Identification of New Natural DNA G-Quadruplex Binders Selected by a Structure-Based Virtual Screening Approach



Abstract
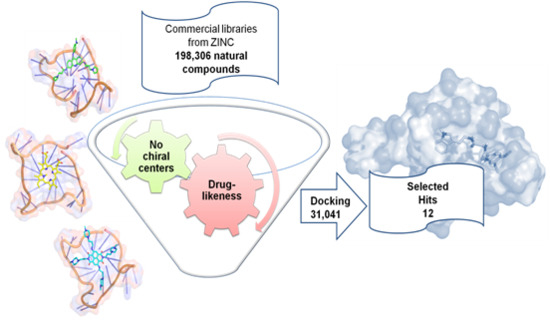
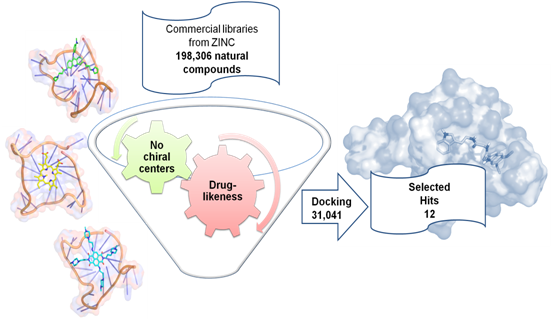
:
1. Introduction
2. Results and Discussion
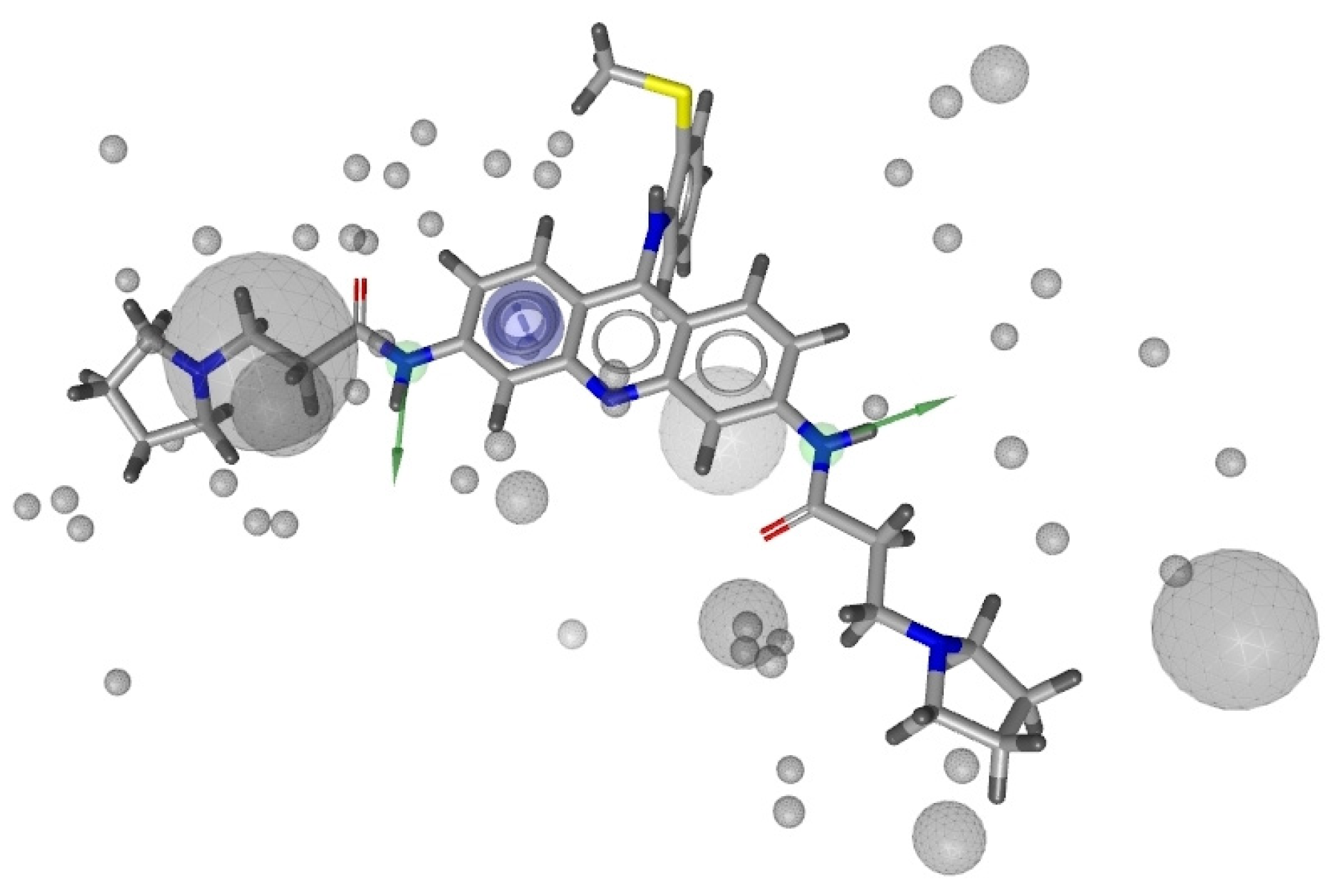
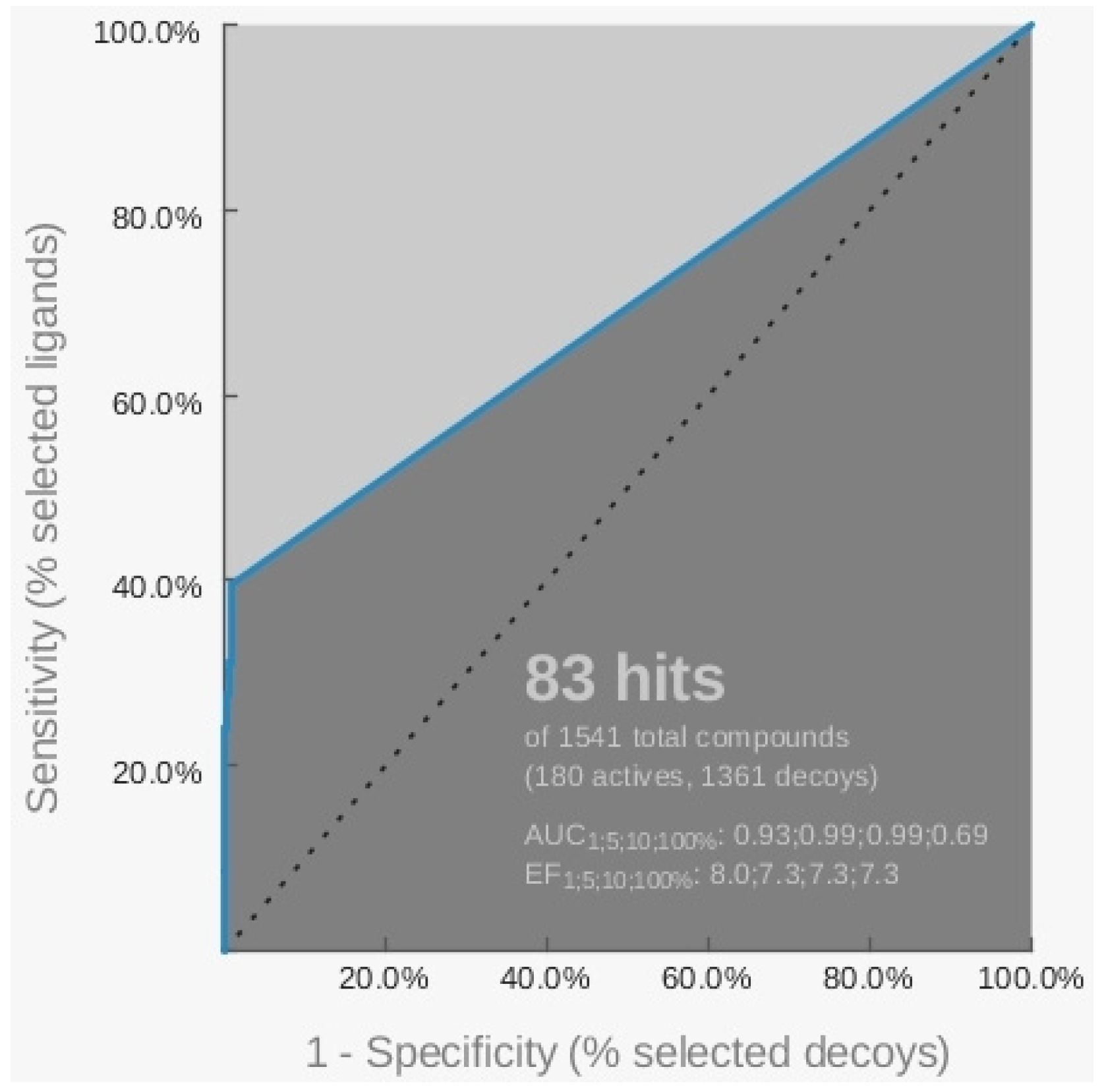
2.1. Pharmacophore Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
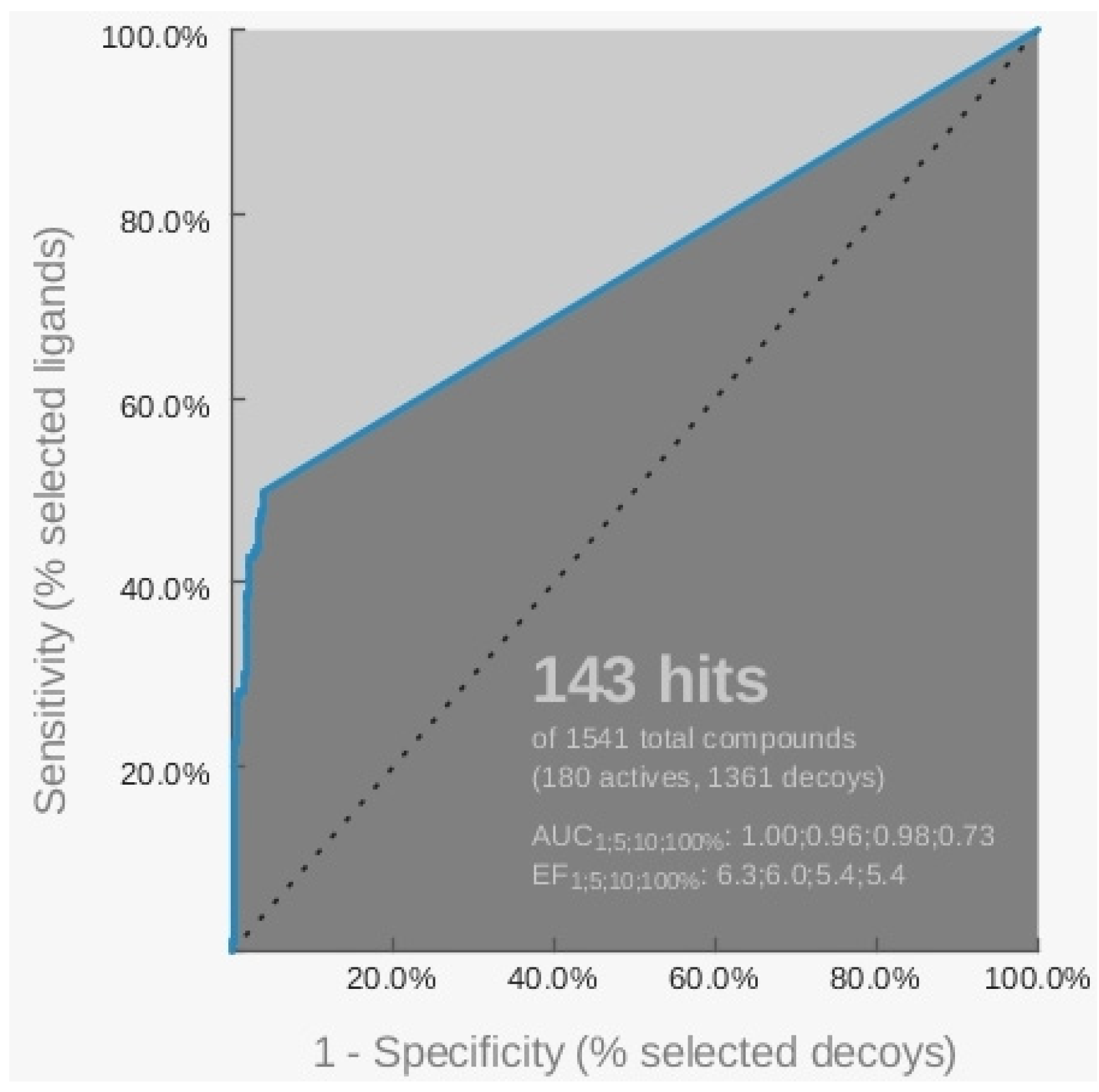
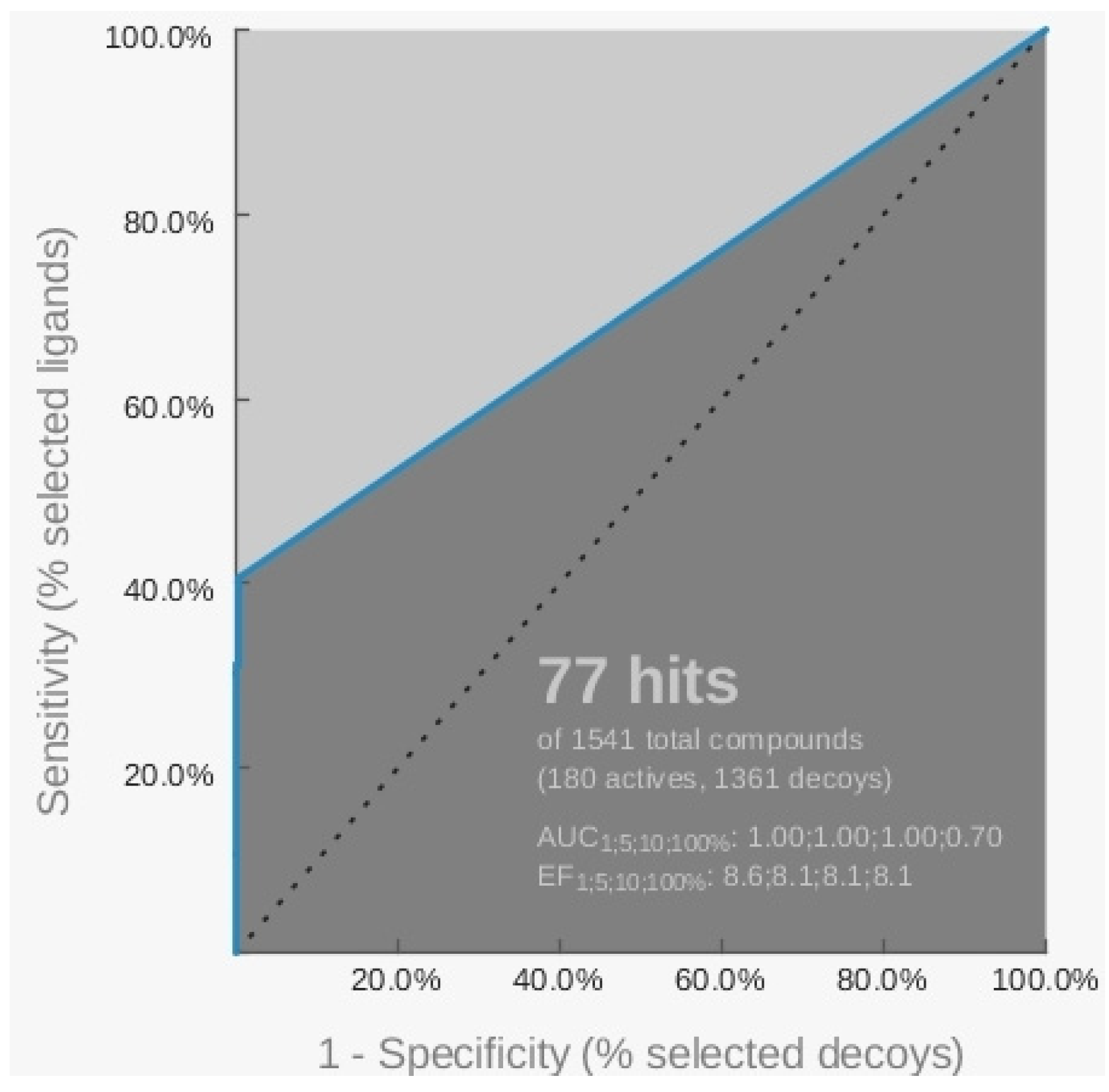{kind=link}
{kind=link}
{kind=link}
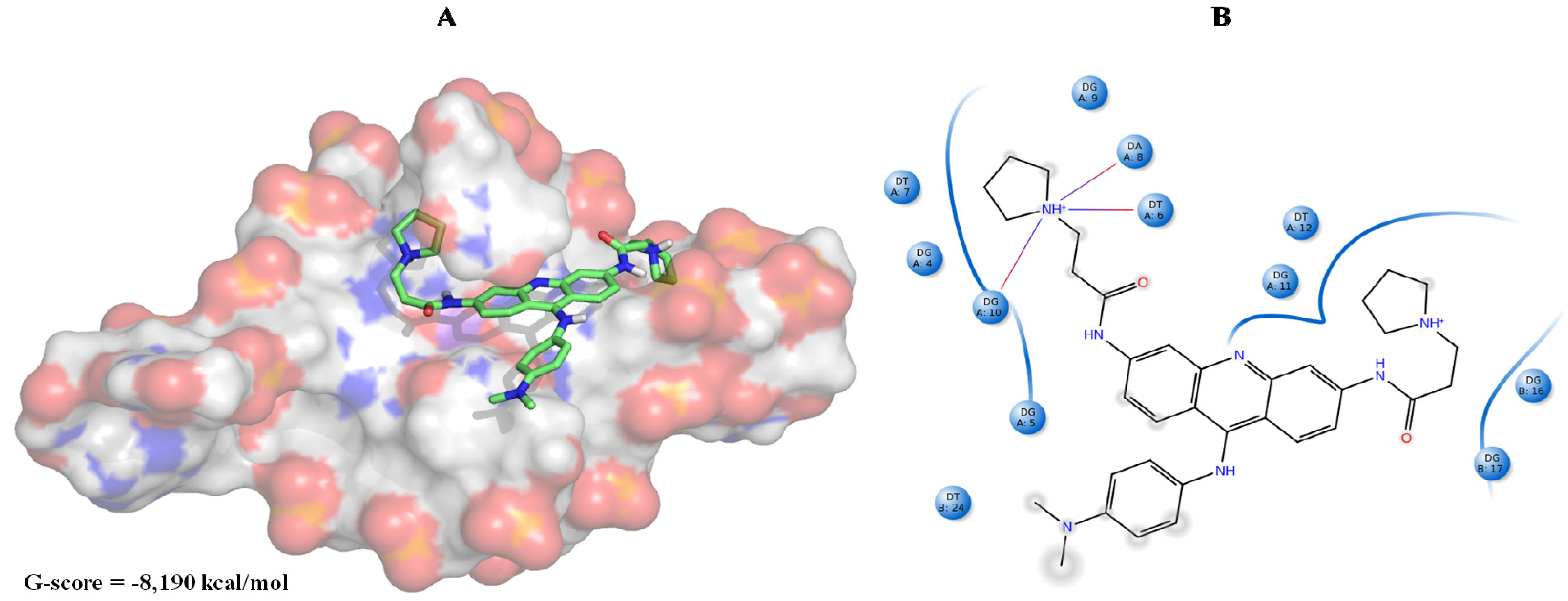
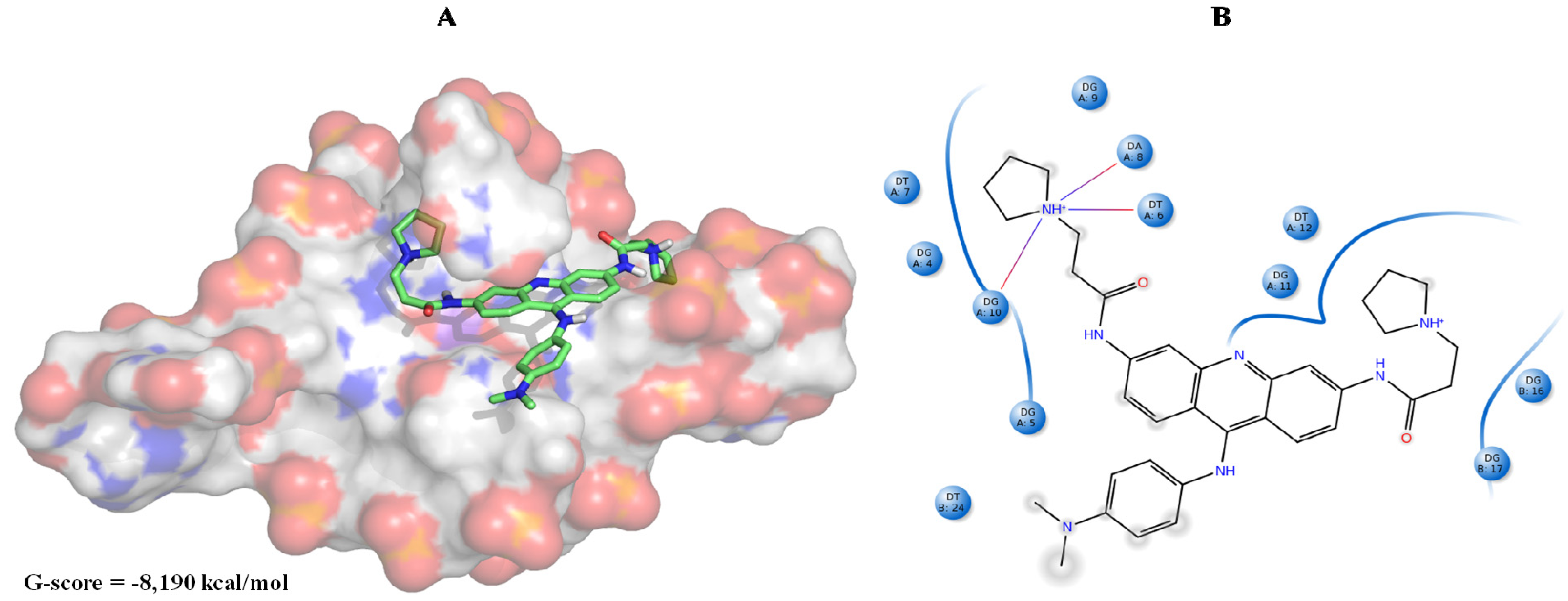
{kind=link}
{kind=link}
| 3CE5 PDB model | |||||
|---|---|---|---|---|---|
| Hypothesis | # Features | Active compounds | % Active | Inactive compounds | % Inactive |
| H_1 | 4 | 42 | 23.3 | 2 | 0.14 |
| H_2 | 3 | 71 | 39.4 | 12 | 1.17 |


| 3SC8 PDB model | |||||
|---|---|---|---|---|---|
| Hypothesis | # Features | Active compounds | % Active | Inactive compounds | % Inactive |
| H_1 | 3 | 73 | 40.5 | 90 | 6.61 |
| H_2 | 4 | 4 | 2.22 | 14 | 1.03 |
| H_3 | 3 | 73 | 40.5 | 4 | 0.29 |
| H_4 | 3 | 73 | 40.5 | 60 | 4.41 |
| H_5 | 3 | 74 | 41.1 | 45 | 3.31 |
| H_6 | 3 | 74 | 41.1 | 26 | 14.4 |
| H_7 | 3 | 74 | 41.1 | 84 | 6.17 |


| 4FXM PDB model | |||||
|---|---|---|---|---|---|
| Hypothesis | # features | Active compounds | % Active | Inactive compounds | % Inactive |
| H_1 | 3 | 17 | 9.4 | 11 | 0.8 |
| H_2 | 3 | 109 | 60.5 | 496 | 36.4 |
| H_3 | 3 | 107 | 59.4 | 476 | 35 |
| H_4 | 3 | 101 | 56.1 | 460 | 33.8 |
| H_5 | 3 | 94 | 52.2 | 240 | 17.6 |
| H_6 | 3 | 94 | 52.2 | 204 | 15 |
| H_7 | 3 | 99 | 55 | 113 | 8.3 |
| H_8 | 3 | 99 | 55 | 136 | 10 |
| H_9 | 3 | 90 | 50 | 73 | 5.63 |


2.2. Virtual Screening of the Natural Database
| Model | Screened ligands |
|---|---|
| 3CE5 | 673 |
| 3SC8 | 73 |
| 4FXM | 4013 |
| Best hits | ||
 |  | 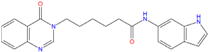 |
| ZINC79190432 −8.451 | ZINC77031588 * −8.509 | ZINC32124244 −8.485 |
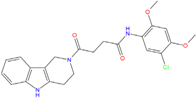 | 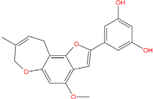 |  |
| ZINC20760949 −8.429 | ZINC14610063 −8.334 | ZINC12902036 −9.082 |
