Four New Glycosides from the Fruit of Xanthium sibiricum Patr.

Abstract
:1. Introduction
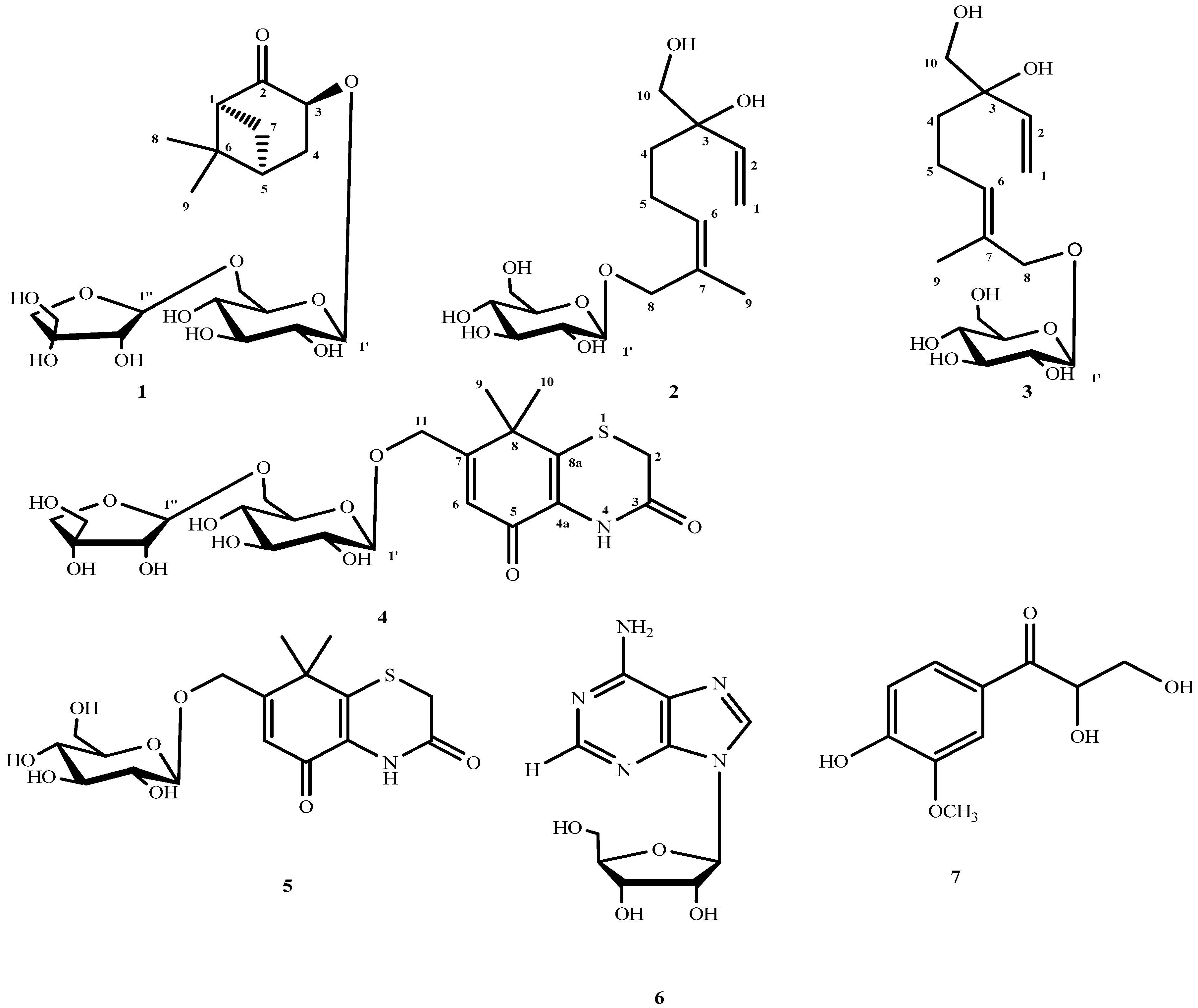
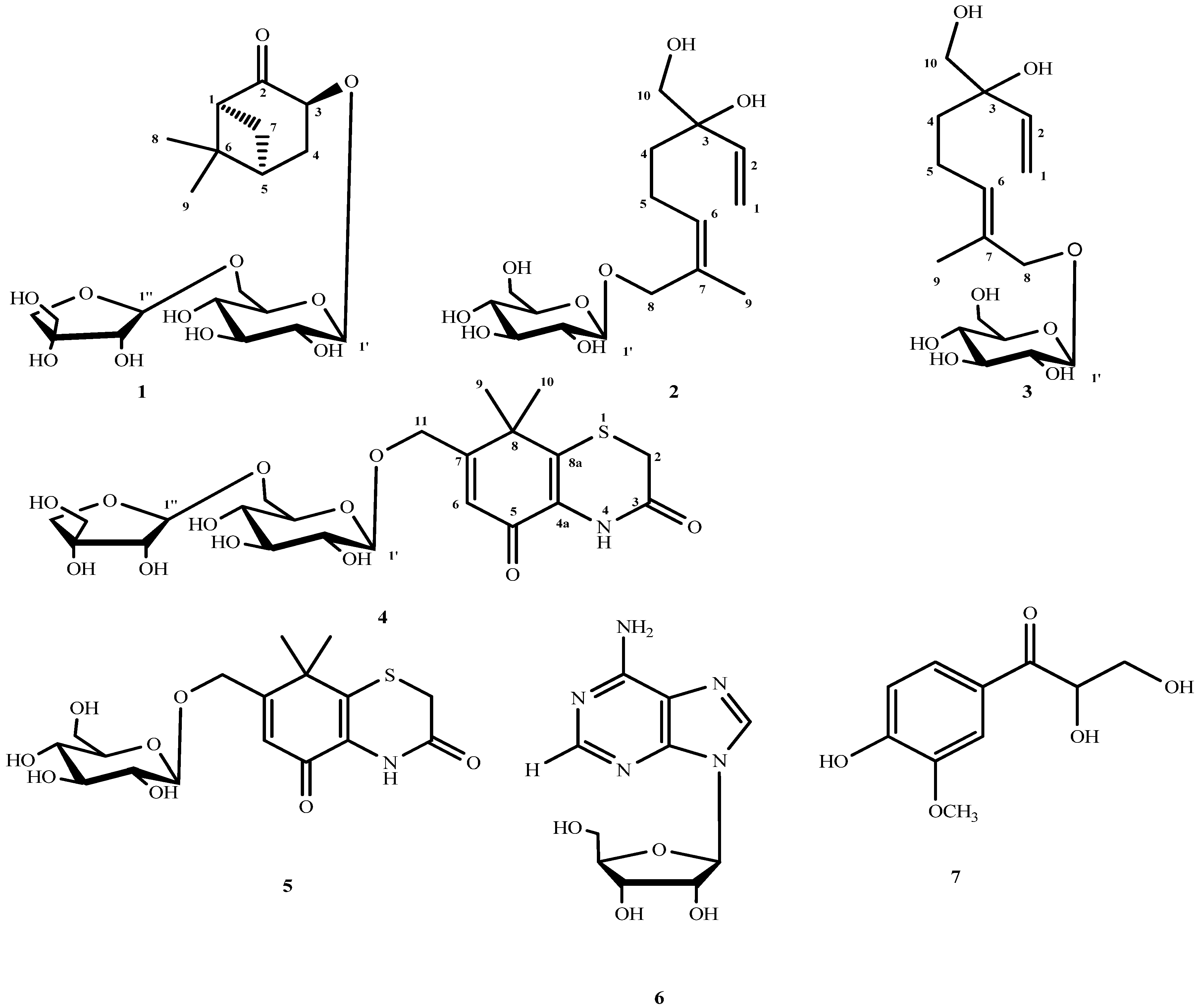
2. Results and Discussion

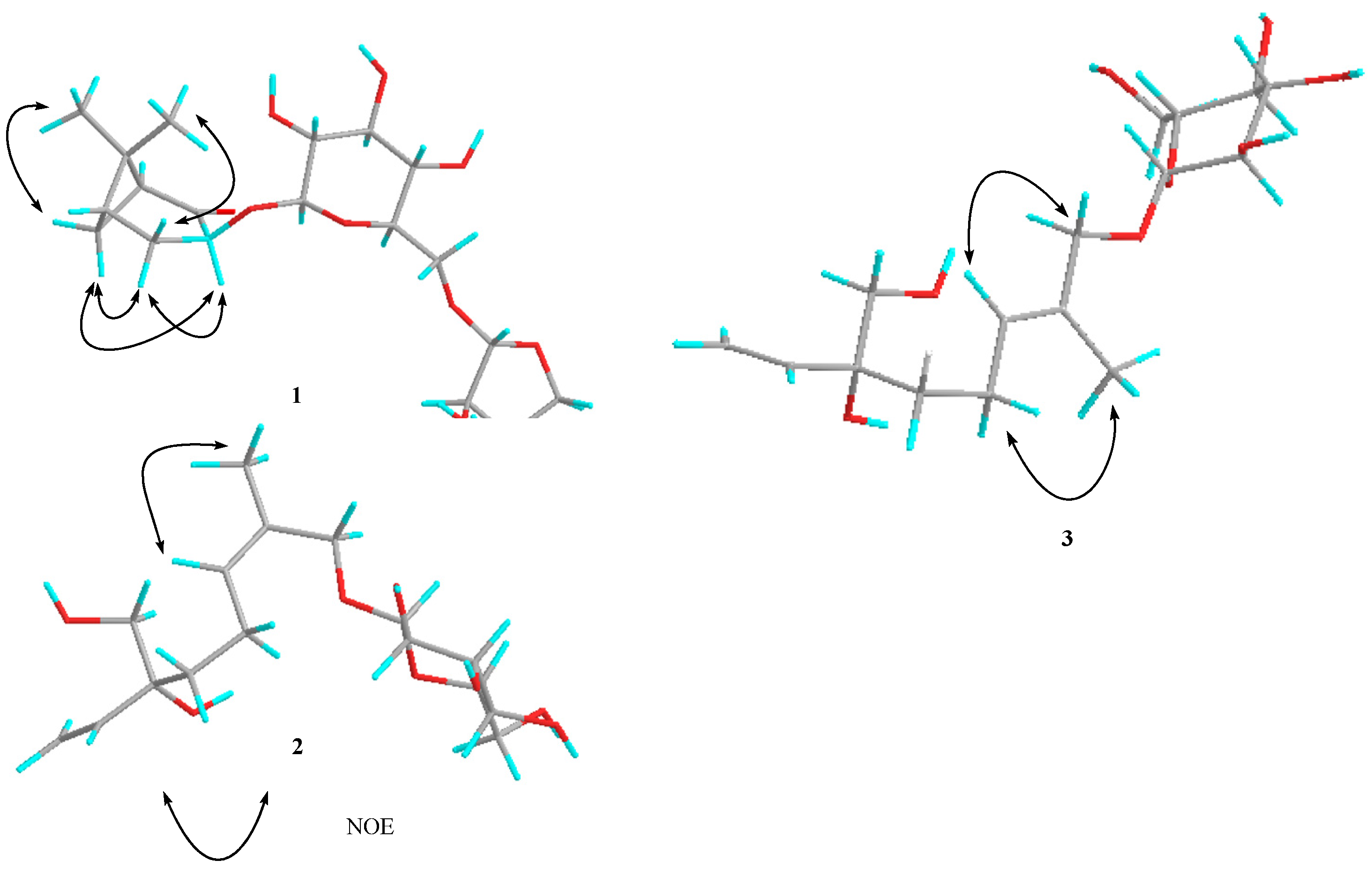

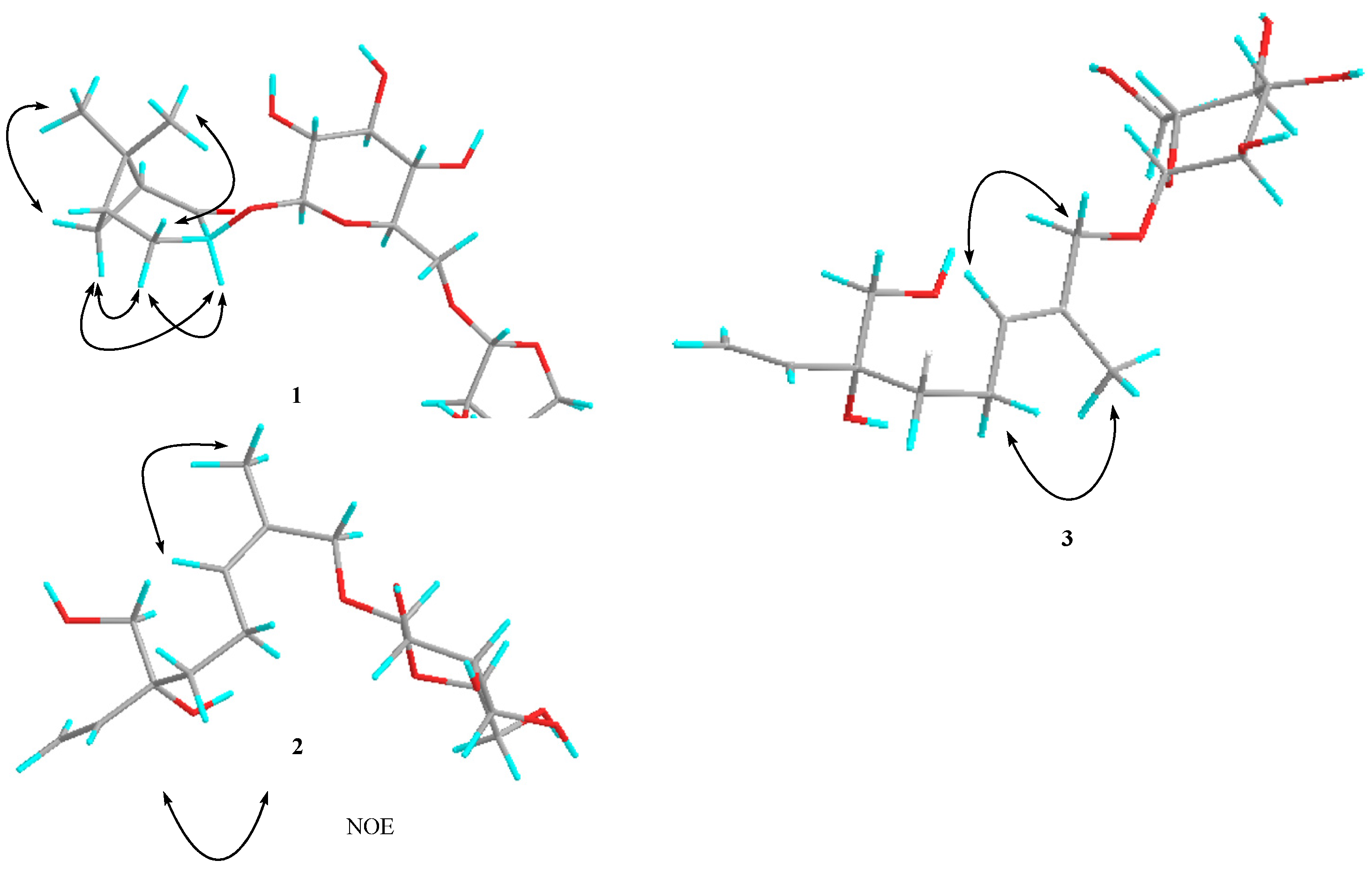
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 2.58 (1H, t, J = 5.4 Hz) | 5.27 (1H, dd, J = 1.7, 17.4 Hz) | 5.30 (1H, dd, J = 1.6, 17.4 Hz) | |
| 5.15 (1H, dd, J = 1.7, 10.9 Hz) | 5.17 (1H, dd, J = 1.6, 10.9 Hz) | |||
| 2 | 5.89 (1H, dd, J = 17.4, 10.9 Hz) | 5.88 (1H, dd, J = 17.4, 10.9 Hz) | 3.48 (2H, brs) | |
| 3 | 4.70 (1H, dd, J = 6.9, 10.6 Hz) | |||
| 4 | 1.90 (1H, dd, J = 6.9, 13.6 Hz) | 1.57 (1H, ddd, J = 9.6, 6.0, 2.4 Hz) | 1.59 (1H, ddd, J = 9.6, 6.0, 2.4 Hz) | |
| 2.77 (1H, ddd, J = 4.6, 10.6, 13.6 Hz) | 1.54 (1H, ddd, J = 9.6, 6.0, 2.4 Hz) | 1.52 (1H, ddd, J = 9.6, 6.0, 2.4 Hz) | ||
