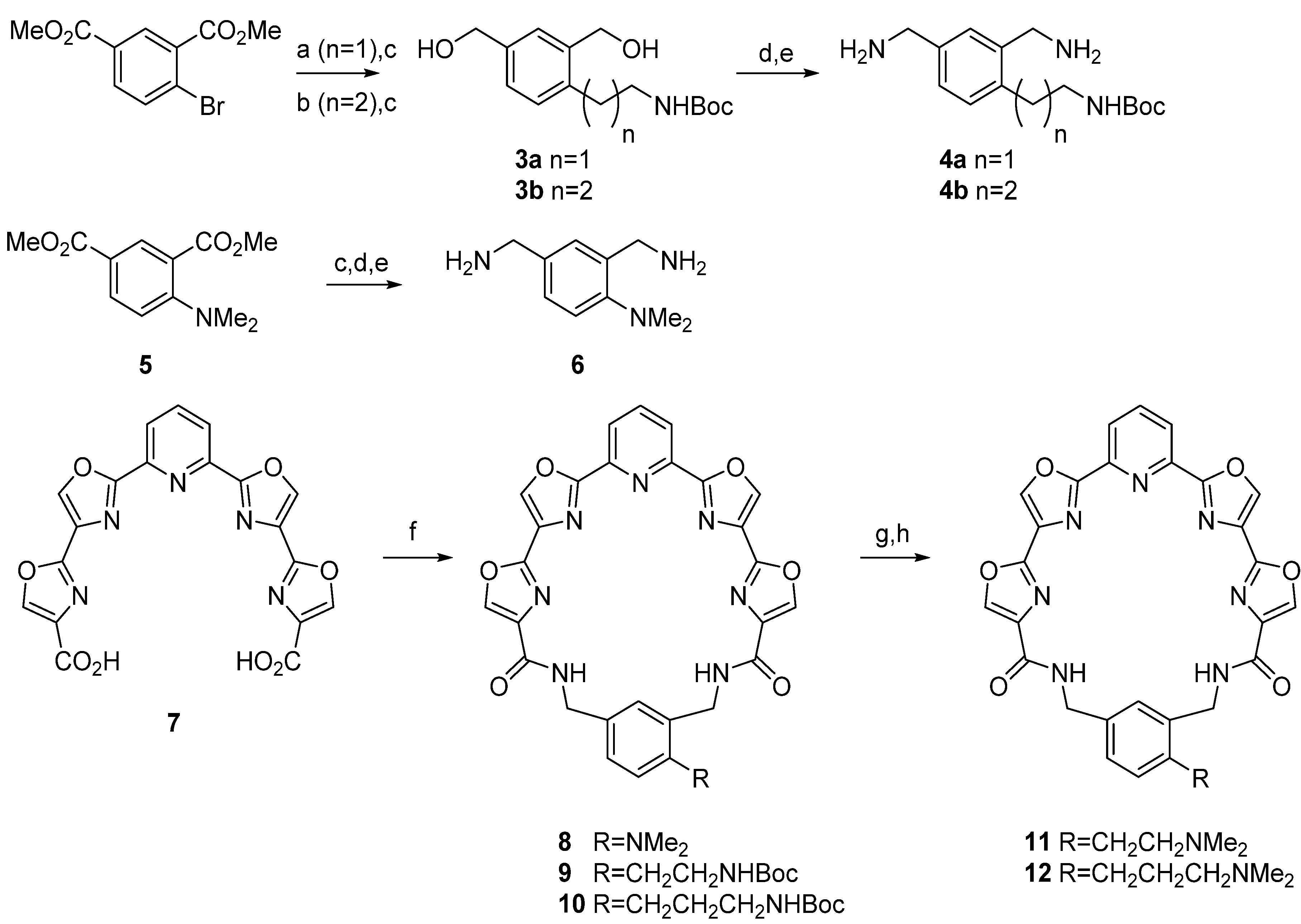
1,3-Bis(hydroxymethyl)-4-[2-[(tert-butoxycarbonyl)amino]ethyl]benzene (3a). Step A. A mixture of dimethyl 4-bromoisophthalate (273 mg, 1 mmol), potassium 2-(tert-butoxycarbonylamino)ethyl trifluoroborate (301 mg, 1.2 mmol), Cs2CO3 (1.08 g, 3.3 mmol), and PdCl2(dppf).CH2Cl2 (49 mg, 0.06 mmol) in toluene 3 mL and water 1 mL was heated overnight at 80 °C under N2 in a sealed tube. The reaction mixture was then cooled to room temperature and saturated aqueous NH4Cl was added. The mixture was extracted with CH2Cl2 and the organic layer was dried over MgSO4 and then the solvent was evaporated under reduced pressure. The crude product was purified by flash chromatography eluting with 0%–20% ethyl acetate in hexane to give 229 mg of a colorless oil that proved to be an inseparable mixture of the desired product and 2,2',4,4'-tetra(carbomethoxy)biphenyl. This mixture was carried on to the next step. Step B. The mixture from above was dissolved in anhydrous THF (10 mL) and cooled to 0 °C under N2 and treated with LiBH4 (80 mg, 6 mmol) followed by EtOH (1 mL). The reaction was allowed to warm to room temperature. After 24 h the reaction mixture was poured into water and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4 and concentrated. Purification was effected by flash chromatography eluting with 0%–5% MeOH/CH2Cl2 to give 3a as a colorless oil; 157 mg, 56% (two steps); 1H-NMR δ 7.14 (s, 1H), 7.02 (d, 1H, J = 8), 6.97 (d, 1H, J = 8), 5.18 (br s, 1H), 4.43 (s, 2H), 4.36 (s, 2H), 3.11 (m, 2H), 2.66 (t, 2H, J = 8), 1.30 (s, 9H); 13C-NMR δ 156.5, 139.4, 139.2, 136.2, 129.9, 127.5, 126.5, 79.4, 64.3, 62.4, 41.6, 32.4, 28.2.
1,3-Bis(hydroxymethyl)-4-[3-[(tert-butoxycarbonyl)amino]propyl]benzene (3b). Step A. A solution of N-Boc allylamine (432 mg, 2.75 mmol) in THF (5 mL) was flushed with nitrogen, treated with 9-BBN (358 mg, 2.75 mmol) and stirred at room temperature for 2 h. Degassed water (0.14 mL) was added and the mixture was then added via cannula to a flask containing dimethyl 4-bromoisophthalate (500 mg, 1.83 mmol), PdCl2(dppf).CH2Cl2 (75 mg, 0.09 mmol), Ph3As (28 mg, 0.09 mmol), Cs2CO3 (1.79 g, 5.49 mmol) and DMF (5 mL) under nitrogen. The reaction was stirred at 60 °C overnight and then the solvents were removed under reduced pressure. The residue was poured into brine and extracted several times with ether. The organic layers were combined, washed with brine, and dried over Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography eluting with 0%–50% EtOAc/hexanes to give a yellow oil; 457 mg, 71%; 1H-NMR δ 8.49 (s, 1H), 8.01 (d, 1H, J = 8), 7.30 (d, 1H, J = 8), 4.90 (s, 1H), 3.86 (s, 6H), 3.13 (m, 2H), 2.99 (t, 2H, J = 8), 1.77 (m, 2H), 1.40 (s, 9H); 13C-NMR δ 166.9, 166.1, 156.0, 148.9, 132.7, 132.6, 131.1, 129.5, 128.1, 78.9, 52.1, 52.0, 40.2, 31.6, 31.5, 28.4; Step B. Prepared using the procedure detailed above for 3a, StepB. Purification was effected by flash chromatography eluting with 0%–5% MeOH/CH2Cl2 to obtain 3b as a colorless oil; 259 mg, 88%; 1H-NMR δ 7.26 (s, 1H), 7.11 (d, 1H, J = 8), 7.07 (d, 1H, J = 8), 5.07 (br s, 1H), 4.52 (s, 2H), 4.47 (s, 2H), ), 4.12 (br s, 2H), 3.05 (t, 2H, J = 4), 2.57, (t, 2H, J = 8), 1.67(m, 2H), 1.42 (s, 9H); 13C-NMR δ 156.4, 138.9, 138.7, 138.6, 129.1, 127.1, 126.4, 79.2, 64.4, 62.2, 40.2, 31.1, 28.9, 28.4.
1,3-Bis(aminomethyl)-4-[2-[(tert-butoxycarbonyl)amino]ethyl]benzene (4a). Step A. A solution of 3a (150 mg, 0.56 mmol) in THF (7.5 mL) was cooled to 0 °C under N2 and treated dropwise with diphenyl phosphorylazide (DPPA) (0.36 mL, 1.68 mmol) followed by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (0.25 mL, 1.68 mmol). Stirring continued at 0 °C for 4 h and the reaction was then allowed to warm overnight to room temperature. The solution was poured into 5% HCl and extracted with EtOAc. The organic layer was separated, washed with brine, dried over Na2SO4, filtered, and evaporated to a brown oil. Purification by flash chromatography eluting with 5%–10% EtOAc/hexane afforded the diazide as a colorless oil; 112 mg, 62%; 1H-NMR δ 7.24 (m, 3H), 4.60 (br s, 1H), 4.35 (s, 2H), 4.27 (s, 2H), 3.29 (m, 2H), 2.80 (t, 2H, J = 8), 1.37 (s, 9); 13C-NMR δ 155.8, 137.1, 134.3, 134.2, 130.8, 129.6, 128.5, 79.4, 54.3, 52.4, 41.4, 32.7, 28.4. Step B. The diazide (90 mg, 0.27 mmol) was dissolved in a mixture of THF and water (5:2) and polymer-supported triphenylphosphine (PS-PPh3) (227 mg, 3 mmol/g) was added. The mixture was stirred at room temperature overnight and then filtered and concentrated. The crude product was re-dissolved in toluene and evaporated several times to give 4a as a yellow oil; 54 mg, 71%; 1H-NMR δ 7.10 (m, 3H), 5.56 (br s, 1H), 3.83 (s, 2H), 3.77 (s, 2H), 3.30 (m, 2H), 2.78 (t, 2H, J = 8), 1.35 (s, 9H); 13C-NMR δ 156.1, 141.7, 135.9, 130.1, 127.4, 126.1, 125.5, 78.9, 46.2, 43.7, 31.9, 30.3, 28.4.
