Endogenous Protection Derived from Activin A/Smads Transduction Loop Stimulated via Ischemic Injury in PC12 Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
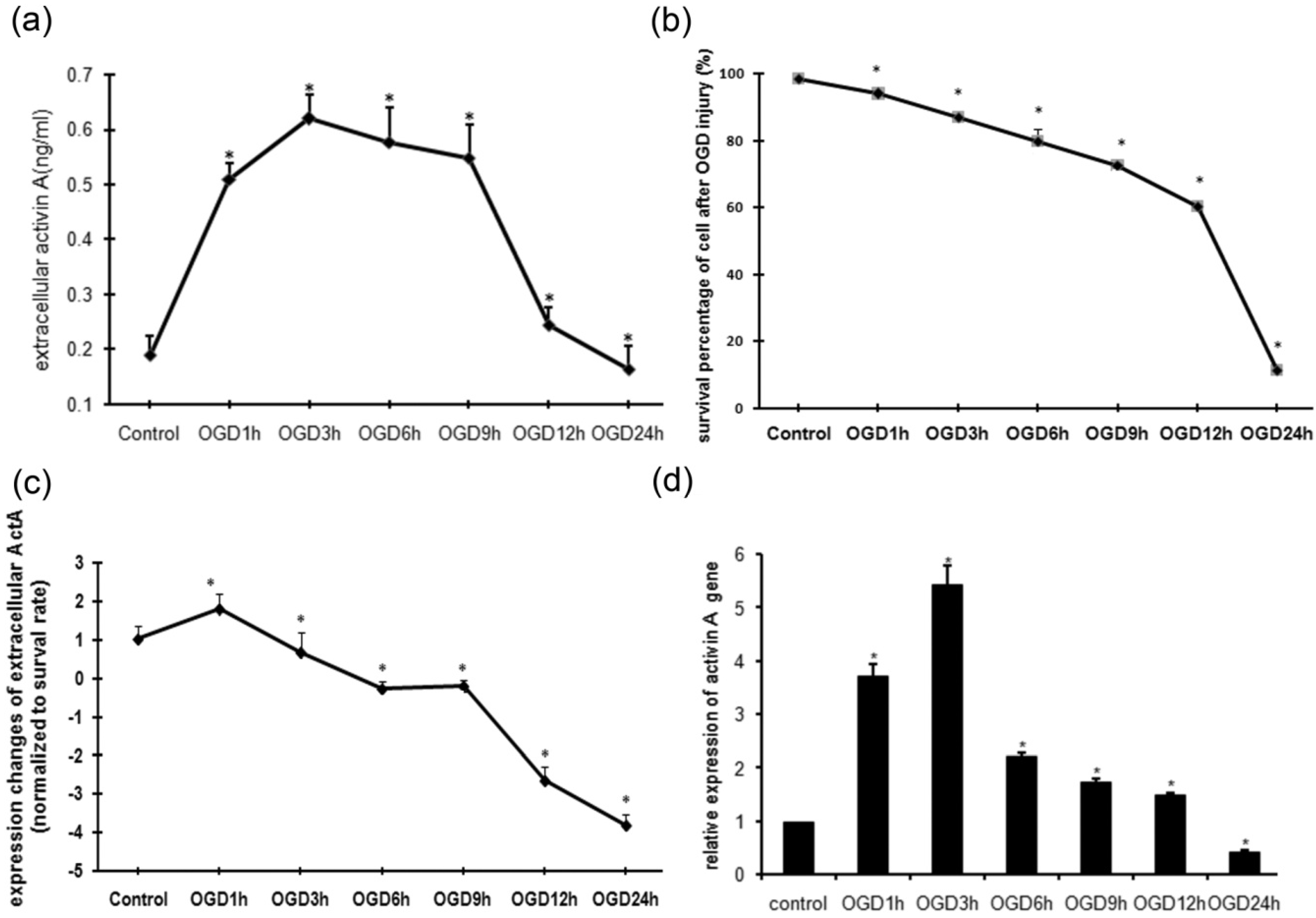
2.1. MTT Assay of PC12 Cells Subjected to OGD Injury
2.2. OGD Injury Up-Regulated the Production of ActA by PC12 Cells

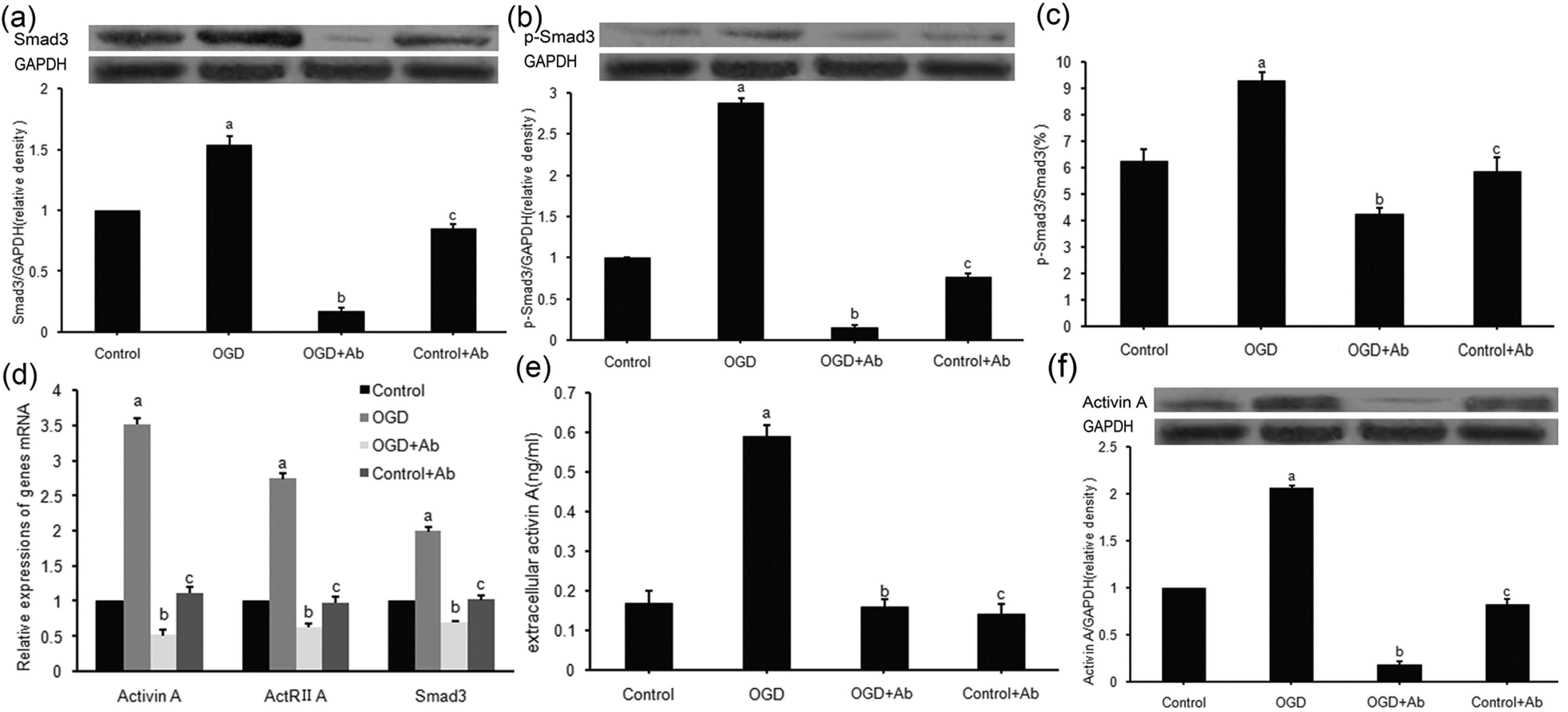
2.3. ActRⅡ-Ab Suppressed both Intracellular Signaling and Extracellular Secretion of ActA in PC12 Cells Subjected to OGD

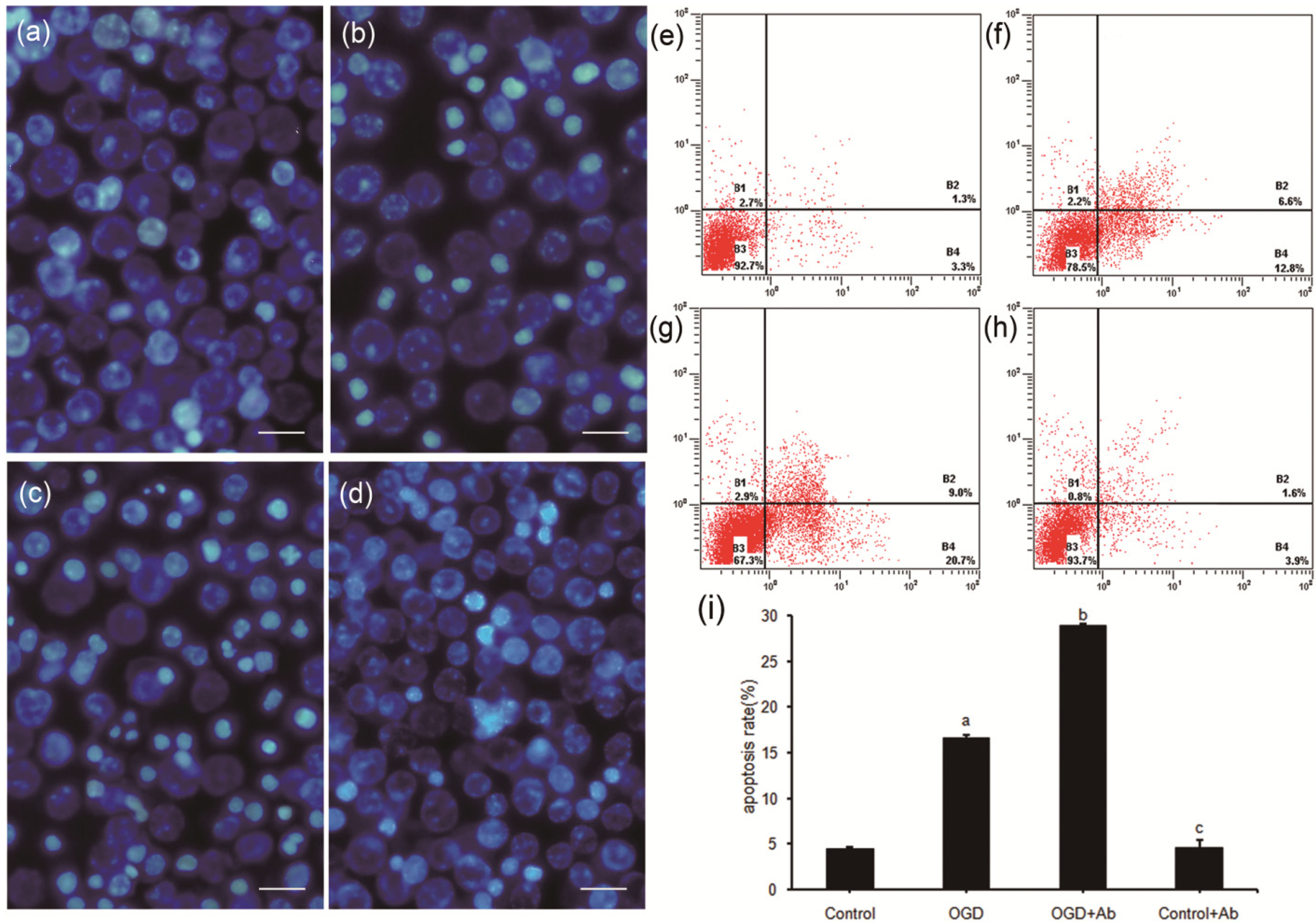
2.4. ActRII-Ab Augments Injury in PC12 Cells Induced by Ischemia (OGD) via Blocking of Activin A Signaling

3. Experimental
3.1. Cell Culture and Oxygen-Glucose Deprivation
3.2. Cell Survival Rate Assay
3.3. Extracellular Level of ActA Assay by ELISA
3.4. Pre-Treatment of ActRⅡ-Ab
3.5. Hoechst33342 Fluorescence Staining
3.6. Flow-Cytometry Analysis
3.7. Real-Time PCR
3.8. Western Blot Analysis
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chiuve, S.E.; Rexrode, K.M.; Spiegelman, D.; Logroscino, G.; Manson, J.E.; Rimm, E.B. Primary prevention of stroke by healthy lifestyle. Circulation 2008, 6, 947–954. [Google Scholar]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischemic stroke: An integrated view. Trends Neurosci. 1999, 9, 391–397. [Google Scholar]