| 5 | 2.22 (1H, m) | 2.05 (1H, m) | 2.06 (1H, m) | |
| 2.15 (1H, m) | 2.16 (1H, m) | |||
| 6 | 5.41 (1H, brt, J = 7.0 Hz) | 5.48 (1H, brt, J = 6.5 Hz) | 6.67 (1H, brs) | |
| 7 | 2.48 (1H, m) | |||
| 1.81 (1H, m) | ||||
| 8 | 0.79 (3H, s) | 4.35 (1H, d, J = 11.4 Hz) | 4.18 (1H, d, J = 11.5 Hz) | |
| 4.20 (1H, d, J = 11.4 Hz) | 4.03 (1H, d, J = 11.5 Hz) | |||
| 9 | 1.36 (3H, s) | 1.76 (3H, s) | 1.68 (3H, s) | 1.47 (1H, s) |
| 10 | 3.40 (2H, d, J = 2.6 Hz) | 3.41 (2H, d, J = 3.2 Hz) | 1.47 (1H, s) | |
| 11 | 4.70 (1H, d, J = 15.8 Hz) | |||
| 4.51 (1H, d, J = 15.8 Hz) | ||||
| Glc-1' | 4.50 (1H, d, J = 7.8 Hz) | 4.21 (1H, d, J = 7.8 Hz) | 4.23 (1H, d, J = 7.8 Hz) | 4.35 (1H, d, J = 7.7 Hz) |
| 2' | 3.24 (1H, m) | 3.15 (1H, dd, J = 8.9, 7.8 Hz) | 3.18 (1H, m) | 3.30 (1H, m) |
| 3' | 3.35 (1H, m) | 3.25 (1H, m) | 3.25 (1H, m) | 3.34 (1H, m) |