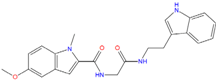 | 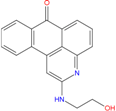 |  |
| ZINC12664647 −8.378 | ZINC12377179 −8.828 | ZINC04252698 * −8.206 |
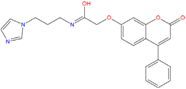 | 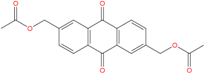 | 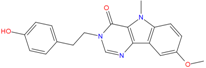 |
| ZINC03985155 −8.609 | ZINC03843477 −8.215 | ZINC02131213 −8.944 |

3. Experimental
3.1. Dataset of Active, Inactive and Decoys Compounds
3.2. Database of Natural Compounds
3.3. Pharmacophore Models Generation and Virtual Screening
3.4. Docking Experiments
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef]
- Allshire, R.C.; Dempster, M.; Hastie, N.D. HumanTelomeres contain at least 3 types of G-rich repeat distributed non-randomly. Nucleic Acids Res. 1989, 17, 4611–4627. [Google Scholar] [CrossRef]
- De Lange, T.; Shiue, L.; Myers, R.M.; Cox, D.R.; Naylor, S.L.; Killery, A.M.; Varmus, H.E. Structure and variability of human chromosome ends. Mol. Cell Biol. 1990, 10, 518–527. [Google Scholar]
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar]
- Wieland, M.; Hartig, J.S. RNA quadruplex-based modulation of gene expression. Chem. Biol. 2007, 14, 757–763. [Google Scholar] [CrossRef]
- Mullen, M.A.; Olson, K.J.; Dallaire, P.; Major, F.; Assmann, S.M.; Bevilacqua, P.C. RNA G-Quadruplexes in the model plant species Arabidopsis thaliana: Prevalence and possible functional roles. Nucleic Acids Res. 2010, 38, 8149–8163. [Google Scholar] [CrossRef]
- Zahler, A.M.; Williamson, J.R.; Cech, T.R.; Prescott, D.M. Inhibition of telomerase by G-quartet DNA structures. Nature 1991, 350, 718–720. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef]
- Hazel, P.; Parkinson, G.N.; Neidle, S. Predictive modelling of topology and loop variations in dimeric DNA quadruplex structures. Nucleic Acids Res. 2006, 34, 2117–2127. [Google Scholar] [CrossRef]
- Laughlan, G.; Murchie, A.; Norman, D.; Moore, M.; Moody, P.; Lilley, D.; Luisi, B. The high resolution crystal structure of a parallel-stranded guanine tetraplex. Science 1994, 265, 520–524. [Google Scholar]
