An Efficient Solvent-Free Synthesis of 2-Hydroxy-2-(trifluoromethyl)-2H-chromenes Using Silica-Immobilized L-Proline

Abstract
:1. Introduction
2. Results and Discussion
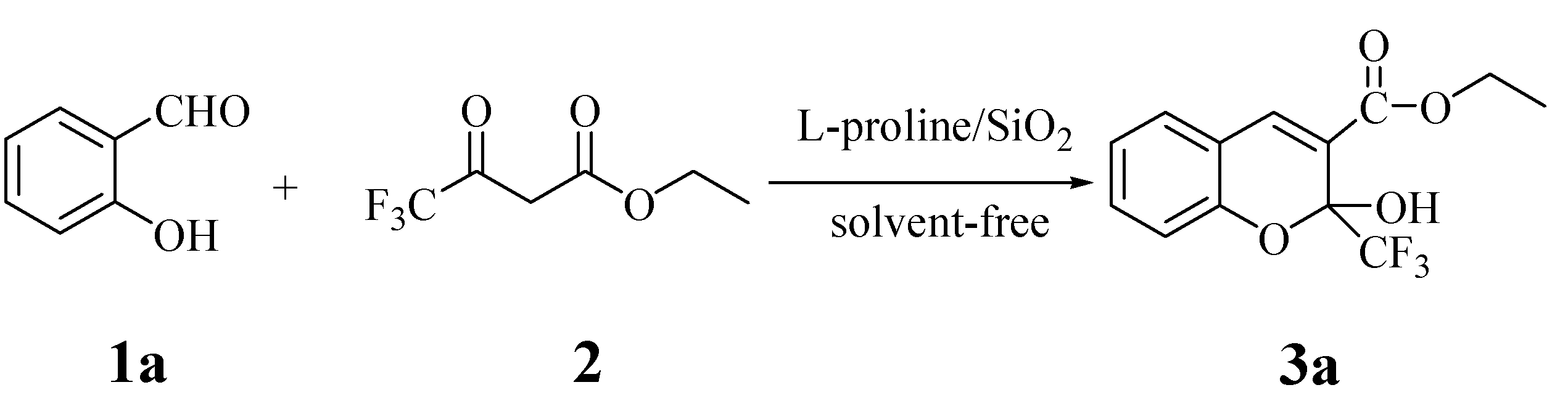
2.1. Synthesis


{kind=link}
{kind=link}
{kind=link}
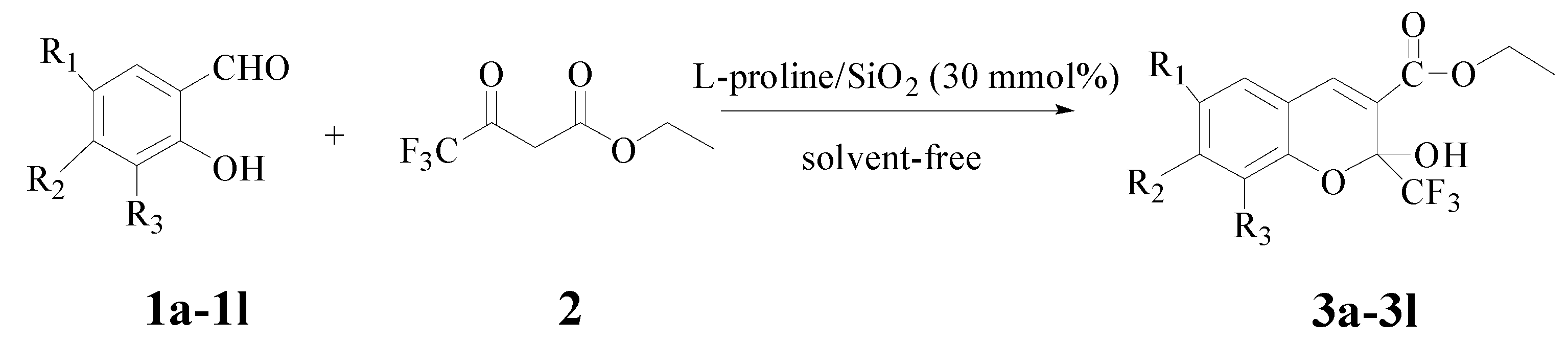{kind=link}
{kind=link}
| Entry | Mol ratio of 1a and 2 | Loading of catalyst (mol%) a | Time | Yield b (%) |
|---|---|---|---|---|
| 1 | 1:1.5 | 0 | 6 h c | 0.5 |
| 2 | 1:1.5 | 0 | 20 min | 0.7 |
| 3 | 1:1.5 | 20 | 6 h c | 69 |
| 4 | 1:1.5 | 10 | 15 min | 38 |
| 5 | 1:1.5 | 20 | 18 min | 80 |
| 6 | 1:1.5 | 30 | 14 min | 82 |
| 7 | 1:1.5 | 40 | 9 min | 82 |
| 8 | 1:1 | 30 | 17 min | 43 |
| 9 | 1:2 | 30 | 14 min | 82 |
| 10 | 1:2.5 | 30 | 15 min | 56 |
| 11 | 1:3 | 30 | 15 min | 38 |
| 12 | 1:1.5 | 30 | 14 min | 82 d |
| 13 | 1:1.5 | 30 | 14 min | 80 e |

| Entry | R1 | R2 | R3 | Product | MWI (126 W) | Without MWI b | ||
|---|---|---|---|---|---|---|---|---|
| Time (min) | Yield a (%) | Time (h) | Yield a (%) | |||||
| 1 | H | H | H | 3a | 12 | 82 | 6 | 75 |
| 2 | Cl | H | H | 3b | 17 | 89 | 6 | 81 |
| 3 | Br | H | H | 3c | 16 | 85 | 6 | 77 |
| 4 | Cl | H | Cl | 3d | 12 | 87 | 8 | 80 |
| 5 | Br | H | Br | 3e | 16 | 88 | 6 | 81 |
| 6 | H | H | OMe | 3f | 12 | 70 | 6 | 69 |
| 7 | H | OMe | H | 3g | 8 | 68 | 8 | 65 |
| 8 | H | H | OEt | 3h | 16 | 72 | 8 | 66 |
| 9 | Me | H | H | 3i | 18 | 81 | 6 | 65 |
| 10 | H | OH | H | 3j | 17 | 66 | 8 | 67 |
| 11 | NO2 | H | H | 3k | 6 | 92 | 2 | 83 |
| 12 | H | -CH=CH-CH=CH- | 3l | 18 | 74 | 8 | 68 | |

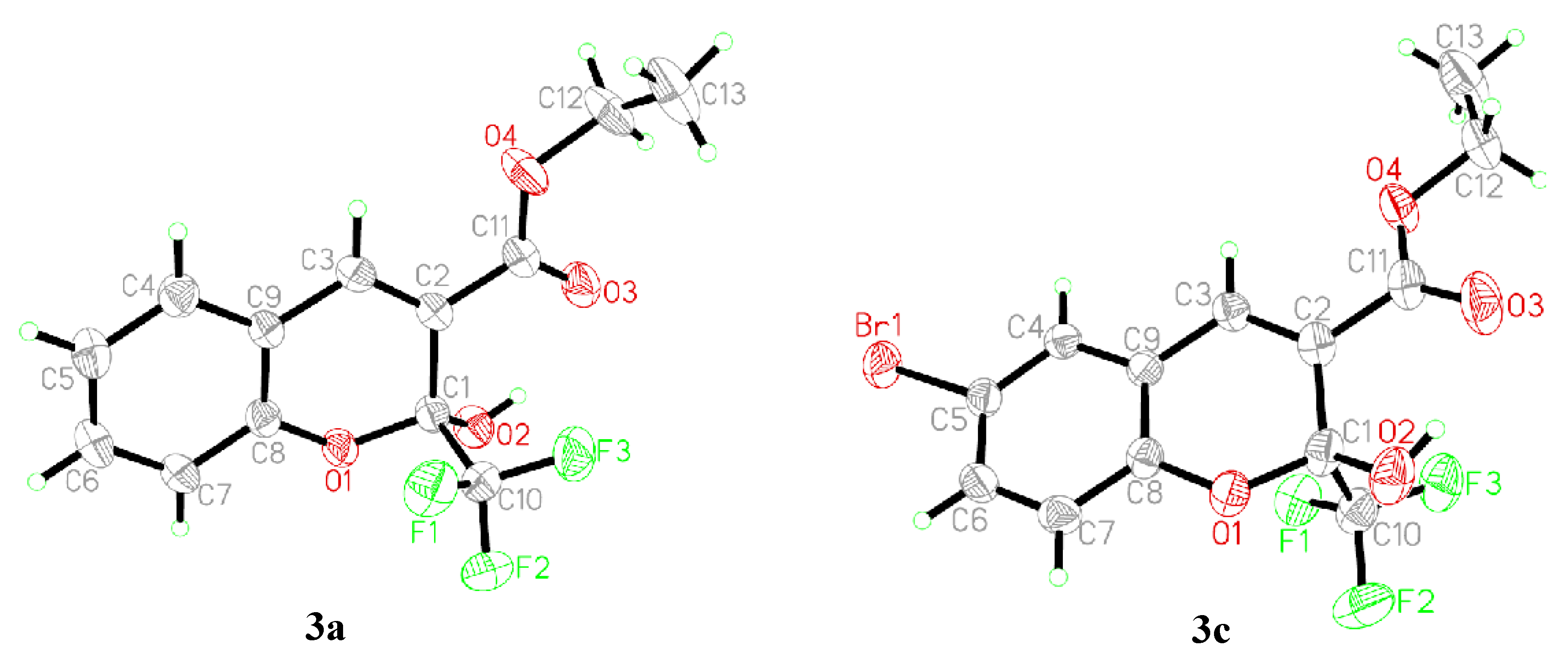
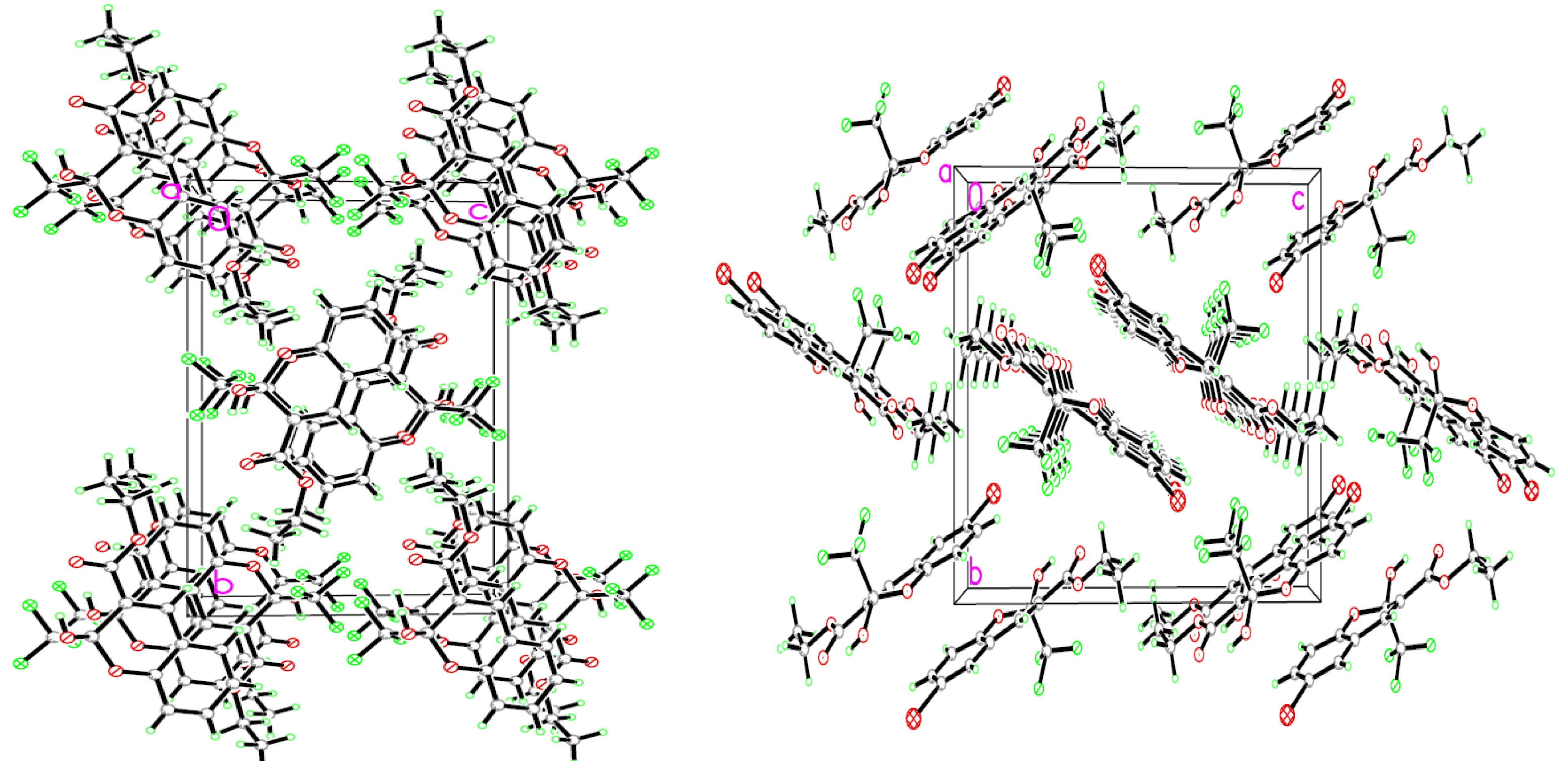
2.2. Structural Characterization of Chromenes 3a–l


| 3a | 3c | |
|---|---|---|
| formula | C13H11F3O4 | C13H10BrF3O4 |
| Fw | 288.22 | 367.12 |