- Ebner, F.H.; Mariotto, S.; Darra, E.; Suzuki, H.; Cavalieri, E. Use of STAT1 inhibitors in the treatment of brain I/R injury and neurodegenerative diseases. Agents Med. Chem. 2011, 11, 2–7. [Google Scholar]
- Taniguchi, H.; Anacker, C.; Suarez-Mier, G.B.; Wang, Q.; Andreasson, K. Function of prostaglandin E2EP receptors in the acute outcome of rodent hypoxic ischemic encephalopathy. Neurosci. Lett. 2011, 31, 185–190. [Google Scholar]
- Ferreira, M.C.; Cavallo, I.K.; Florio, P.; Petraglia, F.; Reis, F.M. Activin beta subunit, follistatin and follistatin-like 3 are expressed in the endometrium of ovary ectomized rats and regulated by estrogen replacement. J. Mol. Histol. 2008, 39, 535–541. [Google Scholar] [CrossRef]
- Kingsley, D.M. The TGF-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 8, 133–146. [Google Scholar] [CrossRef]
- Torres, P.B.; Florio, P.; Galleri, L.; Reis, F.M.; Borges, L.E.; Petraglia, F. Activin A, activin receptor type II, nodal, and cripto mRNA are expressed by eutopic and ectopic endometrium in women with ovarian endometriosis. Reprod. Sci. 2009, 16, 727–733. [Google Scholar] [CrossRef]
- Gressner, O.A. Intracrine signaling mechanisms of activin A and TGF-β. Vitam. Horm. 2011, 85, 59–77. [Google Scholar] [CrossRef]
- Lin, X.; Duan, X.; Liang, Y.Y.; Su, Y.; Wrighton, K.H.; Long, J.; Hu, M.; Davis, C.M.; Wang, J.; Brunicardi, F.C.; et al. PPM1A functions as a Smad phosphatase to terminate TGF beta signaling. Cell 2006, 125, 915–928. [Google Scholar] [CrossRef]