1,3-Bis(aminomethyl)-4-[3-[(tert-butoxycarbonyl)amino]propyl]benzene (4b). Step A. Prepared using the procedure detailed above for 4a, Step A. Purification was achieved by flash chromatography eluting with 0%–15% EtOAc/hexanes. A colorless oil was obtained; 113 mg, 65%; 1H-NMR δ 7.17 (m, 3H), 4.58 (br s, 1H), 4.30 (s, 2H), 4.26 (s, 2H), 3.12 (m, 2H), 2.61 (t, 2H, J = 8), 1.72 (m, 2H), 1.38 (s, 9H); 13C-NMR δ 155.9, 140.4, 133.7, 133.6, 130.1, 129.5, 128.5, 79.2, 54.3, 52.5, 40.3, 31.2, 29.3, 28.4. Step B. Prepared using the procedure detailed above for 4a, Step B. The filtrate was azeotroped with toluene to obtain 4b as a yellow oil; 73 mg, 96%; 1H NMR (CD3OD) δ 7.05 (m, 3H), 3.72 (s, 2H), 3.66 (s, 2H), 2.97 (m, 2H), 2.54 (t, 2H, J = 8), 1.61 (m, 2H), 1.32 (s, 9H); 13C-NMR δ 158.5, 140.8, 140.7, 139.5, 130.6, 128.4, 127.4, 79.9, 46.2, 43.3, 31.0, 30.2, 28.9.
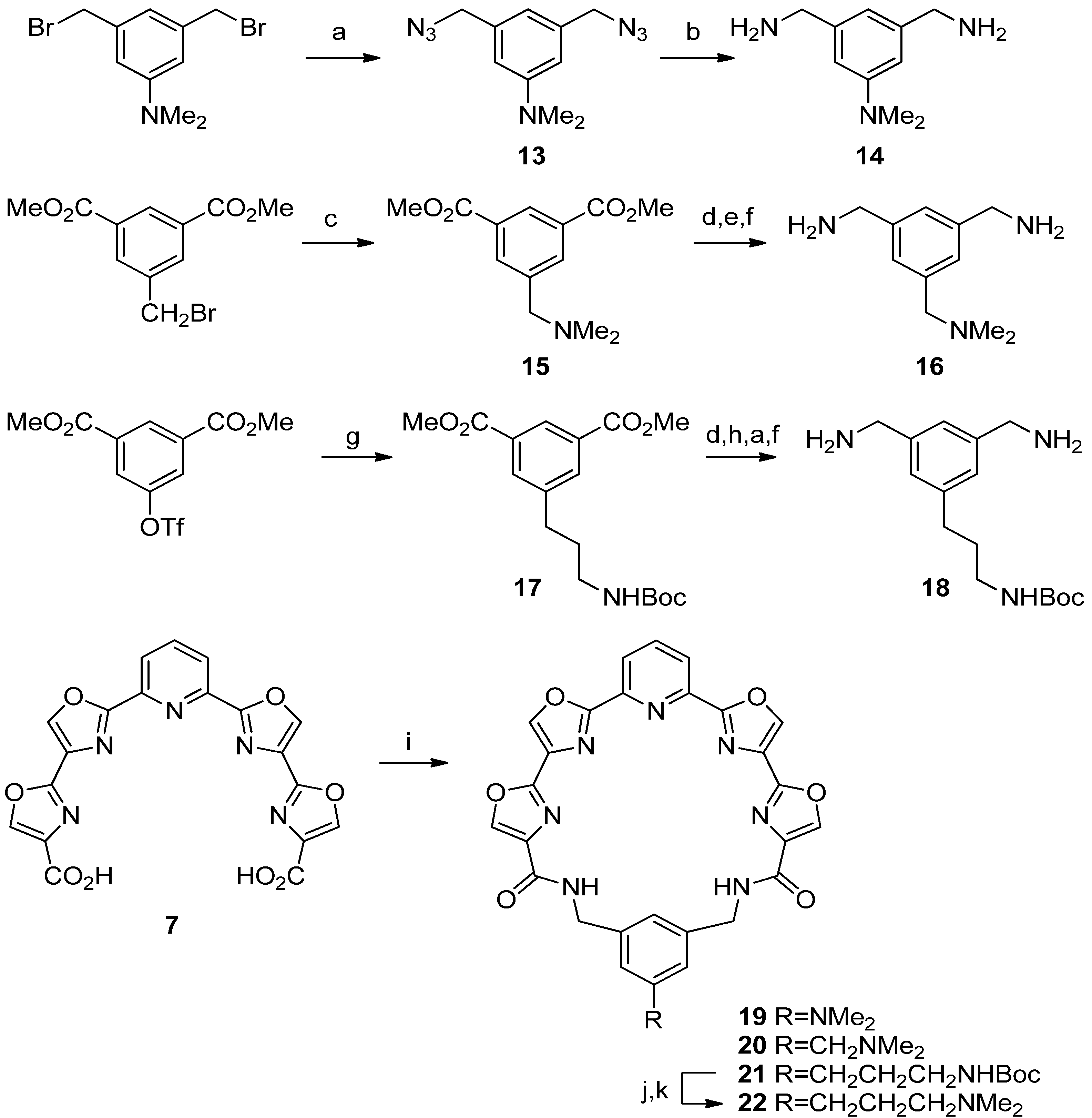
N,N-Dimethyl-2,4-bis(aminomethyl)aniline (
6).
Step A. Prepared from
5 [
27] using the procedure detailed above for
3a,
Step B. Flash chromatography eluting with 0%–4% MeOH/CH
2Cl
2 gave the diol as a yellow oil; 1.18 g, 90%;
1H-NMR δ 7.17 (dd, 1H,
J = 1,8), 7.12 (d, 1H,
J = 1), 7.09 (d, 1H,
J = 8), 4.70 (s, 2H), 4.51 (s, 2H), 2.65 (s, 2H);
13C NMR δ 150.6, 137.5, 135.0, 127.3, 126.9, 120.1, 64.5, 64.3, 44.8.
Step B. Prepared using the procedure detailed above for
4a,
StepA. Flash chromatography eluting with 0-4% EtOAc/hexane gave the diazide as a colorless oil; 287 mg, 44%;
1H-NMR δ 7.32 (d, 1H,
J = 2), 7.28 (dd, 1H,
J = 2,7), 7.18 (d, 1H
, J = 7), 4.52 (s, 2H), 4.34 (s, 2H), 2.73 (s, 6H);
13C-NMR δ 152.9, 130.6, 130.4, 129.9, 128.9, 120.1, 54.3, 50.6, 45.1.
Step C. Prepared using the procedure detailed above for
4a,
Step B. The residue was re-dissolved in toluene and evaporated several times to give
6 as a yellow oil; 161 mg, 83%;
1H-NMR δ 7.18 (d, 1H,
J = 1.6), 7.07 (dd, 1H,
J = 1.6, 8), 6.99 (d, 1H,
J = 8), 3.82 (s, 2H), 3.72 (s, 2H), 2.59 (s, 6H);
13C-NMR δ 150.1, 137.2, 136.2, 126.4, 125.2, 118.6, 44.9, 44.1, 42.1.
Pyridyl tetraoxazole macrocycle with a 4-(N,N-dimethylamino) group on the phenyl ring (
8). Diacid
7 [
21] (121 mg, 0.28 mmol) was suspended in DMF (120 mL) and MnSO
4•H
2O (95 mg, 0.56 mmol) was added. The solution was warmed to 65 °C under N
2 for 20 min and then cooled back to room temperature. EDC (213 mg, 1.1 mmol), HOBt (150 mg, 1.1 mmol), and 2,6-lutidine (239 mg, 2.3 mmol) were added followed by the slow drop-wise addition of a solution of
9 (50 mg, 0.28 mmol) in DMF (5 mL). The solution was stirred at room temperature for 48 h and then the white precipitate was filtered off and purified by flash chromatography eluting with 0%–10% MeOH/CH
2Cl
2 to give macrocycle
8 as a white solid; 27 mg, 17%; mp 268–270 °C;
1H-NMR (CDCl
3 + CD
3OD) δ 8.21 (s, 1H), 8.19 (s, 2H), 8.16 (s, 1H), 7.99 (m, 3H), 7.62 (m, 1H), 7.27 (m, 3H), 7.07 (d, 1H,
J = 8), 4.63 (d, 2H,
J = 4), 4.47 (d, 2H,
J = 4 ), 2.61 (s, 6H);
13C-NMR (CDCl
3 + CD
3OD) δ 160.5, 160.4, 160.1, 159.5, 154.3, 154.2, 153.7, 145.5, 145.4, 140.7, 140.4, 139.2, 138.8, 138.5, 137.5, 137.5, 132.5, 132.1, 131.94, 131.8, 131.1, 130.5, 122.7, 122.6, 121.1, 45.1, 42.9, 40.4; HRMS (ESI)
m/z calcd for C
29H
22N
8O
6 (M+H): 579.1735; found: 579.1737.
Pyridyl tetraoxazole macrocycle with a 4-[2-[(tert-butoxycarbonyl)amino]ethyl]group on the phenyl ring (9). Prepared using the procedure detailed above for 8. White solid; 18 mg, 18%; mp 198–200 °C; 1H-NMR δ 8.32 (s, 2H), 8.27 (s, 2H), 8.05 (m, 3H), 7.41 (s, 1H), 7.25 (d, 1H, J = 8), 7.22 (d, 1H, J = 8), 5.42 (br s, 1H), 4.59 (s, 2H), 4.49 (s, 2H), 3.17 (m, 2H), 2.78 (t, 2H, J = 8), 1.32 (s, 9H); 13C-NMR δ 160.7, 159.9, 154.3, 144.9, 141.5, 141.46, 139.5, 138.9, 138.0, 137.2, 135.3, 132.4, 131.5, 130.9, 130.85, 129.6, 122.8, 79.1, 43.6, 41.2, 38.7, 32.5, 28.9; HRMS (ESI) m/z calcd for C34H30N8O8 (M+Na): 701.2084; found: 701.2075.