- Phillips, K.; Dauter, Z.; Murchie, A.I.H.; Lilley, D.M.J.; Luisi, B. The crystal structure of a parallel-stranded guanine tetraplex at 0.95 Å resolution. J. Mol. Biol. 1997, 273, 171–182. [Google Scholar] [CrossRef]
- Hardin, C.C.; Perry, A.G.; White, K. Thermodynamic and kinetic characterization of the dissociation and assembly of quadruplex nucleic acids. Biopolymers 2000, 56, 147–194. [Google Scholar] [CrossRef]
- Guedin, A.; Gros, J.; Alberti, P.; Mergny, J.L. How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res. 2010, 38, 7858–7868. [Google Scholar] [CrossRef]
- Bugaut, A.; Balasubramanian, S. A sequence-independent study of the influence of short loop lengths on the stability and topology of intramolecular DNA G-quadruplexes. Biochemistry 2008, 47, 689–697. [Google Scholar] [CrossRef]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human telomere, oncogenic promoter and 5′-UTR G-quadruplexes: Diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef]
- Hurley, L.H.; Wheelhouse, R.T.; Sun, D.; Kerwin, S.M.; Salazar, M.; Fedoroff, O.Y.; Han, F.X.; Han, H.; Izbicka, E.; Von Hoff, D.D. G-quadruplexes as targets for drug design. Pharmacol. Ther. 2000, 85, 141–158. [Google Scholar] [CrossRef]
- Neidle, S.; Parkinson, G. Telomere maintenance as a target for anticancer drug discovery. Nat. Rev. Drug Discov. 2002, 1, 383–393. [Google Scholar] [CrossRef]
- De Cian, A.; Lacroix, L.; Douarre, C.; Temime-Smaali, N.; Trentesaux, C.; Riou, J.F.; Mergny, J.L. Targeting telomeres and telomerase. Biochimie 2008, 90, 131–155. [Google Scholar] [CrossRef]
- Punchihewa, C.; Yang, D.Z. Therapeutic Targets and Drugs: G-Quadruplex and G-Quadruplex Inhibitors. In Cancer Drug Discovery and Development: Telomeres and Telomerase in Cancer; Hiyama, K., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 251–280. [Google Scholar]
- Gordon, K.E.; Parkinson, E.K. Analysis of telomerase activity and telomere function in cancer. Methods Mol. Biol. 2004, 281, 333–348. [Google Scholar]
- Guyen, B.; Schultes, C.M.; Hazel, P.; Mann, J.; Neidle, S. Synthesis and evaluation of analogues of 10H-indolo[3,2-b]quinoline as G-quadruplex stabilising ligands and potential inhibitors of the enzyme telomerase. Org. Biomol. Chem. 2004, 2, 981–988. [Google Scholar] [CrossRef]