- Müller, M.R.; Zheng, F.; Werner, S.; Alzheimer, C. Transgenic mice expressing dominant-negativeactivin receptor IB in forebrain neurons reveal novel functions of activin at glutamatergic synapses. J. Biol. Chem. 2006, 281, 76–84. [Google Scholar]
- Wilms, H.; Schwark, T.; Brandenburg, L.O.; Sievers, J.; Dengler, R.; Deuschl, G.; Lucius, R. Regulation of activin A synthesis in microglial cells: pathophysiological implications for bacterial meningitis. J. Neurosci. Res. 2010, 88, 16–23. [Google Scholar] [CrossRef]
- He, J.T.; Mang, J.; Mei, C.L.; Yang, L.; Wang, J.Q.; Xing, Y.; Yang, H.; Xu, Z.X. Neuroprotective effects of exogenous activin a on oxygen-glucose deprivation in PC12 cells. Molecules 2011, 17, 315–327. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, R.; Guo, Y.L.; Mei, Y.W. Effect of neuregulin on apoptosis and expressions of STAT3 and GFAP in rats following cerebral ischemic reperfusion. J. Mol. Neurosci. 2009, 37, 67–73. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, X.; Liang, J.; Han, S.; Wang, Y.; Yin, Y.; Luo, Y.; Li, J. Phosphorylation of p38 MAPK mediates hypoxic preconditioning-induced neuroprotection against cerebral ischemic injury via mitochondria translocation of Bcl-xL in mice. Brain Res. 2013, 29, 78–88. [Google Scholar]