Pyridyl tetraoxazole macrocycle with a 4-[3-[(tert-butoxycarbonyl)amino]propyl]group on the phenyl ring (10). Prepared using the procedure detailed above for 8. Off-white solid; 21.2 mg, 18%; mp 190–192 °C; 1H-NMR δ 8.39 (d, 2H, J = 8), 8.20 (m, 4H), 8.01 (m, 3H), 7.30 (s, 1H), 7.23 (d, 1H, J = 8), 7.16 (d, 1H, J = 8), 4.81 (br s, 1H), 4.59 (s, 2H), 4.48 (s, 2H), 3.07(m, 2H), 2.61 (m, 2H), 1.68 (m, 2H), 1.36 (s, 9H); 13C-NMR δ 160.5, 159.7, 159.4, 156.0, 154.2, 145.3, 140.9, 140.7, 139.0, 138.4, 137.5, 137.2, 135.2, 132.0, 131.9, 131.8, 130.0, 122.8, 122.7, 79.1, 43.7, 41.5, 31.9, 29.6, 29.2, 28.4; HRMS (ESI) m/z C35H32N8O8 (M+H): 693.2425; found: 693.2416.
Pyridyl tetraoxazole macrocycle with a 4-[2-(N,N-dimethylamino)ethyl] group on the phenyl ring (11). Step A. N-Boc derivative 9 (7 mg, 0.0103 mmol) was suspended in 20% HCl (1 mL) and stirred at room temperature for 2 h. The solution was evaporated under reduced pressure to give the amine salt that was used directly for the next step. Step B. The salt from Step A (7 mg, 0.01 mmol) was suspended in 20% MeOH/CH2Cl2 (3 mL) and treated with 37% aqueous formaldehyde (0.5 mL). After stirring for 5 min at room temperature sodium triacetoxyborohydride (24 mg, 0.114 mmol) was added in one portion and stirring was continued overnight. The reaction mixture was partitioned between saturated NaHCO3 and CH2Cl2, and the organic extract was dried over Na2SO4, filtered and evaporated to a solid. Purification was performed by Chromatotron (SiO2, 1 mm rotor) eluting with 1%–20% MeOH/CH2Cl2 + 1% NH4OH to give compound 11 as a pale yellow solid; 1.5 mg, 23%; mp 278–280 °C (dec.); 1H-NMR δ 8.25 (m, 4H), 8.02 (m, 3H), 7.39 (s, 1H), 7.24 (d, 1H, J = 8), 7.19 (d, 1H, J = 8), 4.60 (s, 2H), 4.50 (s, 2H), 2.87 (t, 2H, J = 8), 2.57 (t, 2H, J = 8), 2.23(s, 6H); 13C-NMR δ 160.6, 159.9, 159.6, 154.2, 154.1, 145.1, 141.2, 139.2, 138.9, 138.7, 137.6, 137.5, 135.1, 132.4, 131.7, 130.7, 129.7, 122.7, 60.8, 44.7, 43.6, 41.32, 29.7; HRMS (ESI) m/z calcd for C31H26N8O6 (M+H): 607.2040; found: 607.2048.
Pyridyl tetraoxazole macrocycle with a 4-[3-(N,N-dimethylamino)propyl] group on the phenyl ring (12). Step A. Prepared from 10 using the procedure detailed above for 11, Step A. The product from this reaction was taken directly to the next step without purification. Step B. Prepared using the procedure detailed above for 11, Step B. Off-white solid; 4.8 mg, 61%; mp 202–205 °C; 1H-NMR δ 8.40 (d, 2H, J = 8), 8.22 (m, 4H), 8.01 (m, 3H), 7.33 (s, 1H), 7.25 (d, 1H, J = 8), 7.15 (d, 1H, J = 8), 4.59 (s, 2H), 4.49 (s, 2H), 2.60 (m, 2H) 2.31 (m, 2H), 2.20 (s, 6H), 1.7 (m, 2H); 13C-NMR δ 160.5, 159.6, 159.3, 154.2, 145.4, 140.6, 139.0, 138.4, 137.5, 137.3, 135.0, 131.9, 131.8, 130.7, 130.0, 122.7, 58.9, 44.8, 43.6, 41.5, 30.1, 29.3; HRMS (ESI) calculated for C32H26N8O6 (M+H): 621.2209; found: 621.2204.
N,N-Dimethyl-3,5-bis(azidomethyl)aniline (
13).
N,N-Dimethyl-3,5-bis(bromomethyl)aniline [
28] (100 mg, 0.33 mmol) was dissolved in anhydrous DMF (10 mL) and sodium azide (128 mg, 1.97 mmol) was added. The reaction mixture was placed under argon and heated to 90 °C overnight. After cooling to room temperature, the reaction was poured into water and extracted with CH
2Cl
2. The combined organic extracts were dried with Na
2SO
4 and evaporated under reduced pressure to give diazide
13 as a pale orange oil; 71 mg, 73%;
1H-NMR δ 6.59 (s, 3H), 4.29 (s, 4H), 2.98 (s, 6H);
13C-NMR δ 151.2, 136.9, 115.7, 111.7, 55.2, 40.4; IR 3346, 2876, 2813, 2479, 2098, 1604, 1493, 1443, 1372, 1254, 1165, 1063, 1030, 986, 829, 724 cm
−1.
N,N-Dimethyl-3,5-bis(aminomethyl)aniline (14). Prepared using the procedure detailed above for 4a, Step B. The solvent was removed under reduced pressure to afford 14 as a colorless oil; 30 mg, 53%; 1H-NMR δ 6.57 (s, 3H), 3.81 (s, 4H), 2.96 (s, 6H); 13C-NMR δ 151.2, 144.7, 114.4, 110.2, 47.0, 40.7; IR 3355, 2914, 2360, 1601, 1488, 1442, 1361, 1317, 1230, 1165, 1132, 1061, 995, 834, 700 cm−1; HRMS (ESI) m/z calcd for C10H18N3 (M+H): 180.1501; found: 180.1502.
Dimethyl 5-(N,N-Dimethyl)aminoisophthalate (
15). A solution of dimethyl 5-(bromomethyl)isophthalate [
29] (890 mg, 3.11 mmol) in anhydrous THF (15 mL) was treated at room temperature with dimethylamine (10 mL, 20 mmol, 2M in THF). This was stirred for 30 min during which time a white solid precipitated. The mixture was poured into 1N NaOH and extracted with EtOAc. The organic layer were washed with brine and dried over Na
2SO
4. Concentration under reduced pressure gave
15 as a yellow oil; 761 mg, 97%;
1H-NMR δ 8.59 (s, 1H), 8.19 (s, 2H), 3.95 (s, 6H), 3.51 (s, 2H), 2.26 (s, 6H);
13C-NMR δ 166.3, 140.2, 134.4, 134.3, 130.7, 129.6, 128.8, 63.5, 52.3, 45.4; IR 3434, 2951, 2856, 2820, 2776, 2256, 1728, 1640, 1606, 1456, 1435, 1366, 1329, 1244, 1205, 1148, 1122, 1107, 1043, 1008, 913, 873, 842, 790, 755, 733, 647 cm
−1; HRMS (ESI)
m/z calcd for C
13H
17NO
4 (M+H): 252.1230; found: 252.1237.
1,3-Bis(aminomethyl)-5-(N,N-dimethylamino)methylbenzene (16). Step A. Prepared using the procedure detailed above for 3a, Step B. Concentration gave a yellow oil that was purified by flash chromatography eluting with 1%–20% MeOH/CH2Cl2. The diol was isolated as a white solid; 222 mg, 38%; mp 94–95 °C; 1H-NMR δ 7.42 (s, 1H), 7.27 (s, 2H), 4.75 (s, 4H), 3.99 (s, 2H), 2.53 (s, 6H); 13C-NMR δ 141.6, 132.0, 129.9, 126.0, 67.5, 64.8, 50.0; IR 3396, 3004, 2950, 2370, 2271, 1644, 1525, 1468, 1368, 1168, 1018, 873, 847, 821 cm−1; HRMS (ESI) m/z calcd for C11H17NO2 (M+H): 196.1332; found: 196.1331. Step B. Prepared using the procedure detailed above for 4a, Step A. Purification by flash chromatography eluting with 10%–30% EtOAc/hexane afforded the diazide as a colorless oil; 200 mg, 72%; 1H-NMR δ 7.30 (m, 3H), 4.43 (s, 4H), 4.01 (s, 2H), 2.56 (s, 6H); 13C-NMR δ 136.6, 132.7, 131.7, 125.6, 67.1, 54.1, 50.1; IR 2950, 2372, 2272, 2097, 1693, 1464, 1345, 1246, 1169, 1017, 842, 819 cm−1; HRMS (ESI) m/z calcd for C11H15N7 (M+H): 246.1462; found: 246.1460. Step C. Prepared using the procedure detailed above for 4a, Step B. 16 was obtained as a colorless oil; 77 mg, 100%; 1H-NMR δ 7.37 (s, 1H), 7.22 (s, 2H), 3.82 (m, 6H), 2.49 (s, 6H); 13C-NMR δ 143.6, 130.6, 129.8, 120.1, 67.4, 66.2, 49.7; IR 3314, 2945, 2369, 2318, 2271, 1666, 1605, 1467, 1169, 1017, 819 cm−1; HRMS (ESI) m/z calcd for C11H19N3 (M+H): 194.1652; found: 194.1649.