| crystal system | monoclinic | monoclinic |
| space group | P2(1)/c | P2(1)/c |
| a (Å) | 8.415 (1) | 7.085 (2) |
| b (Å) | 16.604 (3) | 12.831(3) |
| c (Å) | 9.443 (9) | 15.526 (3) |
| β (deg) | 97.51 (3) | 93.52 (3) |
| V (Å3) | 1308.2 (5) | 1408.9 (5) |
| Z | 4 | 4 |
| T (K) | 293 (2) | 293 (2) |
| Dcalc (Mg m−3) | 1.463 | 1.731 |
| μ (mm−1) | 0.135 | 2.964 |
| R1 (I>2σ(I)) | 0.0792 | 0.0492 |
| R1 (all data) | 0.0829 | 0.0651 |
| wR2 (I>2σ(I)) | 0.2261 | 0.1433 |
| wR2 (all data) | 0.2310 | 0.1553 |
| GOOF | 1.052 | 1.074 |
| Crystal | D-H· A | d(D-H) | d(D·A) | D-H· A |
|---|---|---|---|---|
| 3a | O2-H2· O3 | 0.82 | 2.71 (1) | 143 |
| 3c | O2-H2· O3 | 0.82 | 2.64 (1) | 147 |
3. Experimental
3.1. Gereral
3.2. Preparation of Catalyst L-Proline/SiO2
3.3. General Synthetic Procedures for Compounds 3a–3l
3.3.1. Oven Heating Procedure
3.3.2. Microwave Irradiation Procedure
4. Conclusion
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Curini, M.; Cravotto, G.; Epifano, F.; Giannone, G. Chemistry and biological activity of natural and synthetic prenyloxycoumarins. Curr. Med. Chem. 2006, 13, 199–222. [Google Scholar] [CrossRef]
- Monga, P.K.; Sharma, D.; Dubey, A. Comparative Study of Microwave and Conventional Synthesis and Pharmacological activity of Coumarins: A Review. J. Chem. Pharm. Res. 2012, 4, 822–850. [Google Scholar]
- Schweizer, E.E.; Meeder-Nycz, D. Chromenes, Chromanes, Chromones; Ellis, G.P., Ed.; Wiley-Interscience: New York, NY, USA, 1977. [Google Scholar]
- Bedoya, L.M.; Beltrán, M.; Sancho, R.; Olmedo, D.A.; Sánchez-Palomino, S.; del Olmo, E.; López-Pérez, J.L.; Muñoz, E.; San Feliciano, A.; Alcamí, J. 4-Phenylcoumarins as HIV transcription inhibitors. Bioorg. Med. Chem. Lett. 2005, 21, 4447–4450. [Google Scholar]