- Suzuki, S.; Tanaka, K.; Suzuki, N. Ambivalent aspects of interleukin-6 in cerebral ischemia: inflammatory versus neurotrophic aspects. J. Cereb. Blood Flow Metab. 2009, 29, 464–479. [Google Scholar] [CrossRef]
- Mukerji, S.S.; Katsman, E.A.; Wilber, C.; Haner, N.A.; Selman, W.R.; Hall, A.K. Activin is a neuronal survival factor that is rapidly increased after transient cerebral ischemia and hypoxia in mice. J. Cereb. Blood Flow Metab. 2007, 27, 1161–1172. [Google Scholar] [CrossRef]
- Ohnishi, N.; Miyata, T.; Ohnishi, H.; Yasuda, H.; Tamada, K.; Ueda, N.; Mashima, H.; Sugano, K. Activin A is an autocrine activator of rat pancreatic stellate cells: Potential therapeutic role of follistatin for pancreatic fibrosis. Gut 2003, 52, 1487–1493. [Google Scholar] [CrossRef]
- Powers, J.F.; Schelling, K.H.; Brachold, J.M.; Tischler, A.S. Plasticity of pheochromocytoma cell lines from neurofibromatosis knockout mice. Ann. N. Y. Acad. Sci. 2002, 971, 371–378. [Google Scholar] [CrossRef]
- Cho, Y.Y. A novel role of brain-type ACS4 isotype in neuronal differentiation. Biochem. Biophys. Res. Commun. 2012, 419, 505–510. [Google Scholar] [CrossRef]
- Marsell, R.; Einhorn, T.A. The role of endogenous bone morphogenetic proteins in normal skeletal repair. Injury 2009, 40, 4–7. [Google Scholar] [CrossRef]
- Miyazono, K. Positiveand negative regulation of TGF-beta signaling. J. Cell. Sci. 2000, 113, 1101–1109. [Google Scholar]