Dimethyl 5-[3-[(tert-Butoxycarbonyl)amino]propyl]isophthalate (
17). A reusable sealed-tube was equipped with a magnetic stirrer and charged with dimethyl 5-(trifluoromethanesulfonyloxy)isophthalate [
30] (342 mg, 1 mmol), potassium 3-(
tert-butoxycarbonylamino)propyltrifluoroborate (265 mg, 1 mmol), RuPhos (46.7 mg, 0.1 mmol), Cs
2CO
3 (977 mg, 3 mmol), Pd(OAc)
2 (11.2 mg, 0.05 mmol), toluene (3 mL), and water (1 mL) and was sparged with nitrogen for a few minutes and then tightly sealed. The tube was placed into a pre-heated oil bath at 95 °C for 23 h. The tube was allowed to come to room temperature, sat. NH
4Cl (16 mL) was added, and the mixture was extracted with CH
2Cl
2 (3 × 20 mL). The combined extracts were dried over Na
2SO
4, filtered and evaporated under reduced pressure to give the crude product as a yellow oil. Purification by flash chromatography on silica gel eluting with 25% EtOAc/hexanes afforded
17 as a colorless oil; 250 mg, 71%;
1H-NMR δ 8.49 (s, 1H), 8.03 (s, 2H), 4.59 (br s, 1H), 3.92 (s, 6H), 3.14 (t, 2H,
J = 7), 2.73 (t, 2H,
J = 7), 1.83 (tt, 2H,
J = 7,7), 1.42 (s, 9H);
13C-NMR δ 166.3, 155.9, 142.5, 133.7, 130.7, 128.5, 79.3, 52.3, 40.1, 32.7, 31.6, 28.4.
1,3-Bis(aminomethyl)-5-[3-[(tert-butoxycarbonyl)amino]propyl]benzene (18). Step A. Prepared using the procedure detailed above for 3a, Step B. The diol was obtained as a colorless oil; 800 mg, 98%; 1H-NMR δ 7.09 (s, 1H), 7.02 (s, 2H), 4.75 (br s, 1H), 4.53 (s, 4H), 3.47 (br s, 2H), 3.04 (t, 2H, J = 7), 2.57 (t, 2H, J = 7), 1.73 (m, 2H), 1.42 (s, 9H); 13C-NMR δ 156.2, 141.9, 141.4, 126.1, 123.1, 79.4, 64.8, 40.2, 32.8, 31.4, 28.4. Step B. The diol (710 mg, 2.4 mmol) was dissolved in CH2Cl2 (24 mL), treated with Et3N (1.1 mL, 8 mmol), and cooled to 0 °C under a drying tube. Methanesulfonyl chloride (0.62 mL, 8 mmol) was added and the solution was stirred for 5.5 h and then poured into water (25 mL). The organic layer was separated and washed with water (10 mL) and then dried over Na2SO4, filtered and evaporated to give a pale-yellow oil. Flash chromatography on silica gel eluting with 2:1 EtOAc/hexanes afforded the dimesylate as a colorless oil; 270 mg, 25%; 1H-NMR δ 7.30 (s, 1H), 7.27 (s, 2H), 5.22 (s, 4H), 4.66 (br s, 1H), 3.14 (t, 2H, J = 7), 2.99 (s, 6H), 2.67 (t, 2H, J = 7), 1.82 (tt, 2H, J = 7,7), 1.45 (s, 9H); 13C-NMR δ 156.0, 143.4, 134.5, 129.5, 126.4, 79.3, 70.7, 40.0, 38.2, 32.7, 31.6, 28.4. Step C. Prepared using the procedure detailed above for 13. The diazide was obtained as a colorless oil; 180 mg, 87%; 1H-NMR δ 7.11 (s, 3H), 4.56 (br s, 1H), 4.34 (s, 4H), 3.16 (t, 2H, J = 7), 2.68 (t, 2H, J = 7), 1.83 (m, 2H), 1.46 (s, 9H); 13C-NMR δ 156.0, 143.1, 136.3, 128.1, 125.6, 77.3, 54.6, 40.3, 32.9, 31.7, 28.4. Step D. Prepared using the procedure detailed above for 4a, Step B. Diamine 18 is a pale-yellow oil; 140 mg, 90%; 1H NMR δ 7.09 (s, 1H), 7.00 (s, 2H), 4.58 (br s, 1H), 3.83 (s, 4H), 3.13 (m, 2H), 2.62 (t, 2H, J = 7), 1.80 (m, 6H (H2' + 2NH2)), 1.44 (s, 9H); 13C-NMR δ 156.0, 143.6, 142.2, 125.7, 123.6, 79.1, 46.4, 40.2, 33.0, 31.7, 28.4.
Pyridyl tetraoxazole macrocycle with a 5-N,N-dimethylamino group on the phenyl ring (19). Prepared using the procedure detailed above for 8. White solid; 9 mg, 10%; mp 275–280 °C (dec.); 1H-NMR δ 8.32 (m, 6H), 8.10 (m, 3H), 6.90 (s, 1H), 6.71 (s, 2H), 4.56 (s, 4H), 2.93 (s, 6H); 13C-NMR δ 160.8, 160.3, 154.3, 151.7, 145.2, 141.4, 139.4, 138.9, 138.4, 137.7, 131.8, 122.8, 118.3, 113.3, 44.0, 40.6; IR 3417, 1644, 1605, 1439, 1370, 1172, 1112, 926 cm−1; HRMS (ESI) m/z calcd for C29H23N8O6 (M+H): 579.1735; found: 579.1727.
Pyridyl tetraoxazole macrocycle with a 5-(N,N-dimethylaminomethyl) group on the phenyl ring (20). Prepared using the procedure detailed above for 8. White solid; 5 mg, 4%; mp > 300 °C; 1H-NMR δ 8.58 (s, 2H), 8.44 (m, 4H), 8.63 (s, 2H), 8.07 (m, 2H), 3.97 (m, 6H), 3.46 (s, 3H), 3.12 (s, 3H); HRMS (ESI) m/z calcd for C30H24N8O6 (M+H) 593.1892; found: 593.1885.
Pyridyl tetraoxazole macrocycle with a 5-[3-[(tert-butoxycarbonyl)amino]propyl] group on the phenyl ring (21). Prepared using the procedure detailed above for 8. White solid; 40 mg, 43%; 1H-NMR δ 8.27 (s, 2H, H5 oxazole), 8.25 (s, 2H, H5 oxazole), 8.06 (m, 3H, H3-5 pyr), 7.52 (t, 2H, J = 5, lactam NH), 7.23 (s, 1H, H2 phenyl), 7.15 (s, 2H, H4,6 phenyl), 4.59 (br s, 1H, Boc NH), 4.55 (d, 4H, J = 5, CH2NHC=O), 3.10 (m, 2H, H3'), 2.60 (t, 2H, J = 7, H1'), 1.78 (m, 2H, H2'), 1.39 (s, 9H, Me3); 13C-NMR δ 160.5 (C=O lactam), 159.7 (C2,6 pyridine), 156.0 (C=O carbamate), 154.2 (C2 distal oxazole), 145.4 (C2 proximal oxazole), 143.6 (C1,3 phenyl), 142.7 (C5 phenyl), 140.6 (C5 prox. oxazole), 139.1 (C5 distal oxazole), 138.5 (C4 pyr), 137.7 (C4 distal oxazole), 131.8 (C4 (proximal oxazole), 129.5 (C4,6 phenyl), 127.8 (C2 phenyl), 122.7 (C3,5 pyr), 79.1 (CMe3), 43.8 (CH2NHC=O), 40.2 (C3'), 32.8 (C1'), 31.7 (C2'), 28.4 (Me3).
Pyridyl tetraoxazole macrocycle with a 5-[3-(N,N-dimethylamino)propyl] group on the phenyl ring (22). Step A. Prepared from 21 using the procedure detailed above for 11, Step A, but substituting 1:1 TFA/CH2Cl2 for 20% HCl. The trifluoroacetate salt is a white solid; 38 mg, 93%. Step B. The salt was converted into 22 using the procedure detailed above for 11 Step B. White solid; 16 mg, 48%; 1H-NMR δ 8.28 (s, 2H, H5 distal oxazole), 8.19 (s, 2H, H5 proximal oxazole), 7.98 (s, 3H, H3-5 pyridine), 7.21(s, 1H, H2 phenyl), 7.03 (s, 2H, H4,6 phenyl), 4.42 (s, 4H, CH2 macrocycle), 2.49 (t, 2H, J = 7, H1'), 2.41 (t, 2H, J = 7, H3'), 2.24 (s, 6H, Me2N), 1.74 (m, 2H, H2'); 13C-NMR δ 160.6 (C=O), 160.2 (C2,6 pyr), 154.4 (C2 dist. oxaz.), 145.0 (C2 prox. oxaz.), 141.2 (C5 prox. oxaz.), 139.6 (C5 dist. oxaz.), 138.7 (C4 pyr), 137.7 (C4 dist. oxaz.), 131.3 (C4 prox. oxaz.), 129.0 (C4,6 phen), 127.6 (C2 phen), 122.8 (C3,5 pyr.), 58.4 (C3'), 43.9 (Me2N), 43.5 (CH2 macrocycle), 32.8 (C1'), 27.4 (C2'); HRMS (ESI) m/z calcd for C32H28N8O6 (M+H) 621.2210; found: 621.2205.