| 4' | 3.28 (1H, m) | 3.30 (1H, m) | 3.31 (1H, m) | 3.25 (1H, m) |
| 5' | 3.40 (1H, m) | 3.35 (1H, m) | 3.34 (1H, m) | 3.41 (1H, m) |
| 6' | 3.98 (1H, dd, J = 12.5, 2.0 Hz) | 3.87 (1H, dd, J = 12.5, 2.0 Hz) | 3.85 (1H, dd, J = 12.0, 2.2 Hz) | 3.98 (1H, dd, J = 12.9, 1.6 Hz) |
| 3.60 (1H, dd, J = 12.5, 5.0 Hz) | 3.67 (1H, dd, J = 12.5, 5.0 Hz) | 3.65 (1H, dd, J = 12.0, 5.6 Hz) | 3.61 (1H, dd, J = 11.4, 6.5 Hz) | |
| Api-1'' | 5.01 (1H, d, J = 2.5 Hz) | 5.01 (1H, d, J = 2.4 Hz) | ||
| 2'' | 3.89 (1H, d, J = 2.5 Hz) | 3.89 (1H, d, J = 2.4 Hz) | ||
| 3'' | ||||
| 4'' | 3.74 (1H, d, J = 9.6 Hz) | 3.95 (1H, d, J = 9.6 Hz) | ||
| 3.97 (1H, d, J = 9.6 Hz) | 3.74 (1H, d, J = 9.6 Hz) | |||
| 5'' | 3.56 (2H, s) | 3.48 (2H, s) |
| No. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 57.9 | 114.5 | 114.5 | |
| 2 | 215.2 | 142.6 | 142.5 | 29.8 |
| 3 | 78.5 | 76.7 | 76.7 | 164.7 |
| 4 | 32.9 | 38.2 | 37.5 | |