- Shay, J.W.; Gazdar, A.F. Telomerase in the early detection of cancer. J. Clin. Pathol. 1997, 50, 106–109. [Google Scholar] [CrossRef]
- Harley, C.B. Telomerase and cancer therapeutics. Nat. Rev. Cancer 2008, 8, 167–179. [Google Scholar] [CrossRef]
- Shay, J.W.; Keith, W.N. Targeting telomerase for cancer therapeutics. Br. J. Cancer 2008, 98, 677–683. [Google Scholar] [CrossRef]
- Yang, D.; Okamoto, K. Structural insights into G-quadruplexes: Towards new anticancer drugs. Future Med. Chem. 2010, 2, 619–646. [Google Scholar] [CrossRef]
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of human telomerase by a G-quadruplex-interactive compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef]
- Monchaud, D.; Teulade-Fichou, M.P. A hitchhiker’s guide to G-quadruplex ligands. Org. Biomol. Chem. 2008, 6, 627–636. [Google Scholar] [CrossRef]
- Fedoroff, O.Y.; Salazar, M.; Han, H.; Chemeris, V.V.; Kerwin, S.M.; Hurley, L.H. NMR-based model of a telomerase-inhibiting compound bound to G-quadruplex DNA. Biochemistry 1998, 37, 12367–12374. [Google Scholar] [CrossRef]
- Haider, S.M.; Parkinson, G.N.; Neidle, S. Structure of a G-quadruplex–ligand complex. J. Mol. Biol. 2003, 326, 117–125. [Google Scholar] [CrossRef]
- Gavathiotis, E.; Heald, R.A.; Stevens, M.F.; Searle, M.S. Drug recognition and stabilisation of the parallel- stranded DNA quadruplex d(TTAGGGT)4 containing the human telomeric repeat. J. Mol. Biol. 2003, 334, 25–36. [Google Scholar] [CrossRef]
- Campbell, N.H.; Parkinson, G.N.; Reszka, A.P.; Neidle, S. Structural basis of DNA quadruplex recognition by an acridine drug. J. Am. Chem. Soc. 2008, 130, 6722–6724. [Google Scholar]
- Parkinson, G.N.; Cuenca, F.; Neidle, S. Topology conservation and loop flexibility in quadruplex-drug recognition: Crystal structures of inter- and intramolecular telomeric DNA quadruplex-drug complexes. J. Mol. Biol. 2008, 381, 1145–1156. [Google Scholar] [CrossRef]