- Yu, D.; Suzuki, M.; Xie, L.; Morris-Natschke, S.L.; Lee, K.H. Recent progress in the development of coumarin derivatives as potent anti-HIV agents. Med. Res. Rev. 2003, 23, 322–345. [Google Scholar] [CrossRef]
- Xiao, G.Q.; Liang, B.X.; Chen, S.H.; Ou, T.M.; Bu, X.Z.; Yan, M. 3-Nitro-2H-chromenes as a new class of inhibitors against thioredoxin reductase and proliferation of cancer cells. Arch. Pharm. Chem. Life Sci. 2012, 345, 767–770. [Google Scholar] [CrossRef]
- Thomas, N.; Zachariah, S.M. Pharmacological activities of chromene derivatives: An overview. Asian J. Pharm. Clin. Res. 2011, 6, 11–15. [Google Scholar]
- Abdel-Wahab, A.H.F.; Mohamed, H.M.; El-Agrody, A.M.; El-Nassag, M.A.; Bedair, A.H. Synthesis and reactions of some new benzylphthalazin-1-ylaminophenols, 2H-chromene and 5H-chromeno[2,3-d]pyrimidine derivatives with promising antimicrobial activities. Lett. Org. Chem. 2012, 9, 360–367. [Google Scholar] [CrossRef]
- Kidwai, M.; Saxena, S.; Khan, M.K.; Thukral, S.S. Aqua mediated synthesis of substituted 2-amino-4H-chromenes and in vitro study as antibacterial agents. Bioorg. Med. Chem. Lett. 2005, 15, 4295–4298. [Google Scholar] [CrossRef]
- Lago, J.H.; Ramos, C.S.; Casanova, D.C.; Morandim, A.A.; Bergamo, D.C.; Cavalheiro, A.J.; Bolzani, V.S.; Furlan, M.; Guimarães, E.F.; Young, M.C.; et al. Benzoic acid derivatives from Piper species and their fungitoxic activity against Cladosporium cladosporioides and C. sphaerospermum. J. Nat. Prod. 2004, 67, 1783–1788. [Google Scholar] [CrossRef]
- Mukai, K.; Okabe, K.; Hosose, H. Synthesis and stopped-flow investigation of antioxidant activity of tocopherols. Finding of new tocopherol derivatives having the highest antioxidant activity among phenolic antioxidants. J. Org. Chem. 1989, 54, 557–560. [Google Scholar] [CrossRef]