| 4a | 131.0 | |||
| 5 | 41.4 | 22.8 | 22.8 | 177.1 |
| 6 | 45.5 | 131.5 | 130.3 | 123.4 |
| 7 | 24.9 | 132.7 | 132.9 | 167.2 |
| 8 | 21.9 | 67.8 | 75.9 | 43.5 |
| 8a | 143.5 | |||
| 9 | 26.3 | 21.9 | 14.1 | 27.5 |
| 10 | 69.5 | 69.7 | 27.4 | |
| 11 | 67.9 | |||
| Glc-1' | 105.9 | 102.4 | 102.6 | 103.9 |
| 2' | 75.5 | 75.0 | 75.1 | 75.1 |
| 3' | 77.9 | 77.9 | 77.9 | 78.0 |
| 4' | 71.6 | 71.7 | 71.7 | 71.7 |
| 5' | 77.1 | 78.2 | 78.2 | 77.2 |
| 6' | 68.7 | 62.8 | 62.8 | 68.7 |
| Api-1'' | 111.0 | 111.0 | ||
| 2'' | 78.0 | 78.0 | ||
| 3'' | 80.5 | 80.5 | ||
| 4'' | 75.0 | 75.0 | ||
| 5'' | 65.5 | 65.5 |
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
 −10.2 (c 0.26, MeOH) UV λmax (MeOH) nm (log ε): 201(3.18) IR (KBr): 3560, 3495, 2980, 1713, 1075, 1042 cm−1 1H-NMR: Table 1 13C-NMR: Table 2 HRESIMS m/z [M+Na]+ Calcd for C20H33O11Na 471.1842, Found: