General
Unless otherwise noted, reagents and solvents were purchased from commercial suppliers (Aldrich, Milano, Italy and Fluka, Milano, Italy) and were used without purification. The progress of the reaction was monitored by thin-layer chromatography with F254 silica-gel precoated sheets (Merck, Darmstadt, Germany). UV light, ninhydrin ethanolic solution (0.3% w/v) and potassium permanganate solution (10% w/v) were used for detection. Flash chromatography was performed using Merck silica-gel 60 (Si 60, 40–63 μm, 230–400 mesh ASTM). Dichloromethane was dried by distillation over calcium hydride. All reactions were carried out using flame-dried glassware under nitrogen atmosphere. Melting points were determined on a Gallenkamp melting point apparatus and were not corrected. The 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were recorded on a Bruker Avance 400 spectrometer; chemical shifts (δ scale) are reported in parts per million (ppm). 1H-NMR spectra are reported in the following order: multiplicity, approximate coupling constants (J value) in Hertz (Hz) and number of protons; signals were characterized as s (singlet), d (doublet), t (triplet), q (quadruplet), m (multiplet), b (broad). Mass spectra were recorded on an Applied Biosystems API-150 EX system spectrometer equipped with an ESI interface. The final compounds were analyzed on a ThermoQuest Italia (Milano, Italy). A FlashEA 1112 Elemental Analyzer was used for for C, H and N analyses. The percentages found were within ± 0.4% of the theoretical values.
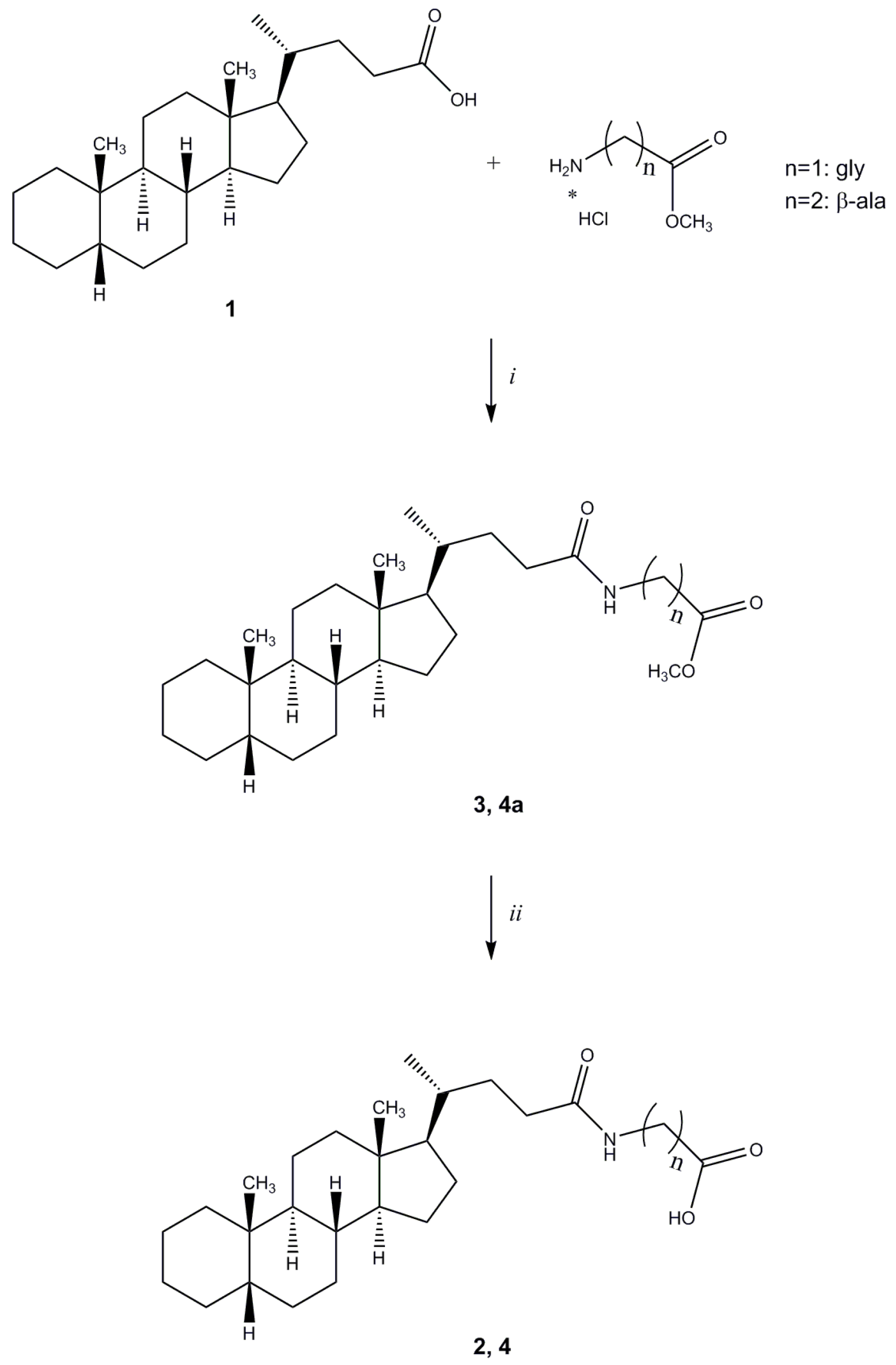
Methyl N-(5β-cholan-24-oyl)glycinate (
3). Compound
3 was synthesized following a modification of a described procedure [
21] in which to a stirred solution of cholanic acid (
1, 0.832 mmol), L-glycine methyl ester hydrochloride (0.915 mmol) and N-methylmorpholine (NMM, 2.080 mmol) in anhydrous CH
2Cl
2 (20 mL) under nitrogen was added
N-(3-dimethylaminopropyl)-
N'-ethylcarbodiimide hydrochloride (EDCI, 0.857 mmol). The reaction mixture was stirred at room temperature overnight and then was diluted with CH
2Cl
2, (30 mL) washed with HCl 2N, brine and dried over anhydrous Na
2SO
4. Evaporation of the solvent under reduced pressure yielded a white solid that was purified by flash chromatography [SiO