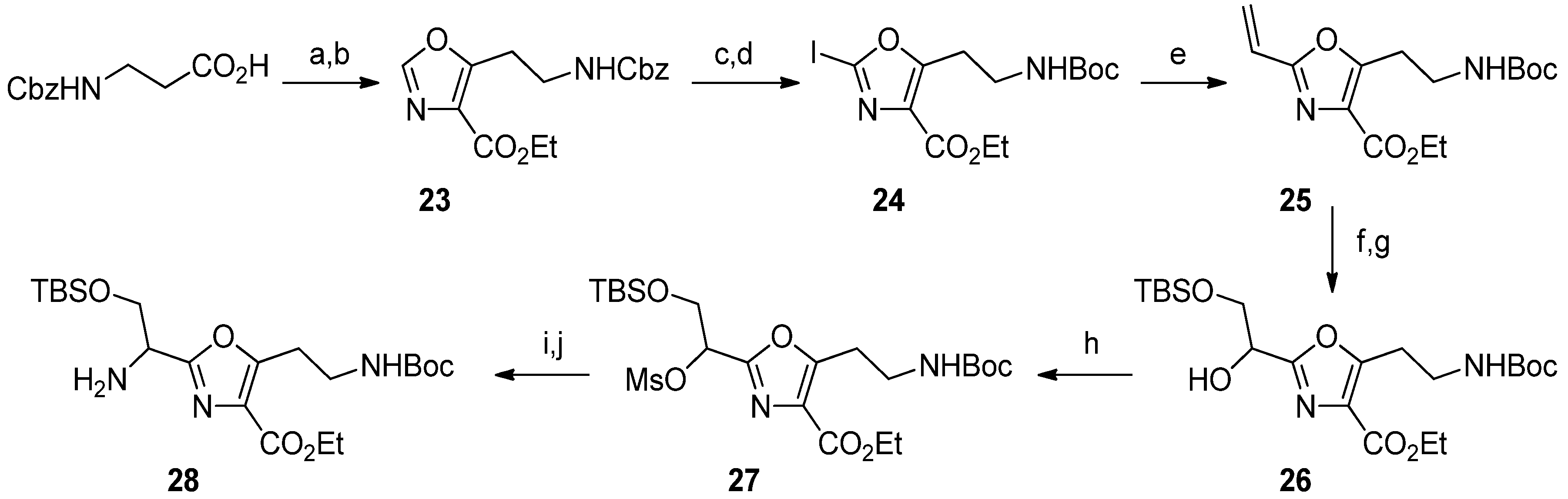
Ethyl 5-[2-[[(Benzyloxy)carbonyl]amino]ethyl]oxazole-4-carboxylate (23). Cbz-β-alanine (6.41 g, 28.7 mmol) was dissolved in anhydrous CH2Cl2 (20 mL) and cooled to 0 °C in an ice bath. It was then treated with oxalyl chloride (5 mL) and stirred at 0 °C for 30 min. The reaction was next warmed to room temperature and stirred for 2.5 h. Removal of solvents in vacuo gave the acid chloride as a colorless oil. This was dissolved in anhydrous DMF (15 mL) and added to a solution of ethyl isocyanoacetate (2.4 mL, 22.1 mmol) and DBU (5 mL, 33.2 mmol) in anhydrous DMF (15 mL) under argon. The dark brown solution was heated to 80 °C for 4.5 h and was then poured into saturated NaHCO3. This was extracted with EtOAc and the combined organic layers were washed with 5% HCl, and brine. After concentration, the resulting brown oil was flash chromatographed on SiO2 with 15%–40% EtOAc in hexane. Oxazole 23 was isolated as a pale orange oil; 3.01 g, 43%; 1H-NMR δ 7.75 (s, 1H), 7.32 (m, 5H), 5.11 (m, 3H), 4.36 (q, 2H, J = 8), 3.55 (m, 2H), 3.28 (t, 2H, J = 8), 1.37 (t, 3H, J = 8); 13C-NMR δ 162.0, 157.0, 156.2, 149.4, 136.5, 128.7, 128.5, 128.4, 128.1, 128.0, 66.7, 61.2, 39.2, 26.6, 14.1; IR 3338, 3131, 3065, 3033, 2982, 2942, 2248, 1716, 1612, 1525, 1455, 1399, 1379, 1349, 1313, 1254, 1184, 1103, 1073, 1038, 912, 840, 788, 776, 736, 699, 647 cm−1; HRMS (ESI) m/z calcd for C16H19N2O5 (M+H): 318.6252; found: 318.6247.
Ethyl 5-[2-[(tert-Butoxycarbonyl)amino]ethyl]-2-iodooxazole-4-carboxylate (24). Step A. Compound 23 (3 g, 9.43 mmol) was dissolved in EtOAc (50 mL) and di-tert-butyl dicarbonate (3.09 g, 14.2 mmol) and 10% Pd/C (300 mg) were added. This was stirred under 1 atm of H2 overnight. The reaction mixture was filtered through Celite while washing with ethyl acetate. This was concentrated in vacuo to give a yellow oil which was flash chromatographed on SiO2 with 10%-40% EtOAc in hexane. The NHBoc product was isolated as a yellow oil; 1.94 g, 72%; 1H-NMR δ 7.82 (s, 1H), 4.92 (br s, 1H), 4.40 (q, 2H, J = 8), 3.48 (m, 2H), 3.27 (t, 2H, J = 8), 1.41 (t, 3H, J = 8); 13C-NMR δ 162.0, 157.3, 155.8, 149.4, 128.3, 79.4, 61.2, 33.7, 28.4, 26.8, 14.3; IR 3366, 3125, 2979, 2936, 2360, 1716, 1612, 1522, 1455, 1393, 1380, 1367, 1349, 1314, 1278, 1252, 1172, 1103, 1074, 1042, 1026, 957, 869, 841, 789, 648 cm−1; HRMS (ESI) m/z calcd for C13H21N2O5 (M+H): 284.6409; found: 284.6407. Step B. The product from the previous reaction (1.94 g, 6.83 mmol) was dissolved in anhydrous THF (10 mL) and placed under argon. The solution was cooled to −42 °C and treated with freshly prepared LiHMDS (19 mL, 15.03 mmol, 0.8 M in THF). The solution became dark yellow in color and was stirred for 20 min. Then a solution of ZnCl2 (30 mL, 15.03 mmol, 0.5 M in THF) was added and a white precipitate formed. The reaction was warmed to 0 °C for 45 min and the solution became clear. Then solid iodine (2.25 g, 8.9 mmol) was added and the reaction stirred for 1 h at room temperature. The reaction mixture was poured into saturated sodium thiosulfate solution to which 25% NH4OH solution had been added. This was extracted with EtOAc and the combined organic layers were dried with brine and Na2SO4. Removal of the solvent in vacuo gave the iodooxazole 24 as an orange oil; 2.5 g, 89%; 1H-NMR δ 4.77 (br s, 1H), 4.38 (q, 2H, J = 8), 3.46 (m, 2H), 3.26 (t, 2H, J = 8), 1.41 (s, 9H); 13C-NMR δ 162.6, 160.9, 155.6, 131.9, 99.9, 79.6, 61.4, 38.7, 28.3, 26.9, 14.3; IR 3367, 2978, 2934, 1698, 1614, 1518, 1494, 1455, 1393, 1367, 1323, 1281, 1250, 1172, 1123, 1076, 1042, 1028, 843, 785, 733 cm−1; HRMS (ESI) m/z calcd for C13H20N2O5I (M+H): 411.0411; found: 411.0412.
Ethyl 5-[2-[(tert-Butoxycarbonyl)amino]ethyl]-2-vinyloxazole-4-carboxylate (25). Oxazole 24 (2.5 g, 6.1 mmol) and tributyl(vinyl)tin (2.7 mL, 9.15 mmol) were dissolved in anhydrous dioxane (20 mL) and placed under argon. Then bis(triphenylphosphine)palladium(II) dichloride (214 mg, 0.31 mmol) was added and the reaction mixture was heated for 4 h at 105 °C. After cooling the solvent was removed in vacuo and the resulting brown oil was flash chromatographed on SiO2 with 10%–30% EtOAc in hexane. Olefin 25 was isolated as a yellow oil; 1.49 g, 79%; 1H-NMR δ 6.59 (dd, 1H, J = 12,16), 6.21 (d, 1H, J = 16), 5.70 (dd, 1H, J = 12), 4.86 (br s, 1H), 4.40 (q, 2H, J = 8), 3.48 (m, 2H), 3.26 (t, 2H, J = 8), 1.41 (s, 9H); 13C NMR δ 162.2, 159.3, 156.8, 155.7, 129.4, 123.2, 122.8, 79.4, 61.1, 38.3, 28.3, 26.8, 14.2; IR 3357, 2978, 2935, 1714, 1607, 1520, 1453, 1393, 1380, 1366, 1326, 1278, 1250, 1176, 1097, 1046, 983, 952, 852, 769, 732 cm−1; HRMS (ESI) m/z calcd for C15H23N2O5 (M+H): 311.1601; found: 311.1600.
Ethyl 5-[2-[(tert-Butoxycarbonyl)amino]ethyl]-2-[[(tert-butyldimethylsilyl)oxy]-1-hydroxyethyl]oxazole-4-carboxylate (26). Step A. AD-mix-β (17 g) and methanesulfonamide (458 mg, 4.81 mmol) were dissolved in a mixture of t-BuOH (150 mL) and water (150 mL) and stirred at room temperature until clear. Then a solution of 25 (1.49 g, 4.81 mmol) in t-BuOH (25 mL) was added. The reaction stirred at room temperature for 16 h and then additional AD-mix-β (3 g) and methanesulfonamide (458 mg, 4.81 mmol) were added and the reaction stirred at room temperature for another 24 h. Then Na2SO3 (22 g) was added and the reaction stirred for 30 minutes. It was next poured into a separatory funnel and the layers were separated. The aqueous layer was extracted with EtOAc and the combined aqueous layers were dried with Na2SO4. The solvent was removed in vacuo to give a pale yellow oil which was purified by flash chromatography eluting with 2%–4% MeOH in CH2Cl2. The diol was obtained as a colorless oil; 901 mg, 55%; 1H-NMR δ 4.84 (m, 2H), 4.38 (q, 2H, J = 8), 4.01 (m, 2H), 3.64 (br s, 1H), 3.47 (m, 3H), 3.21 (m, 2H), 1.37 (m, 12H); 13C-NMR δ 162.4, 162.2, 157.3, 156.0, 129.0, 79.9, 68.4, 65.0, 61.2, 38.7, 28.3, 27.4, 14.4; IR 3406, 2979, 1693, 1520, 1368, 1252, 1168, 1093, 1046 cm−1; HRMS (ESI) m/z calcd for C15H24N2O7Na (M+Na): 367.1476; found: 367.1476. Step B. The diol (900 mg, 2.62 mmol) and imidazole (356 mg, 5.23 mmol) were dissolved in anhydrous DMF (10 mL) and placed under argon. The reaction mixture was cooled to 0 °C and a solution of tert-butyldimethylsilyl chloride (434 mg, 2.88 mmol) in DMF (2 mL) was added dropwise. This was allowed to slowly warm to room temperature and stirred for 24 h. Additional tert-butyldimethylsilyl chloride (120 mg, 0.8 mmol) was added and the reaction stirred at room temperature for 6 h. This was then poured into 5% HCl and extracted with CH2Cl2. The organic extracts were dried with Na2SO4 and concentrated in vacuo to give a colorless oil. This was flash chromatographed on SiO2 with 20%–40% EtOAc in hexane and product 26 was isolated as a colorless oil; 942 mg, 79%; 1H-NMR δ 4.82 (m, 1H), 4.80 (br s, 1H), 4.35 (q, 2H, J = 8), 3.93 (m, 2H), 3.42 (m, 2H), 3.20 (t, 2H, J = 8), 3.12 (d, 1H, J = 4), 1.35 (m, 12H), 0.82 (s, 9H), 0.01 (d, 6H, J = 4); 13C-NMR δ 162.1, 161.2, 157.4, 155.7, 128.7, 79.5, 68.5, 65.3, 61.2, 38.8, 28.4, 25.8, 18.2, 14.2, −5.43; IR 3365, 2955, 2931, 2858, 1717, 1614, 1518, 1463, 1392, 1367, 1326, 1252, 1176, 1127, 1096, 1046, 839, 780 cm−1; HRMS (ESI) m/z calcd for C21H38N2O7SiNa (M+Na): 481.2340; found: 481.2336.