m/z 471.1847. −6.7 (c 0.17, MeOH) UV λmax (MeOH) nm (log ε): 202(2.10) IR (KBr): 3378, 2980, 2830, 1050, 917; cm−1 1H-NMR: Table 1 13C-NMR: Table 2 HRESIMS m/z [M+Na]+ Calcd for C16H28O8Na 371.1682, Found: m/z 371.1680. −8.0 (c 0.19, MeOH) UV λmax (MeOH) nm (log ε): 201(2.10) IR (KBr): 3380, 2979, 2833, 1056, 919; cm−1 1H-NMR: Table 1 13C-NMR: Table 2 HRESIMS m/z [M+Na]+ Calcd for C16H28O8Na 371.1682, Found: m/z 371.1685. −10.7 (c 0.22, MeOH) UV λmax (MeOH) nm (log ε): 201(2.18), 246(2.87) IR (KBr): 3462, 1690; 1658, 1215, 1177, 929 cm−1 1H-NMR: Table 1 13C-NMR: Table 2 HRESIMS m/z [M+H]+ Calcd for C22H32NO12S 534.1645, Found: m/z 534.1650.
−10.2 (c 0.26, MeOH) UV λmax (MeOH) nm (log ε): 201(3.18) IR (KBr): 3560, 3495, 2980, 1713, 1075, 1042 cm−1 1H-NMR: Table 1 13C-NMR: Table 2 HRESIMS m/z [M+Na]+ Calcd for C20H33O11Na 471.1842, Found: m/z 471.1847. −6.7 (c 0.17, MeOH) UV