- Campbell, N.H.; Patel, M.; Tofa, A.B.; Ghosh, R.; Parkinson, G.N.; Neidle, S. Selectivity in ligand recognition of G-quadruplex loops. Biochemistry 2009, 48, 1675–1680. [Google Scholar]
- Burger, A.M.; Dai, F.; Schultes, C.M.; Reszka, A.P.; Moore, M.J.; Double, J.A.; Neidle, S. The G-quadruplex interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res. 2005, 65, 1489–1496. [Google Scholar] [CrossRef]
- Tauchi, T.; Shin-Ya, K.; Sashida, G.; Sumi, M.; Nakajima, A.; Shimamoto, T.; Ohyashiki, J.H.; Ohyashiki, K. Activity of a novel G-quadruplex-interactive telomerase inhibitor, telomestatin (SOT-095), against human leukemia cells: Involvement of ATM-dependent DNA damage response pathway. Oncogene 2003, 22, 5338–5347. [Google Scholar] [CrossRef]
- Salvati, E.; Leonetti, C.; Rizzo, A.; Scarsella, M.; Mottolese, M.; Galati, R.; Sperduti, I.; Stevens, M.F.; D’Incalci, M.; Blasco, M.; et al. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J. Clin. Invest. 2007, 117, 3236–3247. [Google Scholar] [CrossRef]
- Tauchi, T.; Shin-ya, K.; Sashida, G.; Sumi, M.; Okabe, S.; Ohyashiki, J.H.; Ohyashiki, K. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: In vitro and in vivo studies in acute leukemia. Oncogene 2006, 25, 5719–5725. [Google Scholar] [CrossRef]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef]
- Read, M.; Cuesta, J.; Basra, I.; Harrison, J.; Reszka, A.; Gowan, S.; Kelland, L.R.; Neidle, S. Rational design approaches to increase the potency of G-quadruplex mediated telomerase inhibitors. Clin. Cancer Res. 2001, 7, 3797S. [Google Scholar]
- Read, M.; Harrison, R.J.; Romagnoli, B.; Tanious, F.A.; Gowan, S.H.; Reszka, A.P.; Wilson, W.D.; Kelland, L.R.; Neidle, S. Structure-based design of selective and potent G quadruplex-mediated telomerase inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 4844–4849. [Google Scholar] [CrossRef]
- Incles, C.M.; Schultes, C.M.; Kempski, H.; Koehler, H.; Kelland, L.R. A G-quadruplex telomere targeting agent produces p16-associated senescence and chromosomal fusions in human prostate cancer cells. Mol. Cancer Therap. 2004, 3, 1201–1206. [Google Scholar]