2, CH
2Cl
2:EtOH 98:2]. The crude product was re-crystallized from ethanol-water to give
3. Yield: 81%. Mp: 116–119 °C.
1H-NMR (CDCl
3)
δ = 0.63 (s, 3H, CH
3), 0.90–0.92 (m, 7H), 1.00–1.13 (m, 4H), 1.14–1.30 (m, 8H), 1.32–1.42 (m, 7H), 1.53–1.57 (m, 1H), 1.71–1.74 (m, 3H), 1.78–1.87 (m, 3H), 1.92–1.95 (m, 1H), 2.09–2.16 (m, 1H), 2.26–2.33 (m, 1H), 3.75 (s, 3H, OCH
3), 4.04 (d,
J = 5.2 Hz, 2H, CH
2CO), 6.02 (bs, 1H, NH).
13C-NMR (CDCl
3)
δ = 12.06, 18.37, 20.83, 21.34, 24.24, 24.26, 26.55, 27.03, 27.25, 27.51, 28.25, 31.61, 33.26, 35.36, 35.49, 35.89, 37.59, 40.29, 40.51, 41.20, 42.77, 43.73, 52.34, 56.05, 56.61, 170.64 (C=O), 173.78 (C=O). MS (ESI) calc for C
27H
45NO
3: 431.34; found: 432.6 [M+H]
+, 454.6 [M+Na]
+, 470.5 [M+K]
+. Anal. calc for C
27H
45NO
3: C, 75.13; H, 10.51; N, 3.24; found: C, 75.77; H, 10.96; N, 3.11.
N-(5β-cholan-24-oyl)glycine (
2). Compound
2 was synthesized following a modification of a described procedure [
22] in which to a solution of compound
3 (0.563 mmol) in ethanol (15 mL) a solution of sodium hydroxide 15%
w/v (12 mL) was added and the mixture stirred at room temperature for 1 h. Ethanol was removed under reduced pressure and the solution was acidified with concentrated hydrochloric acid until a precipitate was formed. The resulting suspension was suction-filtered and the obtained white residue washed with water. The crude product was crystallized from ethanol-water to give the title compound
2 (94%) as a white solid. Mp: 192–195 °C.
1H-NMR (DMSO-d
6)
δ = 0.60 (s, 3H, CH
3), 0.81–0.88 (m, 7H), 0.94–1.09 (m, 4H), 1.13–1.25 (m, 9H), 1.32–1.34 (m, 6H), 1.49–1.51 (m, 1H), 1.65–1.84 (m, 6H), 1.90–1.92 (m, 1H), 1.97–2.04 (m, 1H), 2.09–2.16 (m, 1H), 3.69 (d,
J = 6.0 Hz, 2H, CH
2), 8.07 (t,
J = 6.0 Hz, 1H, NH), 12.43 (bs, 1H, COOH).
13C-NMR (DMSO-d
6)
δ = 12.34, 18.75, 20.91, 21.28, 24.32, 24.50, 26.62, 26.96, 27.17, 27.51, 28.16, 31.90, 32.51, 35.34, 35.39, 35.86, 37.60, 40.99, 42.74, 43.57, 56.12, 56.53, 171.90 (C=O), 173.36 (C=O). MS (ESI) calc for C
26H
43NO
3: 417.32; found: 416.6 [M−1]
−. Anal. calc for C
26H
43NO
3: C, 74.77; H, 10.38; N, 3.35; found: C, 74.88; H, 10.75; N, 3.32.
Methyl N-(5β-cholan-24-oyl)-β-alaninate (4a). Compound 4a was synthesized following the procedure described for compound 3 using β-alanine methyl ester hydrochloride and purified by flash chromatography [SiO2, CH2Cl2:EtOH 98:2]. The crude product was re-crystallized from ethanol-water to give 4a. Yield: 81%. Mp: 108–111 °C. 1H-NMR (CDCl3) δ = 0.63 (s, 3H, CH3), 0.90–092 (m, 7H), 1.01–1.14 (m, 4H), 1.16–1.32 (m, 8H), 1.34–1.41 (m, 6H), 1.54–1.57 (m, 1H), 1.61–1.65 (m, 1H), 1.72–1.88 (m, 6H), 1.92–1.96 (m, 1H), 2.00–2.08 (m, 1H), 2.18–2.25 (m, 1H), 2.54 (t, J = 6.0 Hz, 2H, CH2), 3.49–3.54 (m, 2H, CH2), 3.70 (s, 3H, OCH3), 6.02 (bs, 1H, NH). 13C-NMR (CDCl3) δ = 12.04, 18.36, 20.83, 21.34, 24.23, 24.27, 26.55, 27.03, 27.25, 27.51, 28.26, 31.74, 33.62, 33.86, 34.72, 35.36, 35.49, 35.89, 37.59, 40.29, 40.51, 42.76, 43.73, 51.78, 56.04, 56.61, 173.25 (C=O), 173.60 (C=O). MS (ESI) calc for C28H47NO3: 445.36; found: 446.5 [M+H]+, 468.5 [M+Na]+, 484.5 [M+K]+. Anal. calc for C28H47NO3: C, 75.46; H, 10.63; N, 3.14; found: C, 75.28; H, 10.56; N, 3.08.