- Bernard, C.B.; Krishanmurty, H.G.; Chauret, D.; Durst, T.; Philogène, B.J.R.; Sánchez-Vindas, P.; Hasbun, C.; Poveda, L.; San Román, L.; Arnason, J.T. Insecticidal defenses of Piperaceae from the neotropics. J. Chem. Ecol. 1995, 21, 801–814. [Google Scholar] [CrossRef]
- Evans, J.M.; Fake, C.S.; Hamilton, T.C.; Poyser, R.H.; Watts, E.A. Synthesis and antihypertensive activity of substituted trans-4-amino-3,4-dihydro-2,2-dimethyl-2H-1-benzopyran-3-ols. J. Med. Chem. 1983, 26, 1582–1589. [Google Scholar] [CrossRef]
- Thompson, R.; Doggrell, S.; Hoberg, J.O. Potassium channel activators based on the benzopyran substructure: Synthesis and activity of the C-8 substituent. Bioorgan. Med. Chem. 2003, 11, 1663–1668. [Google Scholar] [CrossRef]
- O’Hagen, D.; Rzepa, H.S. Some influences of fluorine in bioorganic chemistry. Chem. Commun. 1997. [Google Scholar] [CrossRef]
- Ojima, I.; Taguchi, T.; Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chicester, UK, 2009. [Google Scholar]
- Uneyama, K. Organofluorine Chemistry; Blackwell Publishing Ltd.: Oxford, UK, 2006. [Google Scholar]
- Ojima, I.; McCarthy, J.R.; Welch, J.T. Biomedical Frontiers of Fluorine Chemistry; American Chemical Society: Washington, DC, USA, 1996. [Google Scholar]
- Arnone, A.; Bernardi, R.; Blasco, F.; Cardillo, R.; Resnati, G.; Gerus, I.I.; Kukhar, V.P. Trifluoromethyl vs. methyl ability to direct enantioselection in microbial reduction of carbonyl substrates. Tetrahedron 1998, 54, 2809–2818. [Google Scholar] [CrossRef]
- Kharrat, S.E.; Laurent, P.; Blancou, H. Novel synthesis of 2-(trifluoromethyl)- and 2-(perfluoroalkyl)-2-hydroxy-2H-chromenes and their regiospecific reaction with silyl enol ethers. J. Org. Chem. 2006, 71, 8637–8640. [Google Scholar] [CrossRef]