- Kelland, L.R. Overcoming the immortality of tumour cells by telomere and telomerase based cancer therapeutics – current status and future prospects. Eur. J. Cancer 2005, 41, 971–979. [Google Scholar] [CrossRef]
- Gowan, S.M.; Harrison, J.R.; Patterson, L.; Valenti, M.; Read, M.A.; Neidle, S.; Kelland, L.R. A G-quadruplex-interactive potent small-molecule inhibitor of telomerase exhibiting in vitro and in vivo antitumor activity. Mol. Pharmacol. 2002, 61, 1154–1162. [Google Scholar] [CrossRef]
- Perry, P.J.; Read, M.A.; Davies, R.T.; Gowan, S.M.; Reszka, A.P.; Wood, A.A.; Kelland, L.R.; Neidle, S. 2,7-Disubstituted amidofluorenone derivatives as inhibitors of human telomerase. J. Med. Chem. 1999, 42, 2679–2684. [Google Scholar] [CrossRef]
- Moorhouse, A.D.; Santos, A.M.; Gunaratnam, M.; Moore, M.; Neidle, S.; Moses, J.E. Stabilization of G-quadruplex DNA by highly selective ligands via click chemistry. J. Am. Chem. Soc. 2006, 128, 15972–15973. [Google Scholar] [CrossRef]
- Huang, H.S.; Chen, I.B.; Huang, K.F.; Lu, W.C.; Shieh, F.Y.; Huang, Y.Y.; Huang, F.C.; Lin, J.J. Synthesis and human telomerase inhibition of a series of regioisomeric disubstituted amidoanthraquinones. Chem. Pharm. Bull. 2007, 55, 284–292. [Google Scholar] [CrossRef]
- Ma, D.L.; Lai, T.S.; Chan, F.Y.; Chung, W.H.; Abagyan, R.; Leung, Y.C.; Wong, K.Y. Discovery of a Drug-Like G-Quadruplex Binding Ligand by High-Throughput Docking. ChemMedChem 2008, 3, 881–884. [Google Scholar] [CrossRef]
- Lee, H.M.; Chan, D.S.H.; Yang, F.; Lam, H.Y.; Yan, S.C.; Che, C.M.; Ma, D.L.; Leung, C.H. Identification of natural product Fonsecin B as a stabilizing ligand of c-myc G-quadruplex DNA by high-throughput virtual screening. Chem. Commun. 2010, 46, 4680–4682. [Google Scholar]
- Alcaro, S.; Artese, A.; Costa, G.; Distinto, S.; Ortuso, F.; Parrotta, L. Conformational studies and solvent-accessible surface area analysis of known selective DNA G-Quadruplex binders. Biochimie 2011, 93, 1267–1274. [Google Scholar] [CrossRef]
- Artese, A.; Costa, G.; Distinto, S.; Moraca, F.; Ortuso, F.; Parrotta, L.; Alcaro, S. Toward the design of new DNA G-quadruplex ligands through rational analysis of polymorphism and binding data. Eur. J. Med. Chem. 2013, 68, 139–149. [Google Scholar] [CrossRef]