N-(5β-Cholan-24-oyl)-β-alanine (
4). Compound
4 was synthesized following a modification of a described procedure [
23]. To a solution of compound
4a (0.449 mmol) in ethanol (15 mL) a solution of sodium hydroxide 15%
w/v (12 mL) was added and the mixture stirred at room temperature for 1 h. Ethanol was removed under reduced pressure and the solution was acidified with concentrated hydrochloric acid until a precipitate was formed. The resulting suspension was suction-filtered and the solid residue thus obtained washed with water. The crude product was re-crystallized from ethyl acetate to give the title compound
4. Yield: 93%. Mp: 152–155 °C.
1H-NMR (DMSO-d
6)
δ = 0.59 (s, 3H, CH
3), 0.84–0.88 (m, 7H), 1.03–1.08 (m, 4H), 1.15–1.34 (m, 15H), 1.50–1.58 (m, 1H), 1.65–1.71 (m, 4H), 1.76–1.84 (m, 2H), 1.89–1.96 (m, 2H), 2.01–2.08 (m, 1H), 2.32 (bt,
J = 6.8 Hz, 2H, CH
2), 3.16–3.21 (m, 2H, CH
2), 7.82 (s, 1H, NH), 12.14 (bs, 1H, COOH).
13C-NMR (DMSO-d
6)
δ = 12.31, 18.74, 20.90, 21.28, 24.31, 24.50, 26.62, 26.96, 27.17, 27.51, 28.18, 32.00, 32.73, 34.42, 35.18, 35.39, 35.86, 37.60, 42.73, 43.57, 56.07, 56.53, 173.01 (C=O), 173.37 (C=O). MS (ESI) calc for C
27H
45NO
3: 431.34; found: 430.6 [M−1]
−. Anal. calc for C
27H
45NO
3: C, 75.13; H, 10.51; N, 3.24; found: C, 75.20; H, 10.53; N, 3.18.
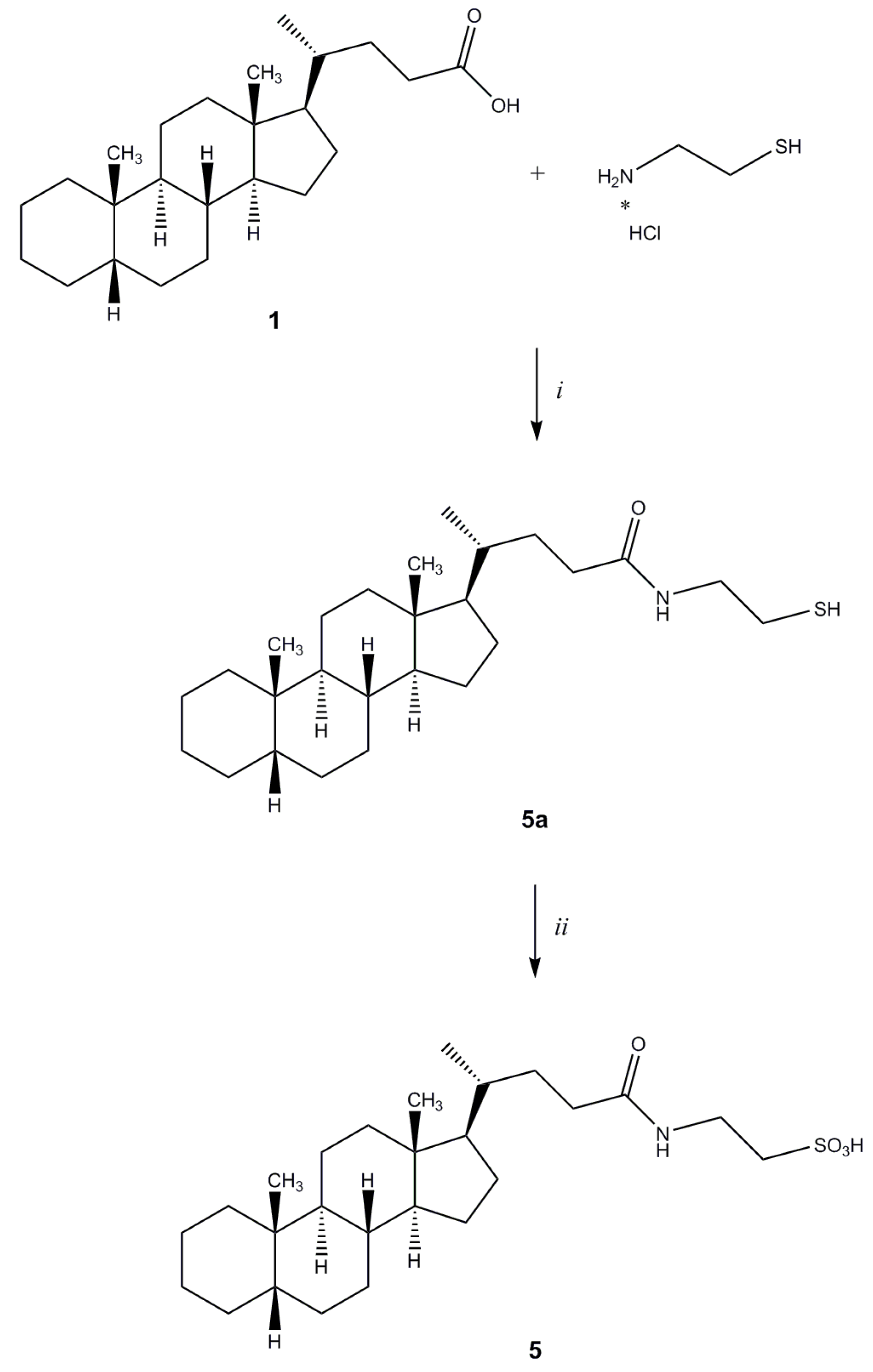
2-[N-(5β-Cholan-24-oyl)amino]ethanethiol (5a). Compound 5a was synthesized following the procedure described for 3 using cysteamine hydrochloride and purified by flash chromatography [SiO2, CH2Cl2:MeOH(NH3) 98:2]. Yield: 36%. 1H-NMR (CDCl3) δ = 0.64 (s, 3H, CH3), 0.84–091 (m, 7H), 1.01–1.41 (m, 20H), 1.57 (m, 1H), 1.72–1.96 (m, 6H), 2.09–2.11 (m, 1H), 2.23–2.26 (m, 1H), 2.39 (m, 1H), 2.64–2.70 (m, 1H, CH2CHH), 3.41–3.46 (m, 1H, CH2CHH), 5.89 (bs, 1H, NH). 13C-NMR (CDCl3) δ = 12.08, 18.40, 20.83, 21.35, 24.26, 24.74, 26.56, 27.04, 27.25, 27.52, 28.29, 30.85, 31.81, 33.63, 35.36, 35.52, 35.90, 37.60, 40.30, 40.52, 42.28, 42.78, 43.74, 56.05, 56.62, 173.86 (C=O). MS (ESI) calc for C26H45NOS: 419.32; found: 420.5 [M+H]+, 442.4 [M+Na]+.