- Wang, J.L.; Carter, J.; Kiefer, J.R.; Kurumbail, R.G.; Pawlitz, J.L.; Brown, D.; Hartmann, S.J.; Graneto, M.J.; Seibert, K.; John, J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part I: The first clinical candidate. Bioorg. Med. Chem. Lett. 2010, 20, 7155–7158. [Google Scholar] [CrossRef]
- Wang, J.L.; Limburg, D.; Graneto, M.J.; Springer, J.; Hamper, J.R.; Liao, S.; Pawlitz, J.L.; Kurumbail, R.G.; Maziasz, T.; Talley, J.J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part II: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010, 20, 7159–7163. [Google Scholar] [CrossRef]
- Wang, J.L.; Aston, K.; Limburg, D.; Ludwig, C.; Hallinan, A.E.; Koszyk, F.; Hamper, B.; Brown, D.; Graneto, M.; Talley, J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part III: The three microdose candidates. Bioorg. Med. Chem. Lett. 2010, 20, 7164–7168. [Google Scholar] [CrossRef]
- Chizhov, D.L.; Sosnovskikh, V.Y.; Pryadeina, M.V.; Burgart, Y.V.; Saloutin, V.I.; Charushin, V.N. The first synthesis of 4-unsubstituted 3-(trifluoroacetyl)coumarins by the Knoevenagel condensation of salicylaldehydes with ethyl trifluoroacetoacetate followed by chromene-coumarin recyclization. Synlett 2008, 2, 281–285. [Google Scholar]
- Li, H.Q.; Cai, L.; Li, J.X.; Hu, Y.X.; Zhou, P.P.; Zhang, J.M. Novel coumarin fluorescent dyes: Synthesis, structural characterization and recognition behavior towards Cu(II) and Ni(II). Dyes Pigments 2011, 91, 309–316. [Google Scholar] [CrossRef]
- Wan, P.; Han, J.W.; Zhao, J.W.; Zhu, S.Z. Regioselective carbon—Carbon bond formation in the reaction of 2-hydroxy-2-(trifluoromethyl)-2H-chromenes with indoles promoted by Lewis acid. Synthesis 2011, 20, 3364–3370. [Google Scholar]