λmax (MeOH) nm (log ε): 202(2.10) IR (KBr): 3378, 2980, 2830, 1050, 917; cm−1 1H-NMR: Table 1 13C-NMR: Table 2 HRESIMS m/z [M+Na]+ Calcd for C16H28O8Na 371.1682, Found: m/z 371.1680. −8.0 (c 0.19, MeOH) UV λmax (MeOH) nm (log ε): 201(2.10) IR (KBr): 3380, 2979, 2833, 1056, 919; cm−1 1H-NMR: Table 1 13C-NMR: Table 2 HRESIMS m/z [M+Na]+ Calcd for C16H28O8Na 371.1682, Found: m/z 371.1685. −10.7 (c 0.22, MeOH) UV λmax (MeOH) nm (log ε): 201(2.18), 246(2.87) IR (KBr): 3462, 1690; 1658, 1215, 1177, 929 cm−1 1H-NMR: Table 1 13C-NMR: Table 2 HRESIMS m/z [M+H]+ Calcd for C22H32NO12S 534.1645, Found: m/z 534.1650.3.4. Acid hydrolysis of 1, 2, 3, and 4
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- The State of Pharmacopoeia Commission of People’s Republic of China. Pharmacopoeia of People’s Republic of China; Chemical Industry Press: Beijing, China, 2010; p. 151.