2-[N-(5β-Cholan-24-oyl)amino]ethanesulfonic Acid (
5). Compound
5 was synthesized following a modification of a described procedure [
24]. To a stirred solution of
5a (0.295 mmol) in CH
2Cl
2 under nitrogen at 0 °C, a solution of peracetic acid in water (40%
w/w, 0.966 mmol) was added dropwise The mixture was allowed to warm to room temperature while stirring over the course of 30 min. Evaporation of the solvent under reduced pressure afforded a white solid that was purified by flash chromatography [SiO
2, CH
2Cl
2:HCOOH:EtOH 76:4:20]. The crude product was re-crystallized from ethanol-water to give
5. Yield: 98%. Mp: 200–203 °C.
1H-NMR (CDCl
3)
δ = 0.59 (s, 3H, CH
3), 0.84–0.88 (m, 7H), 1.01–1.08 (m, 5H), 1.14–1.22 (m, 9H), 1.31–1.34 (m, 6H), 1.51 (m, 1H), 1.61–1.71 (m, 4H), 1.76-1.81 (m, 2H), 1.84–1.94 (m, 2H), 2.00–2.07 (m, 1H), 7.65 (bs, 1H, NH).
13C-NMR (CDCl
3)
δ = 12.35, 18.77, 20.92, 21.30, 24.31, 24.50, 26.62, 26.99, 27.17, 27.53, 28.14, 31.93, 33.11, 35.37, 35.42, 35.91, 35.97, 37.64, 42.77, 43.62, 51.10, 56.09, 56.54, 172.51 (C=O). MS (ESI) calc for C
26H
45NO
4S: 467.31; found: 466.4 [M−1]
−−. Anal. calc for C
26H
45NO
4S: C, 66.77; H, 9.70; N, 2.99; found: C, 66.60; H, 9.11; N, 2.68.
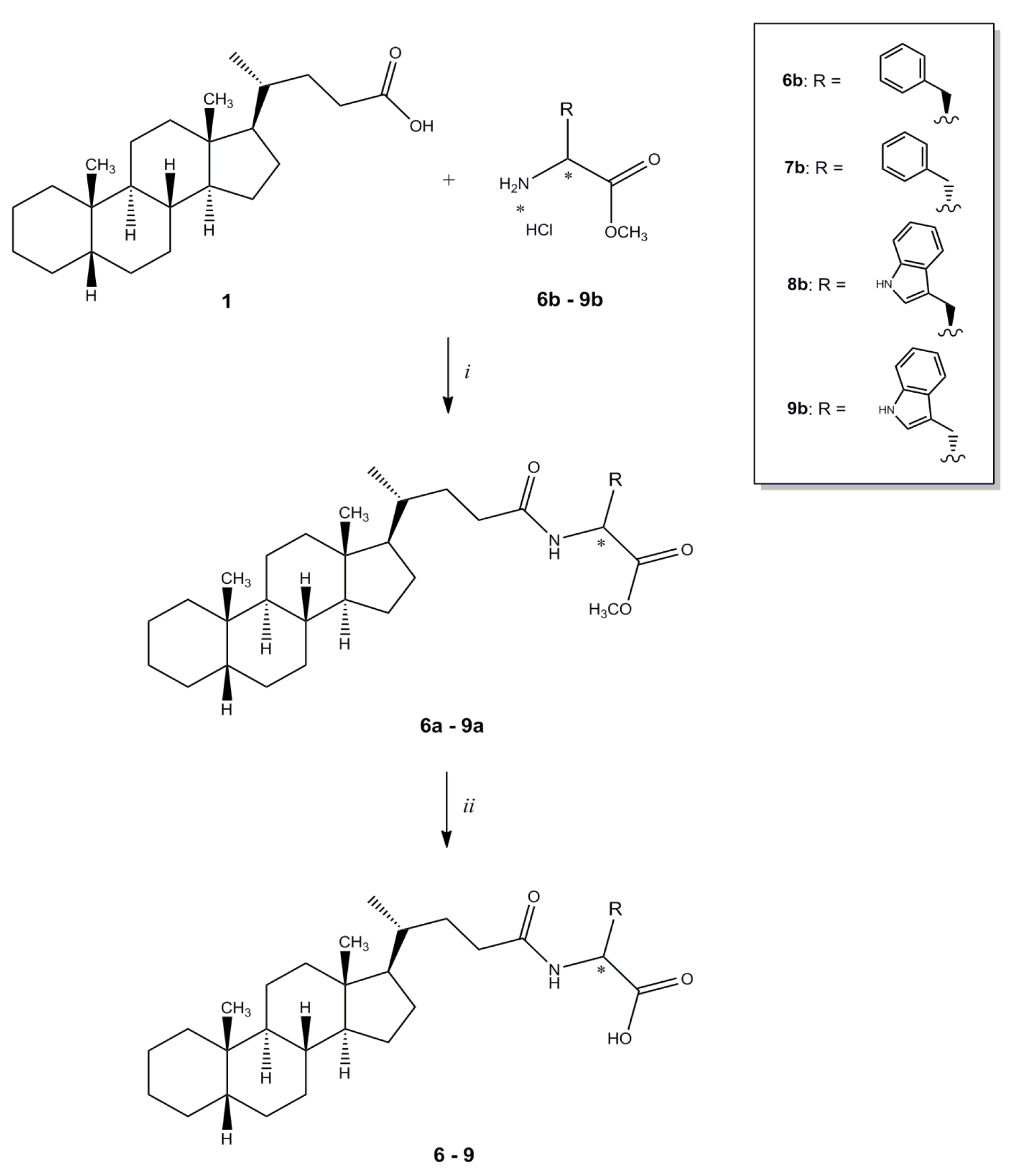
Methyl N-(5β-cholan-24-oyl)-L-phenylalaninate (6a). Compound 6a was synthesized following procedure described for compound 3 starting from L-phenylalanine methyl ester hydrochloride and purified by flash chromatography [SiO2, CH2Cl2:EtOH 98:2]. The crude product was re-crystallized from ethanol-water to give 6a. Yield: 83%. Mp: 78–81 °C. 1H-NMR (CDCl3) δ = 0.63 (s, 3H, CH3), 0.89–0.91 (m, 7H), 1.04–1.08 (m, 4H), 1.13–1.23 (m, 9H), 1.36–1.39 (m, 6H), 1.57 (m, 1H), 1.72–1.95 (m, 7H), 2.03–2.10 (m, 1H), 2.18–2.22 (m, 1H), 3.09 (dd, J = 13.8, 5.7 Hz, 1H, CHCHH), 3.15 (dd, J = 13.8, 5.8 Hz, 1H, CHCHH), 3.73 (s, 3H, OCH3), 4.87–4.92 (m, 1H, CHCH2), 5.86 (d, J = 7.7 Hz, 1H, NH), 7.09 (d, J = 6.4 Hz, 2H, Ar), 7.24-7.30 (m, 3H, Ar). 13C-NMR (CDCl3) δ = 12.07, 18.36, 20.83, 21.35, 24.25, 24.28, 26.56, 27.04, 27.26, 27.52, 28.24, 31.61, 33.40, 35.37, 35.44, 35.89, 37.60, 37.88, 40.29, 40.51, 42.76, 43.73, 52.32, 52.91, 56.01, 56.61, 127.12, 128.56, 129.28, 135.89, 172.21 (C=O), 173.15 (C=O). MS (ESI) calc for C34H51NO3: 521.39; found: 522.6 [M+H]+, 544.5 [M+Na]+. Anal. calc for C34H51NO3: C, 78.26; H, 9.85; N, 2.68; found: C, 78.62; H, 10.27; N, 2.67.