Ethyl 5-[2-[(tert-Butoxycarbonyl)amino]ethyl]-2-[[(tert-butyldimethylsilyl)oxy]-1-(methanesulfonyloxy)ethyl]oxazole-4-carboxylate (27). Prepared using the procedure detailed above for 18, Step B. Concentration in vacuo afforded product 27 as a colorless oil; 958 mg, 97%; 1H-NMR δ 5.58 (dd, 1H, J = 4,8), 4.79 (br s, 1H), 4.32 (q, 2H, J = 8), 4.08 (m, 2H), 3.38 (m, 2H), 3.17 (m, 2H), 3.02 (m, 4H), 1.32 (m, 12H), 0.80 (s, 9H), 0.01 (d, 6H, J = 4); 13C-NMR δ 161.7, 158.3, 156.7, 155.7, 129.2, 79.5, 75.2, 63.3, 61.4, 45.9, 38.8, 28.3, 27.1, 25.7, 18.2, 14.3, −5.38; IR 3407, 2933, 2858, 2251, 1716, 1611, 1513, 1473, 1366, 1253, 1175, 1133, 1094, 1031, 971, 918, 839, 782, 735, 668, 647 cm−1; HRMS (ESI) m/z calcd for C22H41N2O6SiS (M+H): 537.2297; found: 537.2281.
Ethyl 2-[1-Amino-2-[(tert-butyldimethylsilyl)oxy]ethyl]-5-[2-[(tert-butoxycarbonyl)amino]ethyl]oxazole-4-carboxylate (28). Step A. Prepared using the procedure detailed above for 13. Concentration in vacuo afforded the azide as a colorless oil; 813 mg, 94%; 1H-NMR δ 4.76 (br s, 1H), 4.57 (m, 1H), 4.30 (q, 2H, J = 8), 4.01 (m, 2H), 3.38 (m, 2H), 3.16 (t, 2H, J = 8), 1.31 (m, 12H), 0.80 (s, 9H), 0.00 (d, 6H, J = 4); 13C-NMR δ 160.8, 157.1, 156.8, 154.7, 127.9, 78.4, 63.5, 60.3, 58.7, 37.7, 27.3, 25.9, 24.6, 17.1, 13.3, −6.53; IR 3372, 2932, 2858, 2107, 1716, 1613, 1514, 1464, 1366, 1323, 1253, 1175, 1096, 1031, 839, 780, 733 cm−1; HRMS (ESI) m/z calcd for C21H38N5O6Si (M+H): 484.2586; found: 484.2578. Step B. Prepared using the procedure detailed above for 4a, Step B. Product 28 was obtained as a pale yellow oil; 510 mg, 66%; 1H-NMR δ 4.78 (br s, 1H), 4.36 (q, 2H, J = 8), 4.13 (m, 1H), 3.90 (m, 2H), 3.42 (m, 2H), 3.18 (t, 2H, J = 8), 2.29 (br s, 2H), 1.36 (m, 12H), 0.82 (s, 9H), −0.01 (d, 6H, J = 4); 13C-NMR δ 162.3, 157.0, 156.5, 155.7, 128.6, 79.4, 65.9, 61.2, 52.1, 39.9, 28.4, 26.8, 25.8, 18.2, 14.4, −5.44; IR 3379, 2931, 2857, 1716, 1615, 1518, 1463, 1366, 1252, 1174, 1095, 838, 779 cm−1; HRMS (ESI) m/z calcd for C21H40N3O6Si (M+H): 458.2681; found: 458.2671.
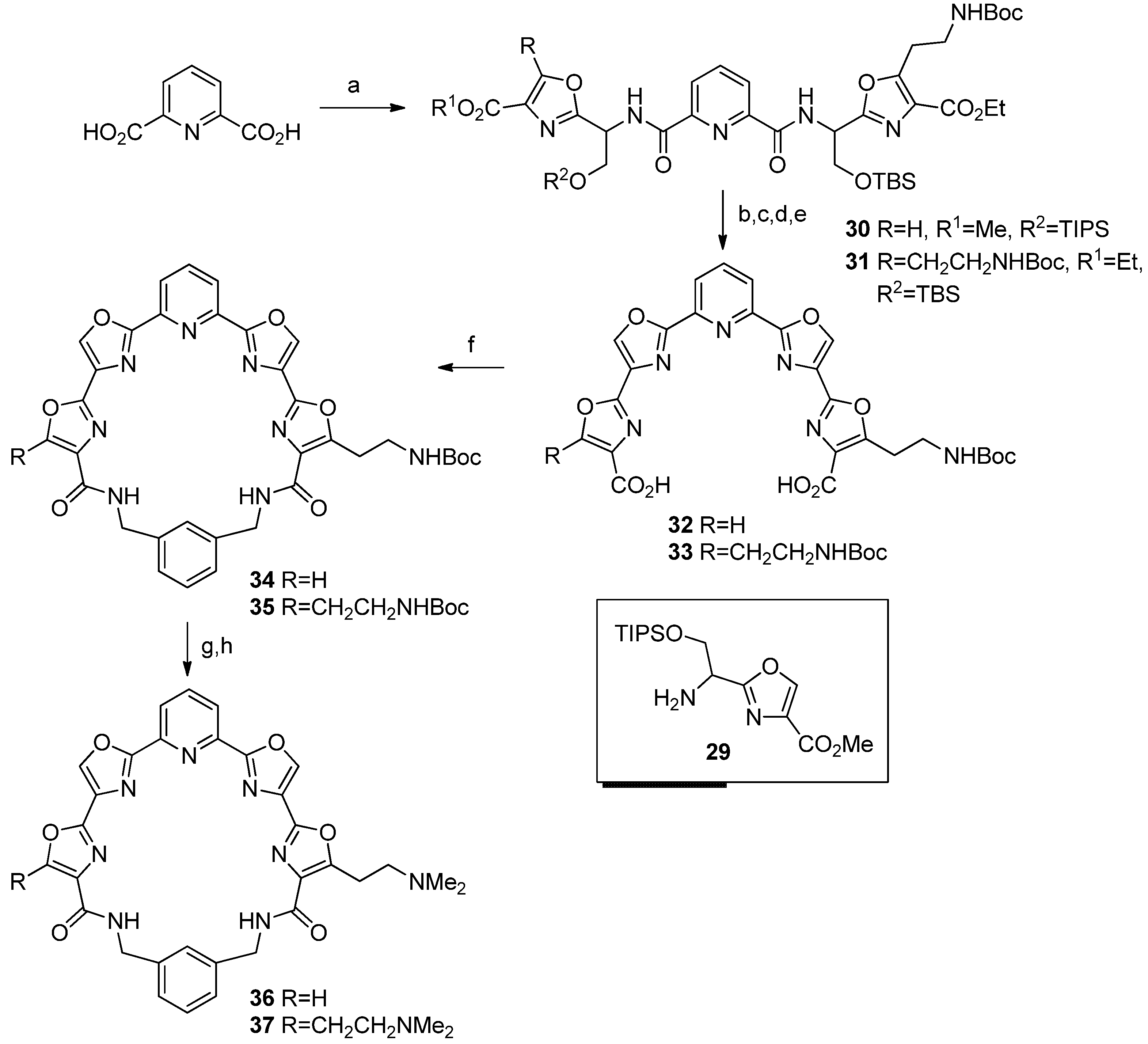
Ethyl 5-[2-[(tert-butoxycarbonyl)amino]ethyl]-2-[2-[(tert-Butyldimethylsilyl)oxy]-1-[[6-[[1-(4-methoxycarbonyl)oxazol-2-yl]-2-[(triiso-propylsilyl)oxy]ethyl]carbamoyl]picolinamido]ethyl]oxazole-4-carboxylate (
30).