- Li, X.J.; Zhao, J.W.; Wang, Z.W.; Han, J.W.; Zhao, J.; Zhu, S.Z. Reactions of 2-(trifluoromethyl)-2-hydroxy-2H-chromenes with thiophenols promoted by Lewis acid. Tetrahedron 2012, 68, 8011–8017. [Google Scholar] [CrossRef]
- Tanaka, K. Solvent-Free Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Hodnett, B.K.; Kybett, A.P.; Clark, J.H.; Smith, K. Supported Reagents and Catalysts in Chemistry; RSC Cambridge: Cambridge, UK, 1998. [Google Scholar]
- Wang, L.M.; Sheng, J.; Tian, H.; Han, J.W.; Fan, Z.Y.; Qian, C.T. A convenient synthesis of α,α'-bis(substituted benzylidene)cycloalkanones catalyzed by Yb(OTf)3 under solvent-free conditions. Synthesis 2004, 18, 3060–3064. [Google Scholar]
- Hou, C.; Zhu, H.; Li, Y.J.; Li, Y.F. Immobilized proline and its derivatives employed in the catalysis of asymmetric organic synthesis. Prog. Chem. 2012, 24, 1729–1741. [Google Scholar]
- Font, D.; Jimeno, C.; Pericàs, M.A. Polystyrene-supported hydroxyproline: An insoluble, recyclable organocatalyst for the asymmetric aldol reaction in water. Org. Lett. 2006, 8, 4653–4655. [Google Scholar] [CrossRef]
- Yang, H.L.; Li, S.W.; Wang, X.Y.; Zhang, F.W.; Zhong, X.; Dong, Z.P.; Ma, J.T. Core-shell silica magnetic microspheres supported proline as a recyclable organocatalyst for the asymmetric aldol reaction. J. Mol. Catal. A Chem. 2012, 363–364, 404–410. [Google Scholar] [CrossRef]
- 34 Li, J.; Yang, G.X.; Qin, Y.Y.; Yang, X.R.; Cui, Y.C. Recyclable Merrifield resin-supported thiourea organocatalysts derived from L-proline for direct asymmetric aldol reaction. Tetrahedron Asymmetry 2011, 22, 613–618. [Google Scholar] [CrossRef]
- Zhao, Y.B.; Zhang, L.W.; Wu, L.Y.; Zhong, X.; Li, R.; Ma, J.T. Silica-supported pyrrolidine-triazole, an insoluble, recyclable organocatalyst for the enantioselective Michael addition of ketones to nitroalkenes. Tetrahedron Asymmetry 2008, 19, 1352–1355. [Google Scholar] [CrossRef]