- Kupershmidt, L.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Blumenfeld, Z. The neuroprotective effect of Activin A and B: Implication for neurodegenerative diseases. J.Neurochem. 2007, 103, 962–971. [Google Scholar] [CrossRef]
- Mei, C.L.; He, J.T.; Mang, J.; Xu, Z.X. Nerve growth factor (NGF) combined with oxygen glucose deprivation (OGD) induces neural ischemia tolerance in PC12 cells. Afr. J. Biochem. Res. 2011, 5, 315–320. [Google Scholar]
- Sample Availability: Samples of all compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mang, J.; Mei, C.-L.; Wang, J.-Q.; Li, Z.-S.; Chu, T.-T.; He, J.-T.; Xu, Z.-X. Endogenous Protection Derived from Activin A/Smads Transduction Loop Stimulated via Ischemic Injury in PC12 Cells. Molecules 2013, 18, 12977-12986. https://doi.org/10.3390/molecules181012977
Mang J, Mei C-L, Wang J-Q, Li Z-S, Chu T-T, He J-T, Xu Z-X. Endogenous Protection Derived from Activin A/Smads Transduction Loop Stimulated via Ischemic Injury in PC12 Cells. Molecules. 2013; 18(10):12977-12986. https://doi.org/10.3390/molecules181012977
Chicago/Turabian StyleMang, Jing, Chun-Li Mei, Jiao-Qi Wang, Zong-Shu Li, Ting-Ting Chu, Jin-Ting He, and Zhong-Xin Xu. 2013. "Endogenous Protection Derived from Activin A/Smads Transduction Loop Stimulated via Ischemic Injury in PC12 Cells" Molecules 18, no. 10: 12977-12986. https://doi.org/10.3390/molecules181012977