Step A. 2,6-Pyridinedicarboxylate (366 mg, 2.2 mmol) and oxazole
29 [
17] (500 mg, 1.46 mmol) were dissolved in anhydrous DMF (5 mL) and placed under argon. The solution was cooled to 0 °C and treated dropwise with a solution of EDC (288 mg, 1.46 mmol), HOBt (197 mg, 1.46 mmol) and 2,6-lutidine (0.34 mL, 2.92 mmol) in DMF (5 mL). The reaction was kept at low temperature for 4 h and then warmed to room temperature and stirred overnight. This was then poured into brine and extracted with EtOAc. The combined organic extracts were washed sequentially with 10% sodium bicarbonate, 5% HCl, water and brine. This was dried with Na
2SO
4 and the solvent was removed under reduced pressure to give a colorless oil. The residue was flash chromatographed on SiO
2 eluting with 15%–50% EtOAc/hexane to give 196 mg of the amide as a colorless oil, 27%;
1H-NMR δ 9.28 (d, 1H,
J = 12), 8.37 (d, 1H,
J = 8), 8.31 (d, 1H,
J = 8), 8.15 (s, 1H), 8.00 (m, 1H), 5.60 (m, 1H), 4.20 (m, 2H), 3.80 (s, 3H), 0.88 (m, 21H);
13C-NMR δ 165.6, 164.1, 163.4, 161.1, 149.1, 146.1, 144.0, 139.1, 133.3, 127.5, 126.4, 64.4, 52.1, 49.7, 17.7, 11.7; IR 3316, 2945, 2867, 1748, 1685, 1584, 1525, 1456, 1345, 1252, 1203, 1114, 1072, 1000, 918, 882, 848, 801, 734, 684, 642 cm
−1; HRMS (ESI)
m/z calcd. for C
23H
34N
3O
7Si (M+H): 492.2161; found: 492.2145
. Step B. The amide from above (196 mg, 0.4 mmol) and oxazole
28 (182 mg, 0.4 mmol) were treated as detailed above in
stepA. Flash chromtography on SiO
2 eluting with 10%–40% EtOAc/hexane gave the product
30 as a colorless oil, 245 mg, 66%;
1H-NMR δ 8.37 (d, 1H,
J = 4), 8.35 (m, 3H), 8.25 (s, 1H), 8.21 (s, 1H), 8.01 (m, 1H), 5.60 (m, 1H), 5.47 (m, 1H), 4.84 (m, 1H), 4.35 (q, 2H,
J = 8), 4.25 (m, 2H), 4.13 (m, 2H), 3.81 (s, 3H), 3.31 (m, 2H), 3.15 (m, 2H), 1.22 (m, 12H), 0.99 (m, 21H), 0.78 (s, 9H), −0.01 (m, 6H);
13C-NMR δ 171.1, 163.6, 163.4, 163.0, 162.9, 161.9, 161.3, 160.4, 157.2, 155.8, 148.5, 144.2, 138.9, 133.4, 129.0, 125.7, 79.3, 64.2, 63.6, 60.3, 52.1, 50.2, 49.9, 38.7, 28.2, 25.6, 21.0, 17.8, 14.2, 11.8, −5.50; IR 3337, 2945, 2866, 1719, 1683, 1583, 1524, 1464, 1444, 1366, 1324, 1252, 1172, 1115, 1000, 919, 882, 840, 780, 733, 646 cm
−1; HRMS (ESI)
m/z calcd for C
44H
70N
6O
12Si
2Na (M+Na): 953.4482; found: 953.4459.
Diethyl 2,2'-[[(Pyridine-2,6-dicarbonyl)bis(azanediyl)]bis[2-[(triisopropylsilyl)oxy]ethane-1,1-diyl]]bis[5-[2-[(tert-butoxycarbonyl)amino]ethyl]oxazole-4-carboxylate] (31). 2,6-Pyridinedicarboxylic acid (37 mg, 0.22 mmol), amine 28 (200 mg, 0.44 mmol) were treated as detailed above for 30, Step A. Product 31 was obtained as a colorless oil; 195 mg, 85%; 1H-NMR δ 8.36 (m, 2H), 8.02 (m, 1H), 5.52 (m, 1H), 4.36 (m, 4H), 4.13 (m, 2H), 3.42 (m, 2H), 3.17 (m, 2H), 1.30 (m, 24H), 0.80 (s, 18H), 0.00 (s, 12H); 13C-NMR δ 163.2, 160.8, 157.1, 156.0, 148.6, 138.9, 127.1, 125.7, 79.4, 63.6, 61.0, 49.8, 38.7, 28.3, 26.9, 25.6, 21.3, 18.0, -5.2; IR 3343, 2931, 2858, 1716, 1615, 1525, 1463, 1392, 1367, 1348, 1325, 1253, 1175, 1122, 1094, 1032, 1003, 919, 840, 780, 735 cm−1; HRMS (ESI) m/z calcd for C49H79N7O14Si2Na (M+Na): 1068.5116; found: 1068.5117.
5-[2-[(tert-Butoxycarbonyl)amino]ethyl]-2'-[6-[4-carboxy-[2,4'-bioxazol]-2'-yl]pyridine-2-yl]-[2,4'-bioxazole]-4-carboxylic acid (32). Step A. 30 (245 mg, 0.26 mmol) was dissolved in anhydrous THF (10 mL) and pyridine (1 mL) and HF-pyridine complex (0.3 mL) was added. The reaction was stirred at room temperature overnight and was then poured into saturated sodium bicarbonate solution. This was extracted with CH2Cl2 and dried with Na2SO4. Removal of solvent under vacuum gave 174 mg of the diol as a colorless oil, 100%; 1H-NMR δ 8.17 (m, 4H), 7.78 (m, 1H), 5.58 (m, 2H), 5.12 (s, 1H), 4.27 (m, 6H), 3.83 (s, 3H), 3.44 (m, 2H), 3.21 (t, 2H, J = 8), 1.36 (s, 9H), 1.08 (t, 3H, J = 8); 13C-NMR δ 161.2, 161.7, 161.4, 160.9, 160.2, 158.6, 157.7, 155.9, 149.5, 148.1, 144.4, 138.5, 136.2, 132.8, 128.1, 125.3, 79.4, 70.3, 62.0, 52.1, 38.4, 29.2, 28.3, 25.0, 22.6, 17.1; IR 3333 (br), 2954, 2250, 1720, 1678, 1617, 1582, 1530, 1442, 1367, 1347, 1324, 1275, 2248, 1174, 1114, 1000, 963, 915, 846, 805, 774, 734, 705, 646 cm−1; HRMS (ESI) m/z calcd for C29H36N6O12Na (M+Na): 683.2283; found: 683.2262. Step B. The diol (174 mg, 0.26 mmol) was dissolved in anhydrous CH2Cl2 (7 mL) and placed under argon. The flask was cooled to −78 °C and the solution was treated with DAST (87 μL, 0.66 mmol). The reaction stirred at low temperature for 4 h and then solid K2CO3 (81 mg, 0.66 mmol) was added. The reaction was warmed to room temperature and poured into saturated sodium bicarbonate solution. This was extracted with CH2Cl2 and the combined organic extracts were dried with Na2SO4. The solvent was removed in vacuo to give the bis(oxazoline) 153 mg as a yellow oil, 93%; 1H-NMR δ 8.26 (m, 3H), 7.94 (t, 1H, J = 8), 5.63 (m, 2H), 4.92 (m, 5H), 4.38 (m, 2H), 3.92 (s, 3H), 3.46 (m, 2H), 3.24 (t, 2H, J = 8), 1.41 (s, 9H); 13C-NMR δ 165.1, 164.9, 163.0, 161.9, 161.3, 160.6, 158.1, 155.7, 146.3, 146.1, 144.8, 137.6, 133.4, 128.9, 126.8, 126.7, 79.3, 71.3, 68.4, 64.0, 61.2, 52.2, 42.1, 38.7, 29.6, 28.3, 14.3; IR 3381, 2977, 2931, 1716, 1639, 1582, 1518, 1460, 1366, 1345, 1322, 1249, 1176, 1144, 1112, 1033, 978, 918, 833, 804, 734 cm−1; HRMS (ESI) m/z calcd for C29H32N6O10Na (M+Na): 647.2072; found: 647.2054. Step C. The bis(oxazoline) (153 mg, 0.25 mmol) was dissolved in anhydrous CH3CN and placed under argon. The flask was cooled to 0 °C and the solution was treated drop-wise sequentially with DBU (156 μL 1.04 mmol) and BrCCl3 (123 μL, 1.25 mmol). The reaction was gradually warmed to room temperature and stirred overnight. A white solid precipitated and was filtered and washed with CH3CN. The solid was dried to give 91 mg of the tetraoxazole diester as a white solid, 60%; mp 222 °C (dec); 1H-NMR δ 8.42 (s, 1H), 8.37 (s, 1H), 8.31 (m, 3H), 8.07 (t, 1H, J = 8), 4.85 (s, 1H), 4.42 (m, 2H), 5.31 (s, 3H), 3.56 (m, 2H), 3.35 (m, 2H), 1.42 (s, 9H); 13C NMR δ 162.0, 161.3, 160.8, 160.5, 157.3, 155.4, 153.3, 145.6, 145.5, 144.0, 141.1, 138.5, 134.5, 131.5, 131.3, 129.9, 124.4, 124.2 79.5, 61.4, 54.4, 38.9, 28.4, 27.0, 14.4; IR 3356, 3111, 2978, 1719, 1574, 1523, 1453, 1367, 1325, 1253, 1158, 1098, 1045, 998, 971, 926, 824, 780, 734, 712 cm−1. Step D. The diester (71 mg, 0.13 mmol) was suspended in a mixture of THF (30 mL) and water (3 mL) and lithium hydroxide (12 mg, 0.29 mmol) was added. The reaction was refluxed for 30 min and then stirred at room temperature overnight. THF was removed under vacuum and 5% HCl was added to the remaining solution. A white solid precipitated and was filtered and washed with water. The solid was dried by azeotroping with toluene 3 times to give 50 mg of diacid 32, as a white solid, 67%; mp 225–226 °C; HRMS (ESI) m/z calcd for C26H22N6O10Na (M+Na): 601.1290; found: 601.1284.