N-(5β-Cholan-24-oyl)-L-phenylalanine (6). Compound 6 was synthesized following the procedure described for compound 2 starting from compound 6a. The crude product was re-crystallized from ethanol-water to give 5. Yield: 97%. Mp: 189–192 °C. 1H-NMR (DMSO-d6) δ = 0.57 (s, 3H, CH3), 0.81–0.82 (m, 4H), 0.88 (s, 3H, CH3), 0.99–1.04 (m, 6H), 1.12–1.18 (m, 8H), 1.25–1.34 (m, 6H), 1.48–1.59 (m, 2H), 1.68–1.80 (m, 5H), 1.87–1.97 (m, 1H), 2.02–2.05 (m, 1H), 2.82 (dd, J = 13.6, 10.0 Hz, 1H, CHCHH), 3.02 (dd, J = 14.0, 4.8 Hz, 1H, CHCHH), 4.34–4.40 (m, 1H, CHCH2), 7.15–7.26 (m, 5H, Ar), 8.08 (d, J = 8.4 Hz, 1H, NH). 13C-NMR (DMSO-d6) δ = 12.32, 18.72, 20.90, 21.28, 24.31, 24.51, 26.62, 26.96, 27.17, 27.51, 28.11, 31.90, 32.50, 35.30, 35.39, 35.86, 37.20, 37.60, 42.70, 43.57, 53.78, 56.05, 56.52, 126.76, 128.53, 129.52, 138.26, 173.03 (C=O), 173.66 (C=O). MS (ESI) calc for C33H49NO3: 507.37; found: 506.6 [M−H]−−. Anal. calc for C33H49NO3: C, 78.06; H, 9.73; N, 2.76; found: C, 78.34; H, 9.40; N, 2.76.
Methyl N-(5β-cholan-24-oyl)-D-phenylalaninate (7a). Compound 7a was synthesized following the procedure described for compound 3 starting from D-phenylalanine methyl ester hydrochloride and purified by flash chromatography [SiO2, CH2Cl2:C2H5OH 99:1]. The crude product was re-crystallized from ethanol-water to give 7a. Yield: 90%. Mp: 82–85 °C. 1H-NMR (CDCl3) δ = 0.63 (s, 3H, CH3), 0.88–0.91 (m, 7H), 1.04–1.13 (m, 5H), 1.16–1.23 (m, 8H), 1.36–1.38 (m, 6H), 1.57 (m, 1H), 1.65 (m, 1H), 1.72–1.85 (m, 6H), 1.93–1.96 (m, 1H), 2.05–2.09 (m, 1H), 2.20–2.24 (m, 1H), 3.09 (dd, J = 13.84, 5.68 Hz, 1H, CHCHH), 3.15 (dd, J = 13.84, 5.84 Hz, 1H, CHCHH ), 3.73 (s, 3H, OCH3), 4.87–4.92 (m, 1H, CHCH2), 5.86 (d, J = 7.6 Hz, 1H, NH), 7.08–7.09 (m, 2H, Ar), 7.24–7.30 (m, 3H, Ar). 13C-NMR (CDCl3) δ = 12.09, 18.33, 20.84, 21.35, 24.24, 24.28, 26.56, 27.04, 27.26, 27.53, 28.24, 31.60, 33.47, 35.37, 35.48, 35.90, 37.60, 37.91, 40.30, 40.52, 42.77, 43.74, 52.30, 52.90, 56.07, 56.61, 127.11, 128.55, 129.28, 135.89, 172.18 (C=O), 173.05 (C=O). MS (ESI) calc for C34H51NO3: 521.39; found: 522.5 [M+H]+, 544.3 [M+Na]+. Anal. calc for C34H51NO4: C, 78.26; H, 9.85; N, 2.68; found: C, 78.62; H, 10.27; N, 2.67.