- Jiangsu New Medical College. Dictionary of Chinese Materia Medica; Shanghai Scientific Technological Publishers: Shanghai, China, 1985; pp. 1071–1072. [Google Scholar]
- Kinghorn, A.D.; Farnsworth, N.R.; Soejarto, D.D.; Cordell, G.A.; Pezutto, J.M.; Udeani, G.O.; Wani, M.C.; Wall, M.E.; Navarro, H.A.; Kramer, R.A.; et al. Novel strategies for the discovery of plant-derived anti-cancer agents. Pure Appl. Chem. 1999, 71, 1611–1618. [Google Scholar] [CrossRef]
- Sato, Y.; Oketani, H.; Yamada, T.; Singyouchi, K.; Ohtsubo, T.; Kihara, M.; Shibata, H.; Hi, T. A xanthanolide with potent anti-bacterial activity against methicillin-resistant Staphylococcus aureus. J. Pharm. Pharmacol. 1997, 49, 1042–1044. [Google Scholar] [CrossRef]
- Saxena, V.K.; Mondal, S.K. A xanthanolide from Xanthium strumarium. Phytochemistry 1994, 35, 1080–1082. [Google Scholar] [CrossRef]
- Turgut, M.; Alhan, C.C.; Gürgöze, M.K.A.; Doğan, Y.; Tekatli, M.; Akpolat, N.; Aygün, A.D. Carboxyatractyloside poisoning in humans. Ann. Trop. Paediatr. 2005, 25, 125–134. [Google Scholar] [CrossRef]
- West, P.L.; McKeown, N.J.; Hendrickson, R.G. Muscle spasm associated with therapeutic use of Cang Er Zi Wan. Clin. Toxicol. (Phila.) 2010, 48, 380–384. [Google Scholar] [CrossRef]
- Chen, B.; Ma, L.; Wang, X.; Shen, Y.; Jia, X. Simultaneous determination of 5 phenolic acids in fried Fructus xanthii from different production sites and its dispensing granules by using ultra-pressure liquid chromatography. Pharmacogn. Mag. 2013, 9, 103–108. [Google Scholar] [CrossRef]
- Kan, S.; Chen, G.; Han, C.; Chen, Z.; Song, X.; Ren, M.; Jiang, H. Chemical constituents from the roots of Xanthium sibiricum. Nat. Prod. Res. 2011, 25, 1243–1249. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Li, F.; Liu, X.; Chen, B.; Tang, Y.X.; Wang, M.K. Cytotoxic sesquiterpene lactones from aerial parts of Xanthium sibiricum. Planta Med. 2013, 79, 661–665. [Google Scholar] [CrossRef]
- Zhang, L.; Tao, L.; Ruan, J.; Li, W.; Wu, Y.; Yan, L.; Zhang, F.; Fan, F.; Zheng, S.; Wang, A.; Lu, Y. Xanthatin induces G2/M cell cycle arrest and apoptosis in human gastric carcinoma MKN-45 cells. Planta Med. 2012, 78, 890–895. [Google Scholar] [CrossRef]
- Ma, Y.T.; Huang, M.C.; Hsu, F.L.; Chang, H.F. Thiazinedione from xanthium strumarium. Phytochemistry 1998, 48, 1083–1085. [Google Scholar] [CrossRef]
- Roussakis, C.; Chinou, I.; Vayas, C.; Harvala, C.; Verbist, J.F. Cytotoxic activity of xanthatin and the crude extracts of Xanthium strumarium. Planta Med. 1994, 60, 473–474. [Google Scholar] [CrossRef]
- Dai, Y.H.; Cui, Z.; Li, J.L.; Wang, D. A new thiaziedione from the fruits of Xanthium sibiricum. J. Asian Nat. Prod. Res. 2008, 10, 343–347. [Google Scholar]
- Ahuja, M.; Nigam, S. Chemical examination of essential oil from the leaves of Xanthium strumarium (Linn.). Flavour Ind. 1970, 1, 627–630. [Google Scholar]
- Taher, H.A.; Ubiergo, G.O.; Talenti, E.C.J. Constituents of the essential of Xanthium cavanillesii. J. Nat. Prod. 1985, 48, 857. [Google Scholar] [CrossRef]