- Artese, A.; Parrotta, L.; Alcaro, S.; Ortuso, F.; Costa, G.; Sissi, C. Molecular recognition of human telomeric DNA by phenanthroline-based G-quadruplex ligands. Open J. Med. Chem. 2013, 3, 41–49. [Google Scholar] [CrossRef]
- Alcaro, S.; Artese, A.; Iley, J.N.; Maccari, R.; Missailidis, S.; Ortuso, F.; Ottanà, R.; Ragazzon, P.; Vigorita, M.G. Tetraplex DNA specific ligands based on the fluorenone-carboxamide scaffold. Bioorg. Med. Chem. Lett. 2007, 17, 2509–2514. [Google Scholar] [CrossRef]
- Alcaro, S.; Artese, A.; Iley, J.N.; Missailidis, S.; Ortuso, F.; Parrotta, L.; Pasceri, R.; Paduano, F.; Sissi, C.; Trapasso, F.; et al. Rational design, synthesis, biophysical and antiproliferative evaluation of fluorenone derivatives with DNA G-quadruplex binding properties. Chem. Med. Chem. 2010, 5, 575–583. [Google Scholar]
- Doria, F.; Nadai, M.; Folini, M.; di Antonio, M.; Germani, L.; Percivalle, C.; Sissi, C.; Zaffaroni, N.; Alcaro, S.; Artese, A.; et al. Hybrid ligand-alkylating agents targeting telomeric G-quadruplex structures. Org. Biomol. Chem. 2012, 10, 2798–2806. [Google Scholar] [CrossRef]
- Alcaro, S.; Costa, G.; Distinto, S.; Moraca, F.; Ortuso, F.; Parrotta, L.; Artese, A. The polymorphisms of DNA G-quadruplex investigated by docking experiments with telomestatin enantiomers. Curr. Pharm Des. 2012, 18, 1873–1879. [Google Scholar] [CrossRef]
- Alcaro, S.; Musetti, C.; Distinto, S.; Casatti, M.; Zagotto, G.; Artese, A.; Parrotta, L.; Moraca, F.; Costa, G.; Ortuso, F.; et al. Identification and characterization of new DNA G-quadruplex binders selected by a combination of ligand and structure-based virtual screening approaches. J. Med. Chem. 2013, 56, 843–855. [Google Scholar] [CrossRef]
- Truchon, J.F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.P.; Bertrand, H.O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- The Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB). Available online: http://www.rcsb.org/pdb (accessed on 18 September 2013).