N-(5β-cholan-24-oyl)-D-phenylalanine (7). Compound 7 was synthesized following the procedure described for 2 using 7a. The crude product was re-crystallized from ethanol-water to give 7. Yield: 97%. Mp: 195–198 °C. 1H-NMR (DMSO-d6) δ = 0.56 (s, 3H, CH3), 0.79–0.81 (m, 3H), 0.83–0.88 (m, 4H), 0.99-1.04 (m, 5H), 1.15–1.18 (m, 9H), 1.31–1.32 (m, 5H), 1.50–1.55 (m, 2H), 1.68–1.96 (m, 7H), 2.04–2.07 (m, 1H), 2.82 (dd, J = 13.52, 8.96 Hz, 1H, CHCHH), 3.03 (dd, J = 13.64, 4.68 Hz, 1H, CHCHH), 4.31–4.32 (m, 1H, CHCH2), 7.14–7.22 (m, 5H, Ar), 7.86 (d, J = 7.64 Hz, 1H, NH). 13C-NMR (CDCl3) δ = 12.33, 18.64, 20.91, 21.28, 24.31, 24.51, 26.62, 26.95, 27.17, 27.51, 28.14, 31.92, 32.75, 35.30, 35.38, 35.84, 37.51, 37.60, 42.70, 43.56, 54.46, 56.22, 56.50, 126.49, 128.34, 129.65, 138.78, 172.52 (C=O), 173.96 (C=O). MS (ESI) calc for C33H49NO3: 507.37; found: 506.5 [M−H]−−. Anal. calc for C33H49NO4: C, 78.06; H, 9.73; N, 2.76; found: C, 78.34; H, 9.40; N, 2.76.
Methyl N-(5β-cholan-24-oyl)-L-tryptophanate (8a). Compound 8a was synthesized following the procedure described for compound 3 starting from L-tryptophan methyl ester hydrochloride and purified by flash chromatography [SiO2, CH2Cl2:EtOH 98:2], yielding compound 8a as a pale-yellow oil, which was used in the next step without further purification. Yield: 98%. 1H-NMR (DMSO-d6) δ = 0.62 (s, 3H, CH3), 0.83–0.91 (m, 7H), 0.99–1.09 (m, 5H), 1.12–1.30 (m, 9H), 1.35–1.38 (m, 6H), 1.55–1.57 (m, 1H), 1.73–2.09 (m, 7H), 2.18–2.26 (m, 1H), 3.32 (dd, J = 5.2, 2.1 Hz, 2H, CHCH2), 3.70 (s, 3H, OCH3), 4.96 (dt, J = 7.8, 5.4 Hz, 1H, CHCH2), 6.00 (bd, J = 8.1 Hz, 1H, NH), 6.97 (d, J = 2.4 Hz, 1H, Ar), 7.12 (td, J = 7.2, 1.2 Hz 1H, Ar), 7.19 (td, J = 7.2, 1.2 Hz 1H, Ar), 7.36 (d, J = 8.1 Hz, 1H, Ar), 7.53 (d, J = 8.1 Hz, 1H, Ar), 8.32 (bs, 1H, NH). 