- Yang, G.Y.; Wang, C.X.; Fan, S.F.; Zhao, L.J.; Wang, D.; Xu, C.L. Study on the cyclization methods of 3-[1-(phenylhydrazono)ethyl]-chromen-2-ones. Synth. Commun. 2013, 43, 1263–1269. [Google Scholar] [CrossRef]
- Yang, G.Y.; Yang, J.T.; Wang, C.X.; Fan, S.F.; Xie, P.H. Microwave-assisted synthesis of 3-methyl-1-phenylchromeno[4,3-c]pyrazol-4(1H)-ones under solvent-free conditions. Heterocycles 2013, 87, 1327–1336. [Google Scholar] [CrossRef]
- Wen, L.L.; Zhang, H.H.; Lin, H.; Shen, Q.L.; Lu, L. A facile synthetic route to 2-trifluoromethyl-substituted polyfunctionalized chromenes and chromones. J. Fluorine Chem. 2012, 133, 171–177. [Google Scholar] [CrossRef]
- Smart and Saint, Area Detector Control, Integration Software; Siemens Analytical X-ray Systems, Inc.: Madison, WI, USA, 1996.
- Sheldrick, G.M. SHELXTL V5.1, Software Reference Manual; Bruker AXS, Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, C.; Yang, G.; Wang, C.; Fan, S.; Xie, L.; Gao, Y. An Efficient Solvent-Free Synthesis of 2-Hydroxy-2-(trifluoromethyl)-2H-chromenes Using Silica-Immobilized L-Proline. Molecules 2013, 18, 11964-11977. https://doi.org/10.3390/molecules181011964
Xu C, Yang G, Wang C, Fan S, Xie L, Gao Y. An Efficient Solvent-Free Synthesis of 2-Hydroxy-2-(trifluoromethyl)-2H-chromenes Using Silica-Immobilized L-Proline. Molecules. 2013; 18(10):11964-11977. https://doi.org/10.3390/molecules181011964
Chicago/Turabian StyleXu, Cuilian, Guoyu Yang, Caixia Wang, Sufang Fan, Lixia Xie, and Ya Gao. 2013. "An Efficient Solvent-Free Synthesis of 2-Hydroxy-2-(trifluoromethyl)-2H-chromenes Using Silica-Immobilized L-Proline" Molecules 18, no. 10: 11964-11977. https://doi.org/10.3390/molecules181011964