- MacLeod, J.K.; Moeller, P.D.; Franke, F.P. Two toxic kaurene glycosides from the burrs of Xanthium pungens. J. Nat. Prod. 1990, 53, 451–455. [Google Scholar] [CrossRef]
- Han, T.; Li, H.L.; Zhang, Q.Y.; Han, P.; Zheng, H.C.; Rahman, K.; Qin, L.P. Bioactivity-guided fractionation for anti-inflammatory and analgesic properties and constituents of Xanthium strumarium L. Phytomedicine 2007, 14, 825–829. [Google Scholar] [CrossRef]
- An, H.J.; Jeong, H.J.; Lee, E.H.; Kim, Y.K.; Hwang, W.J.; Yoo, S.J.; Hong, S.H.; Kim, H.M. Xanthii fructus inhibits inflammatory responses in LPS-stimulated mouse peritoneal macrophages. Inflammation 2004, 28, 263–270. [Google Scholar] [CrossRef]
- Zheng, X.K.; Bi, Y.F.; Feng, W.S. Study on the chemical constituents of Selaginellatamariscina (Beauv.) Spring. Acta Pharm. Sin. 2004, 39, 266–268. [Google Scholar]
- Beate, B.; Peter, W. Isolation and characterization of novel benzoates, cinnamates, flavonoids, and lignans from Riesling wine and screening for antioxidant activity. J. Agric. Food Chem. 2001, 49, 2788–2798. [Google Scholar] [CrossRef]
- Pierre, L.; Gilles, B. Efficient conversion of (1R,5R)-(+)-α-pinene to (1S,5R)-(-)-nopinone. J. Org. Chem. 1986, 51, 1362–1365. [Google Scholar] [CrossRef]
- Masateru, O.; Fumie, S.; Syoko, S.; Chika, T.; Hitoshi, Y. Three new steroid glycosides from the underground parts of Trillium kamtschaticum. Chem. Pharm. Bull. 2007, 55, 1093–1096. [Google Scholar] [CrossRef]
- Qiu, Y.K.; Chen, Y.J.; Pei, Y.P. Constituents with radical scavenging effect from Opuntiadillenii: Structures of new α-pyrones and flavonol glycoside. Chem. Pharm. Bull. 2002, 50, 1507–1510. [Google Scholar] [CrossRef]
- Wang, Z.B.; Gao, H.Y.; Xu, F.M.; Wu, L.J. Three new compounds from the leaves of Acanthopanaxseticosus Harms. Chin. Chem. Lett. 2010, 21, 702–705. [Google Scholar] [CrossRef]
- Isao, K.; Masahiro, S.; Fumi, H.; Zhou, J.L.; My, R.J.L. Apioglycyrrhizin and araboglycyrrhizin, two new sweet oleanene-type triterpeneoligoglycosides from the root of glycyrrhizainflata. Chem. Pharm. Bull. 1989, 37, 551–553. [Google Scholar] [CrossRef]
- Nakamura, S.; Li, X.Z.; Matsuda, H.; Yoshikawa, M. Bioactive constituents from Chinese natural medicines. XXVIII.1)Chemical structures of acyclic alcohol glycosides from the roots of Rhodiola crenulata. Chem. Pharm. Bull. 2008, 56, 536–540. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–4 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, H.; Yang, L.; Liu, C.; Hou, H.; Wang, Q.; Wang, Z.; Yang, B.; Kuang, H. Four New Glycosides from the Fruit of Xanthium sibiricum Patr. Molecules 2013, 18, 12464-12473. https://doi.org/10.3390/molecules181012464
Jiang H, Yang L, Liu C, Hou H, Wang Q, Wang Z, Yang B, Kuang H. Four New Glycosides from the Fruit of Xanthium sibiricum Patr. Molecules. 2013; 18(10):12464-12473. https://doi.org/10.3390/molecules181012464
Chicago/Turabian StyleJiang, Hai, Liu Yang, Chang Liu, Hui Hou, Qiuhong Wang, Zhibin Wang, Bingyou Yang, and Haixue Kuang. 2013. "Four New Glycosides from the Fruit of Xanthium sibiricum Patr." Molecules 18, no. 10: 12464-12473. https://doi.org/10.3390/molecules181012464