13C-NMR (DMSO-d6) δ = 12.05, 18.35, 20.83, 21.35, 24.24, 24.28, 26.57, 27.04, 27.26, 27.53, 27.65, 28.22, 31.51, 33.45, 35.37, 35.43, 35.89, 37.60, 40.28, 40.52, 42.75, 43.74, 52.33, 52.93, 56.00, 56.60, 110.17, 111.28, 118.59, 119.72, 122.25, 122.67, 127.76, 136.11, 172.33 (C=O), 173.33 (C=O). MS (ESI) calc for C36H52N2O3: 560.40; found: 561.7 [M+H]+, 583.6 [M+Na]+, 599.5 [M+K]+.
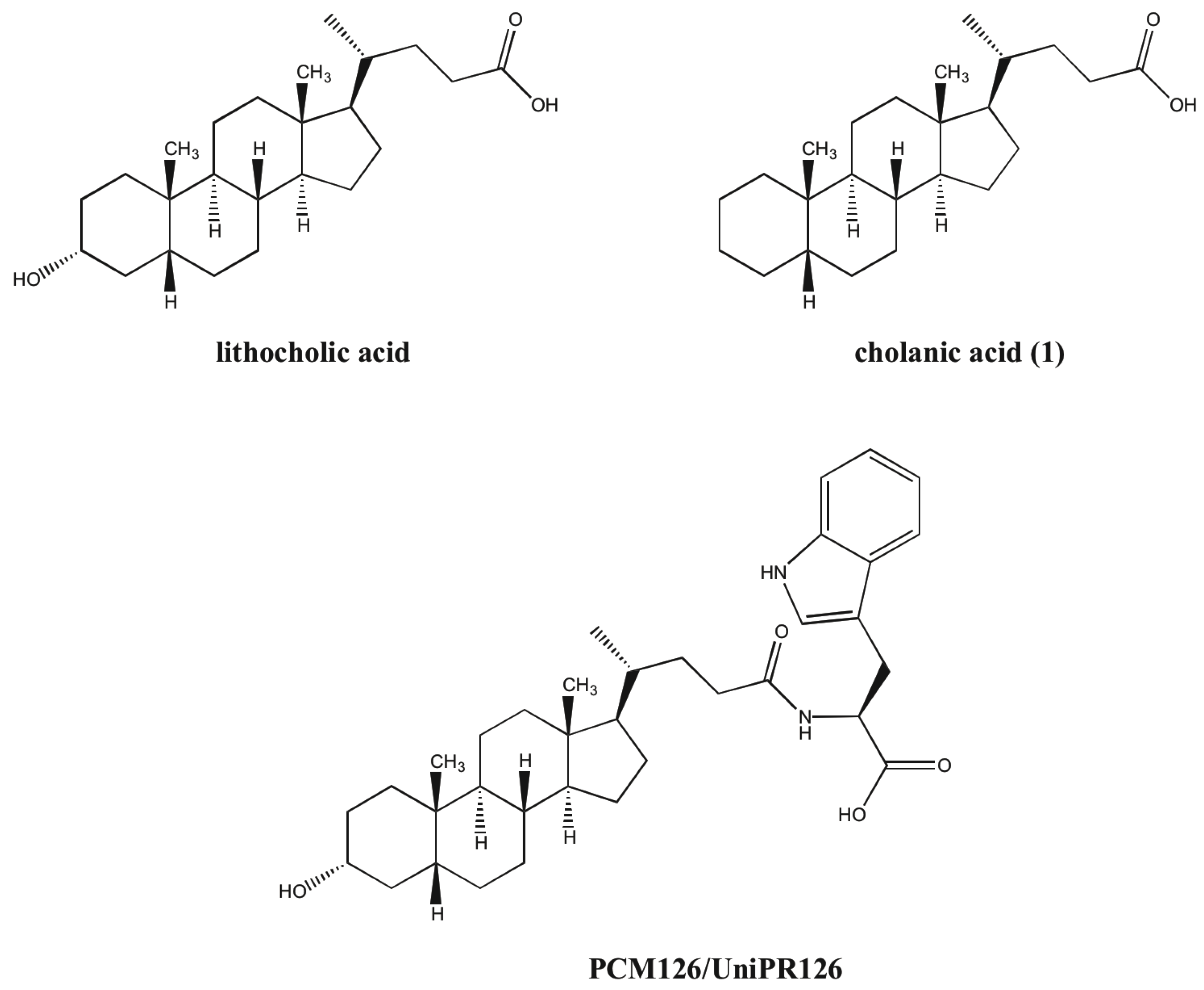
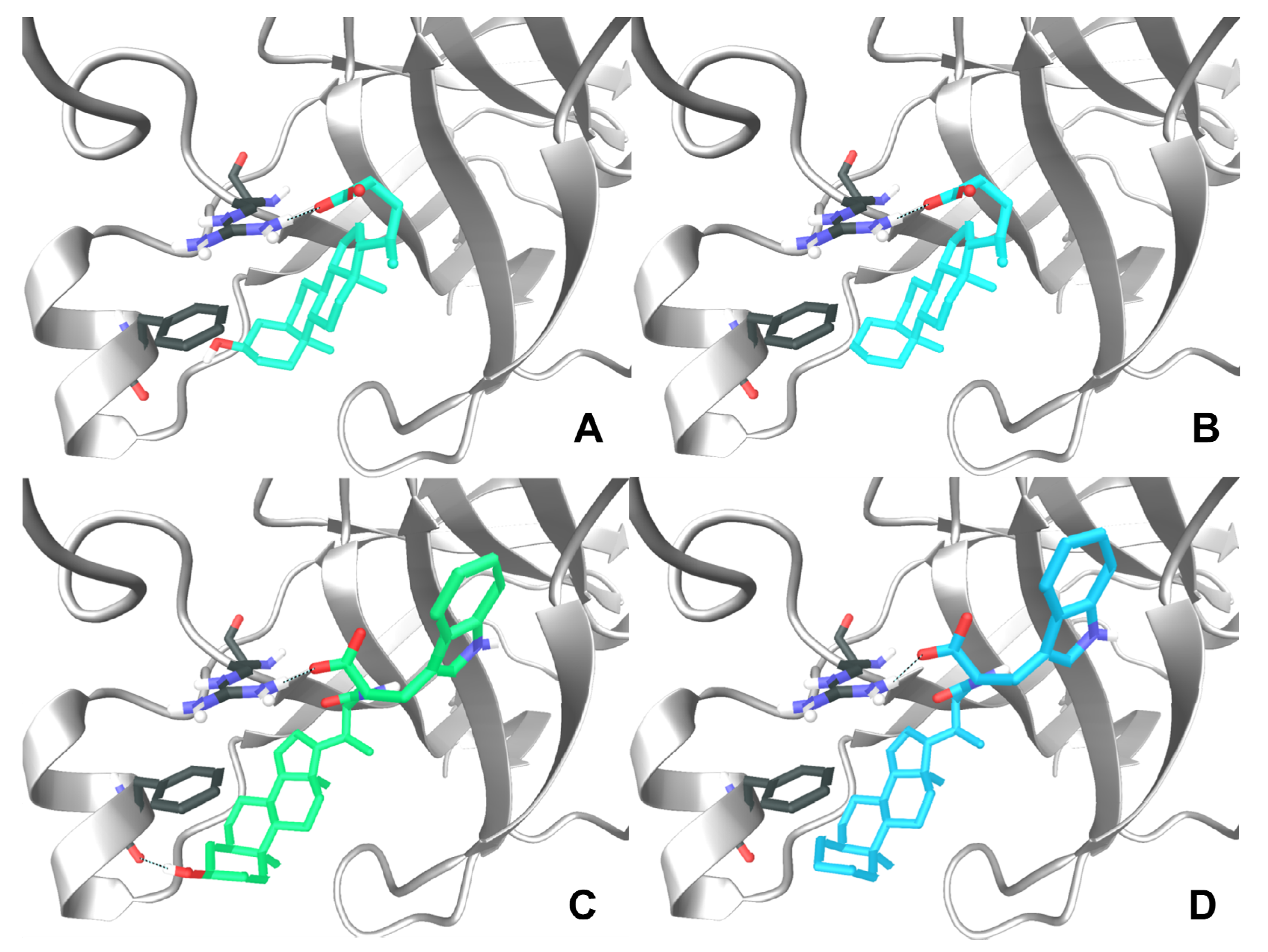
N-(5β-cholan-24-oyl)-L-tryptophan (8). Compound 8 was synthesized following the procedure described for 2 using 8a. The crude product was re-crystallized from ethanol-water to give 8. Yield: 80%. Mp: 98–101 °C. 1H-NMR (DMSO-d6) δ = 0.57 (s, 3H, CH3), 0.80–0.88 (m, 7H), 0.97–1.37 (m, 19H), 1.48–1.57 (m, 2H), 1.68–1.92 (m, 7H), 1.98–2.06 (m, 1H), 2.97 (dd, J = 14.4, 5.6 Hz, 1H, CHCHH), 3.19 (dd, J = 14.4, 5.2 Hz, 1H, CHCHH), 4.12 (q, J = 5.6 Hz, 1H, CHCH2), 6.87 (t, J = 7.6 Hz, 1H, Ar), 6.97 (t, J = 7.2 Hz, 1H, Ar), 7.02 (d, J = 1.6 Hz, 1H,, Ar), 7.17 (d, J = 7.2 Hz, 1H, NH), 7.26 (d, J = 8.0 Hz, 1H, Ar), 7.46 (d, J = 8.0 Hz, 1H, Ar). 13C-NMR (DMSO-d6) δ = 12.33, 18.77, 20.90, 21.28, 24.33, 24.51, 26.62, 26.96, 27.17, 27.52, 28.05, 28.15, 31.91, 33.19, 35.40, 35.46, 35.86, 37.61, 42.70, 43.58, 55.45, 56.07, 56.48, 111.42, 112.13, 118.17, 119.06, 120.72, 123.64, 128.74, 136.32, 171.70 (C=O), 174.85 (C=O). MS (ESI) calc for C35H50N2O3: 546.38; found: 545.7 [M−H]−. Anal. calc for C35H50N2O3 • 0.2 H2O: C, 76.37; H, 9.23; N, 5.09; found: C, 76.07; H, 9.61; N, 4.78.
d-Tryptophan methyl ester hydrochloride (
9b)
. Compound
9b was synthesized following a described procedure [
25] starting from
d-tryptophan.
Methyl N-(5β-cholan-24-oyl)-D-tryptophanate (9a). Compound 9a was synthesized following the procedure described for compound 3 starting from D-tryptophan methyl ester hydrochloride 9b and purified by flash chromatography [SiO2, CH2Cl2:C2H5OH 98:2], to furnish compound 9a as a pale-yellow oil. Yield: 81%. 