2,2'-(Pyridine-2,6-diyl)bis[5-[(tert-butoxycarbonyl)amino]ethyl]-[2,4'-bioxazole]-4-carboxylic acid (33). Step A. Prepared using the procedure detailed above for 32 Step A. The diol was obtained as a yellow oil, 134 mg, 89%; 1H-NMR δ 8.60 (s, 2H), 7.67 (m, 1H), 5.50 (m, 1H), 4.23 (m, 4H), 3.45 (m, 2H), 3.15 (m, 2H), 1.36 (m, 24H); 13C-NMR δ 163.6, 161.9, 157.8, 156.0, 155.9, 148.5, 125.3, 123.8, 79.5, 62.9, 61.2, 53.5, 37.7, 28.3, 27.1, 14.2; IR 3339, 2978, 2934, 2248, 1716, 1616, 1529, 1445, 1367, 1347, 1325, 1281, 1250, 1174, 1092, 1047, 1000, 917, 846, 788, 733, 706 cm−1; HRMS (ESI) m/z calcd for C37H51N7O14Na (M+Na): 840.3386; found: 840.3383. Step B. Prepared using the procedure detailed above for 32 Step B. The bis(oxazoline) was obtained as an orange oil; 116 mg, 91%; 1H-NMR δ 8.25 (d, 2H, J = 8), 7.93 (t, 1H, J = 8), 5.61 (t, 2H, J = 8), 4.91 (m, 6H), 4.39 (q, 4H, J = 8), 3.46 (m, 4H), 3.24 (t, 4H, J = 8), 1.41 (m, 24H); 13C-NMR δ 164.9, 162.0, 160.6, 158.1, 155.7, 146.2, 137.6, 128.9, 126.8, 79.5, 71.3, 64.0, 61.2, 38.7, 28.3, 18.9; IR 3364, 2977, 1713, 1520, 1458, 1366, 1250, 1175, 1093, 1031, 921, 844, 733 cm−1; HRMS (ESI) m/z calcd for C37H47N7O12Na (M+Na): 804.3175; found: 804.3167. Step C. Prepared using the procedure detailed above for 32 Step C. This was purified by flash chromatography eluting with 1%–4% MeOH/CH2Cl2. The tetra-oxazole diester was isolated as a white solid; 58 mg, 50%; mp 245–247 °C; 1H-NMR (CDCl3 + CD3OD) δ 8.55 (s, 2H), 8.43 (d, 2H, J = 4), 8.08 (t, 1H, J = 8), 5.13 (br s, 2H), 4.44 (d, 4H, J = 8), 3.56 (m, 4H), 3.36 (m, 4H), 1.42 (m, 24H); 13C-NMR (CDCl3 + CD3OD) δ 161.8, 160.4, 157.3, 155.7, 153.2, 145.5, 140.6, 138.4, 131.4, 129.7, 124.2, 79.3, 61.3, 38.1, 28.3, 27.0, 14.3; IR 3355, 2978, 1710, 1639, 1524, 1452, 1367, 1250, 1171, 1089, 1048, 926, 733 cm−1; HRMS (ESI) m/z calcd for C37H43N7O12Na (M+Na): 800.2862; found: 800.2853. Step D. Prepared using the procedure detailed above for 32 Step D. Diacid 33 was obtained as a white solid; 35 mg, 65%; mp 250–252 °C (dec.); 1H-NMR (CDCl3 + CD3OD) δ 8.28 (s, 2H), 8.11 (d, 2H, J = 4), 7.82 (t, 1H, J = 8), 5.44 (br s, 2H), 3.22 (m, 4H), 3.05 (m, 4H), 1.13 (s, 18H); 13C-NMR (CDCl3 + CD3OD) δ 168.2, 165.1, 161.9, 157.8, 157.7, 150.3, 145.3, 143.3, 136.4, 134.8, 128.9, 83.6, 43.6, 35.1, 19.1; IR 3439, 2977, 2253, 2127, 1702, 1525, 1453, 1392, 1366, 1342, 1281, 1250, 1173, 1026, 926, 824, 762, 710 cm−1; HRMS (ESI) m/z calcd for C33H35N7O12Na (M+Na): 744.2236; found: 744.2229.
Pyridyl tetraoxazole macrocycle with a single 2-[[(tert-butoxy)carbonyl]amino]ethyl side chain (34). Prepared from 32 using the procedure detailed above for 8. Flash chromatography on SiO2 eluting with 1%–6% MeOH/CH2Cl2 gave macrocycle 34 as a white solid, 8 mg, 20%; mp 144–145 °C; 1H-NMR δ 8.45 (m, 3H), 8.27 (m, 3H), 8.08 (m, 2H), 7.49 (m, 5H), 5.29 (s, 1H), 4.58 (m, 4H), 3.55 (m, 2H), 3.37 (t, 2H, J = 8), 1.43 (s, 9H); 13C-NMR δ 161.0, 160.5, 160.3, 159.7, 156.1, 154.3, 145.6, 145.5, 140.5, 139.0, 138.4, 138.5, 137.7, 123.5, 131.9, 130.9, 129.9, 129.6, 129.5, 128.8, 122.6, 79.5, 43.8, 39.2, 29.0, 28.4; IR 3390, 2959, 2929, 2859, 1727, 1666, 1594, 1516, 1463, 1366, 1274, 1170, 1122, 1073, 991, 777, 738, 709 cm−1; HRMS (ESI) m/z calcd for C34H30N8O8Na (M+Na): 701.2079; found: 701.2069.
Pyridyl tetraoxazole macrocycle with two 2-[[(tert-butoxy)carbonyl]amino]ethyl side chains (35). Prepared from 33 using the procedure detailed above for 8. Flash chromatography eluting with 1–5% MeOH/CH2Cl2 to give macrocycle 35 as a white solid; 11 mg, 28%; mp 170–173 °C; 1H-NMR δ 8.24 (s, 2H), 8.06 (d, 2H, J = 8), 7.58 (s, 1H), 7.37 (m, 4H), 5.26 (s, 2H), 4.57 (d, 4H, J = 4), 3.55 (m, 4H), 3.37 (t, 4H, J = 8), 1.43 (s, 18H); 13C-NMR δ 161.1, 160.4, 156.1, 153.7, 152.2, 145.5, 138.8, 138.4, 137.9, 131.9, 131.5, 129.8, 129.4, 122.7, 79.3, 43.8, 39.2, 28.4, 26.4; IR 3322, 2976, 1695, 1646, 1525, 1440, 1366, 1284, 1250, 1170, 1093, 1047, 992, 926, 780, 734, 707 cm−1; HRMS (ESI) m/z calcd for C41H43N9O10Na (M+Na): 844.3025; found: 844.3024.
Pyridyl tetraoxazole macrocycle with a single 2-(N,N-dimethylamino)ethyl side chain (36). Step A. Prepared from 34 using the procedure detailed above for 22 Step A. The crude product was washed with hexane and dried to give 6 mg of the salt as a white solid, 100%; mp 170 °C (dec); 1H-NMR (DMSO-d6) δ 9.17 (s, 1H), 9.06 (s, 1H), 8.85 (s, 1H), 8.26 (m, 4H), 7.93 (m, 3H), 7.36 (m, 3H), 4.49 (m, 4H), 3.46 (m, 2H), 3.25 (m, 2H); IR 2958, 2918, 2855, 2351, 1726, 1671, 1650, 1595, 1540, 1507, 1441, 1266, 1178, 1111, 986, 915, 833, 800, 707 cm−1; HRMS (free-base form) (ESI) m/z calcd for C29H22N8O6Na (M+H): 579.1735; found: 579.1758. Step B. Prepared using the procedure detailed above for 11 Step B. Product 36 was obtained as a white solid; 4 mg, 100%; mp 150–155 °C; 1H NMR (CDCl3 + CD3OD) δ 8.36 (s, 1H), 8.35 (s, 1H), 8.33 (s, 1H), 8.11 (m, 3H), 7.37 (m, 4H), 4.60 (m, 4H), 3.40 (m, 2H), 2.46 (s, 6H); 13C-NMR (CDCl3 + CD3OD) δ 161.0, 141.5, 139.3, 137.8, 129.5, 129.2, 129.1, 123.0, 122.9, 57.0, 44.9, 43.6, 23.9; IR 3395, 2964, 2918, 2849, 2351, 1732, 1661, 1655, 1595, 1545, 1514, 1463, 1447, 1370, 1321, 1266, 1173, 1108, 992, 926, 921, 822, 734, 722 cm−1; HRMS (ESI) m/z calcd for C31H26N8O6 607.2048; found: 607.2069.
Pyridyl tetraoxazole macrocycle with two 2-(N,N-dimethylamino)ethyl side chains (37). Step A. Prepared from 35 using the procedure detailed above for 22 Step A. The bis(trifluoroacetate) salt was obtained as a white solid; 11 mg, 100%; mp 260 °C (dec.); 1H-NMR (CD3OD) δ 8.63 (s, 2H), 8.57 (s, 4H), 7.53 (s, 1H), 7.30 (s, 4H), 4.48 (s, 4H), 3.42 (s, 4H); IR 3405, 2964, 2926, 2855, 1653, 1529, 1452, 1266, 1025, 992, 827, 800 cm−1; HRMS (ESI) m/z calcd for C31H28N9O6 (M+H): 622.2142; found: 622.2140. Step B. Prepared using the procedure detailed above for 11 Step B. Bis(dimethylamino) product 37 was obtained as a white solid; 6 mg, 100%; mp 180–184 °C; 1H-NMR (CDCl3 + CD3OD) δ 8.31 (s, 2H), 8.08 (s, 3H), 7.52 (s, 1H), 7.35 (s, 3H), 5.32 (s, 4H), 2.80 (t, 4H, J = 8), 2.39 (m, 16H); 13C-NMR (CDCl3 + CD3OD) δ 162.5, 160.9, 145.4, 139.0, 129.4, 129.1, 122.8, 64.5, 44.9, 43.7, 23.9; HRMS (ESI) m/z calcd for C35H36N9O6 (M+H): 678.2789; found: 678.2793.
Temperature-Dependent Spectrophotometry. Temperature-dependent absorption experiments were conducted on an AVIV Model 14DS Spectrophotometer (Aviv Biomedical, Lakewood, NJ, USA) equipped with a thermoelectrically controlled cell holder. Quartz cells with a path length of 1.0 cm were used for all the absorbance studies. Temperature-dependent absorption profiles were acquired at either 260 nm (for ST duplex DNA) or 295 nm (for hTel quadruplex DNA) with a 5 s averaging time. The temperature was raised in 0.5 °C increments, and the samples were allowed to equilibrate for 1 min at each temperature setting. In the quadruplex melting studies, the hTel concentration was 5 µM in strand (120 μM in nucleotide). When present in these quadruplex studies, the drug concentrations were 20 µM. In the duplex melting studies the ST DNA concentrations were 15 µM base pair (30 μM in nucleotide) and, when present, the drug concentrations were 15 µM. The buffer for all the UV melting experiments contained 10 mM potassium phosphate (pH 7.5) and sufficient KCl (132 mM) to bring the total K+ concentration to 150 mM. Prior to their use in the UV melting experiments, all nucleic acid solutions were preheated at 90 °C for 5 min and slowly cooled to room temperature over a period of 4 hr.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}