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Collie, G.W.; Promontorio, R.; Hampel, S.M.; Micco, M.; Neidle, S.; Parkinson, G.N. Structural basis for telomeric G-quadruplex targeting by naphthalene diimide ligands. J. Am. Chem. Soc. 2012, 134, 2723–2731. [Google Scholar]
- Nicoludis, J.M.; Miller, S.T.; Jeffrey, P.D.; Barrett, S.P.; Rablen, P.R.; Lawton, T.J.; Yatsunyk, L.A. Optimized end-stacking provides specificity of n-methyl mesoporphyrin IX for human telomeric G-quadruplex DNA. J. Am. Chem. Soc. 2012, 134, 20446–20456. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprint: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]
- McCormick, J.L.; McKee, T.C.; Cardellina, J.H.; Boyd, M.R. HIV inhibitory natural products. 26. Quinoline alkaloids from Euodia roxburghiana. J. Nat. Prod. 1996, 59, 469–471. [Google Scholar] [CrossRef]
- Bai, L.P.; Ho, H.M.; Ma, D.L.; Yang, H.; Fu, W.C.; Jiang, Z.H. Aminoglycosylation can enhance the G-quadruplex binding activity of epigallocatechin. PLoS One 2013, 8, e53962. [Google Scholar]
- Alves, D.S.; Perez-Fons, L.; Estepa, A.; Micol, V. Membrane-related effects underlying the biological activity of the anthraquinones emodin and barbaloin. Biochem. Pharmacol. 2004, 68, 549–561. [Google Scholar] [CrossRef]
- Perry, P.J.; Reszka, A.P.; Wood, A.A.; Read, M.A.; Gowan, S.M.; Dosanjh, H.S.; Trent, J.O.; Jenkins, T.C.; Kelland, L.R.; Neidle, S. Human telomerase inhibition by regioisomeric disubstituted amidoanthracene-9,10-diones. J. Med. Chem. 1998, 41, 4873–4884. [Google Scholar] [CrossRef]
- Moore, M.J.; Schultes, C.M.; Cuesta, J.; Cuenca, F.; Gunaratnam, M.; Tanious, F.A.; Wilson, W.D.; Neidle, S. Trisubstituted acridines as G-quadruplex telomere targeting agents. Effects of extensions of the 3,6- and 9-side chains on quadruplex binding, telomerase activity, and cell proliferation. J. Med. Chem. 2006, 49, 582–599. [Google Scholar] [CrossRef]
- Neidle, S.; Thurston, D.E. Chemical approaches to the discovery and development of cancer therapies. Nat. Rev. Cancer 2005, 5, 285–296. [Google Scholar] [CrossRef]
- Herraiz, T.; Galisteo, J. Tetrahydro-beta-carboline alkaloids occur in fruits and fruit juices. Activity as antioxidants and radical scavengers. J. Agric. Food Chem. 2003, 51, 7156–7161. [Google Scholar] [CrossRef]
- Moura, D.J.; Richter, M.F.; Boeira, J.M.; Pêgas Henriques, J.A.; Saffi, J. Antioxidant properties of beta-carboline alkaloids are related to their antimutagenic and antigenotoxic activities. Mutagenesis 2007, 22, 293–302. [Google Scholar] [CrossRef]
- Wang, F.X.; Deng, A.J.; Li, M.; Wei, J.F.; Qin, H.L.; Wang, A.P. (3S)-1,2,3,4-Tetrahydro-β-carboline-3-carboxylic acid from Cichorium endivia. L induces apoptosis of human colorectal cancer HCT-8 cells. Molecules 2006, 38, 232–240. [Google Scholar]
- Wan, Y.Y.; Du, Y.M.; Yang, F.X.; Xu, Y.; Chen, R.Z.; Kennedy, J.F. Purification and characterization of hydrosoluble components from the sap of Chinese lacquer tree Rhus vernici fera. Int. J. Biol. Macromol. 2006, 38, 232–240. [Google Scholar] [CrossRef]
- Moon, D.O.; Kim, M.O.; Lee, J.D.; Choi, Y.H.; Kim, G.Y. Butein suppresses c-Myc-dependent transcription and Akt-dependent phosphorylation of hTERT in human leukemia cells. Cancer Lett. 2009, 286, 172–179. [Google Scholar] [CrossRef]
- Sasaki, S.; Ehara, T.; Sakata, I.; Fujino, Y.; Harada, N.; Kimura, J.; Nakamura, H.; Maeda, M. Development of novel telomerase inhibitors based on a bisindole unit. Bioorg. Med. Chem. Lett. 2001, 11, 583–585. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Weininger, D.; Weininger, A.; Weininger, J. L. SMILES. 2. Algorithm for generation of unique SMILES notation. J. Chem. Inf. Model. 1989, 29, 97–101. [Google Scholar] [CrossRef]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hen-drickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Ambinter Natural Products. Available online: http://www.ambinter.com (accessed on July 2013).
- AnalytiCon Discovery NP. Available online: http://www.ac-discovery.com (accessed on 18 September 2013).
- IBScreen NP. Available online: http://www.ibscreen.com (accessed on 18 September 2013).
- Indofine Natural Products. Available online: http://www.indofinechemical.com (accessed on 18 September 2013).
- Molecular Diversity Preservation International. Available online: http://www.mdpi.org (accessed on 18 September 2013).
- Nubbe Natural Products. Available online: http://nubbe.iq.unesp.br (accessed on 18 September 2013).
- Princeton NP. Available online: http://www.princetonbio.com (accessed on 18 September 2013).
- Selleck BioChemicals NP. Available online: http://www.selleckbio.com (accessed on 18 September 2013).
- Specs Natural Products. Available online: http://www.specs.net (accessed on 18 September 2013).
- TCM Database@Taiwan. Available online: http://tcm.cmu.edu.tw (accessed on 18 September 2013).
- UEFS Natural Products. Available online: http://www.uefs.br (accessed on 18 September 2013).
- ChemAxon. Available online: http://www.chemaxon.com (accessed on 18 September 2013).
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Data Bank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- Maestro Graphics User Interface, version 9.4; Schrödinger, LLC: New York, NY, USA, 2013.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Artese, A.; Costa, G.; Ortuso, F.; Parrotta, L.; Alcaro, S. Identification of New Natural DNA G-Quadruplex Binders Selected by a Structure-Based Virtual Screening Approach. Molecules 2013, 18, 12051-12070. https://doi.org/10.3390/molecules181012051
Artese A, Costa G, Ortuso F, Parrotta L, Alcaro S. Identification of New Natural DNA G-Quadruplex Binders Selected by a Structure-Based Virtual Screening Approach. Molecules. 2013; 18(10):12051-12070. https://doi.org/10.3390/molecules181012051
Chicago/Turabian StyleArtese, Anna, Giosuè Costa, Francesco Ortuso, Lucia Parrotta, and Stefano Alcaro. 2013. "Identification of New Natural DNA G-Quadruplex Binders Selected by a Structure-Based Virtual Screening Approach" Molecules 18, no. 10: 12051-12070. https://doi.org/10.3390/molecules181012051