1H-NMR (DMSO-d6) δ = 0.61 (s, 3H, CH3), 0.85–0.91 (m, 7H), 1.02–1.38 (m, 20H), 1.53–1.58 (m, 1H), 1.72–2.04 (m, 7H), 2.15–2.27 (m, 1H), 3.31 (d, J = 5.3 Hz, 2H, CHCH2), 3.68 (s, 3H, OCH3), 4.96 (dt, J = 7.8, 5.3 Hz, 1H, CHCH2), 6.95 (d, J = 1.9 Hz, 1H, Ar), 7.10 (td, J = 6.9, 1.2 Hz 1H, Ar), 7.18 (td, J = 6.9, 1.2 Hz 1H, Ar), 7.34 (d, J = 7.9 Hz, 1H, Ar), 7.52 (d, J = 7.7 Hz, 1H, Ar), 8.43 (bs, 1H, NH). 13C-NMR (DMSO-d6) δ = 12.05, 18.40, 20.90, 21.42, 24.30, 24.35, 26.63, 27.11, 27.33, 27.60, 27.74, 28.27, 31.57, 33.50, 35.43, 35.52, 35.96, 37.67, 40.35, 40.58, 42.82, 43.81, 52.39, 53.02, 56.06, 56.65, 110.06, 111.41, 118.59, 119.72, 122.25, 122.83, 127.80, 136.23, 172.63 (C=O), 173.44 (C=O). MS (ESI) calc for C36H52N2O3: 560.40; found: 561.6 [M+H]+, 583.4 [M+Na]+, 599.5 [M+K]+.
N-(5β-cholan-24-oyl)-D-tryptophan (9). Compound 9 was synthesized following the procedure described for 2 using compound 9a. The crude product was re-crystallized from ethanol-water to give 9. Yield: 77%. Mp: 141–145 °C. 1H-NMR (DMSO-d6) δ = 0.56 (s, 3H, CH3), 0.82–0.88 (m, 7H), 1.00–1.34 (m, 19H), 1.47–1.62 (m, 2H), 1.69–1.98 (m, 7H), 2.05–2.09 (m, 1H), 2.97 (dd, J = 14.4, 8.4 Hz, 1H, CHCHH), 3.15 (dd, J = 14.4, 4.6 Hz, 1H, CHCHH), 4.38–4.43 (m, 1H, CHCH2), 6.95 (t, J = 7.2 Hz, 1H, Ar), 7.04 (t, J = 7.4 Hz, 1H, Ar), 7.10 (s, 1H, Ar), 7.31 (d, J = 8.0 Hz, 1H, Ar), 7.51 (d, J = 7.6 Hz, 1H, Ar), 7.94 (bd, J = 7.6 Hz, 1H, NH), 10.8 (s, 1H, COOH). 13C-NMR (DMSO-d6) δ = 11.81, 18.23, 20.42, 20.81, 23.84, 24.05, 26.15, 26.48, 26.69, 27.04, 27.18, 27.64, 31.37, 32.17, 34.84, 34.93, 35.39, 37.13, 42.23, 43.09, 53.18, 55.65, 56.03, 110.25, 111.27, 118.21, 120.77, 123.41, 127.36, 136.04, 172.36, 173.80. MS (ESI) calc for C35H50N2O3: 546.38; found: 545.5 [M−H]−. Anal. calc for C35H50N2O3 • 0.5 H2O: C, 75.63; H, 9.25; N, 5.04; found: C, 75.58; H, 9.46; N, 4.58.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

