Peptide Conjugation via CuAAC ‘Click’ Chemistry

Abstract
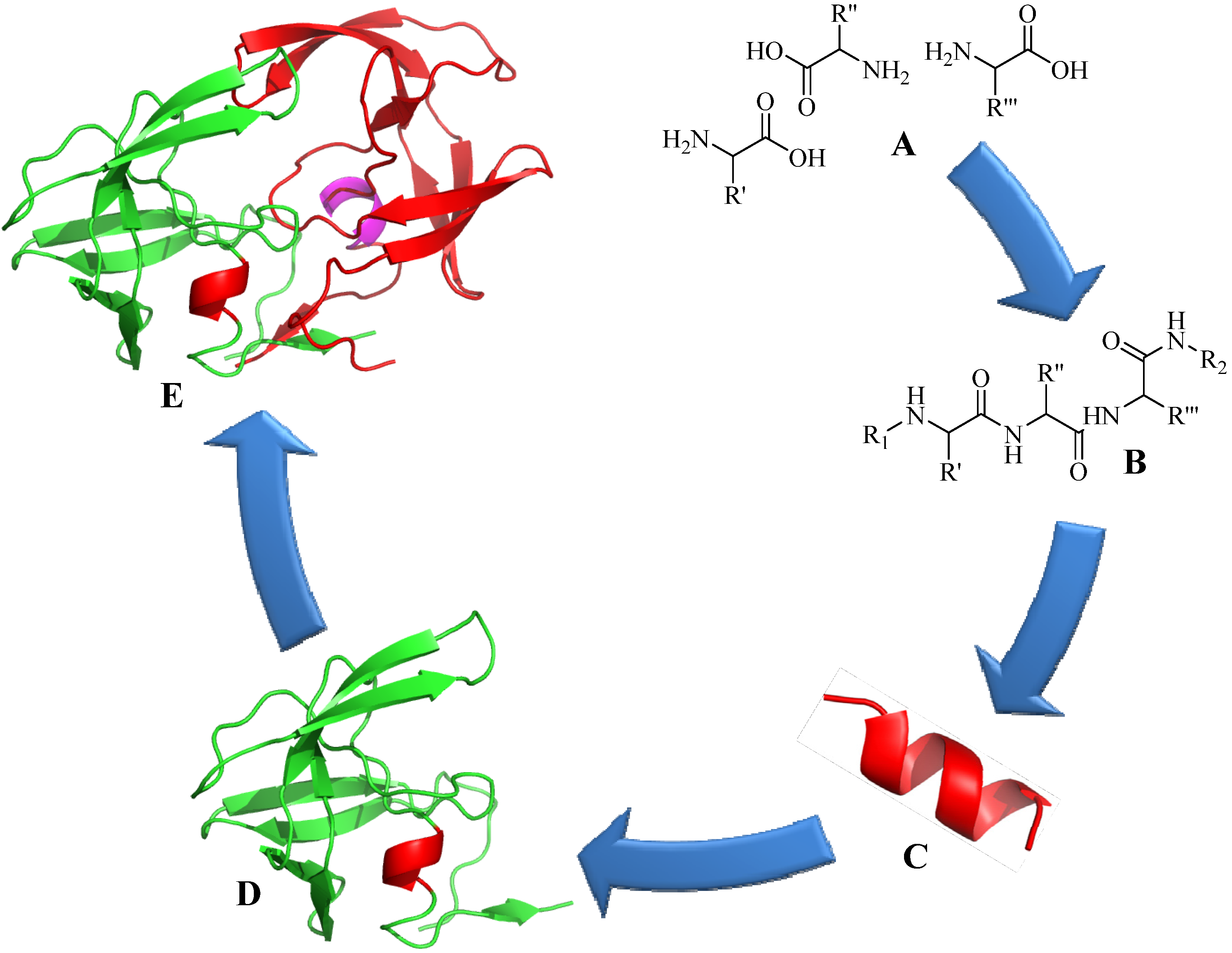
:1. Introduction
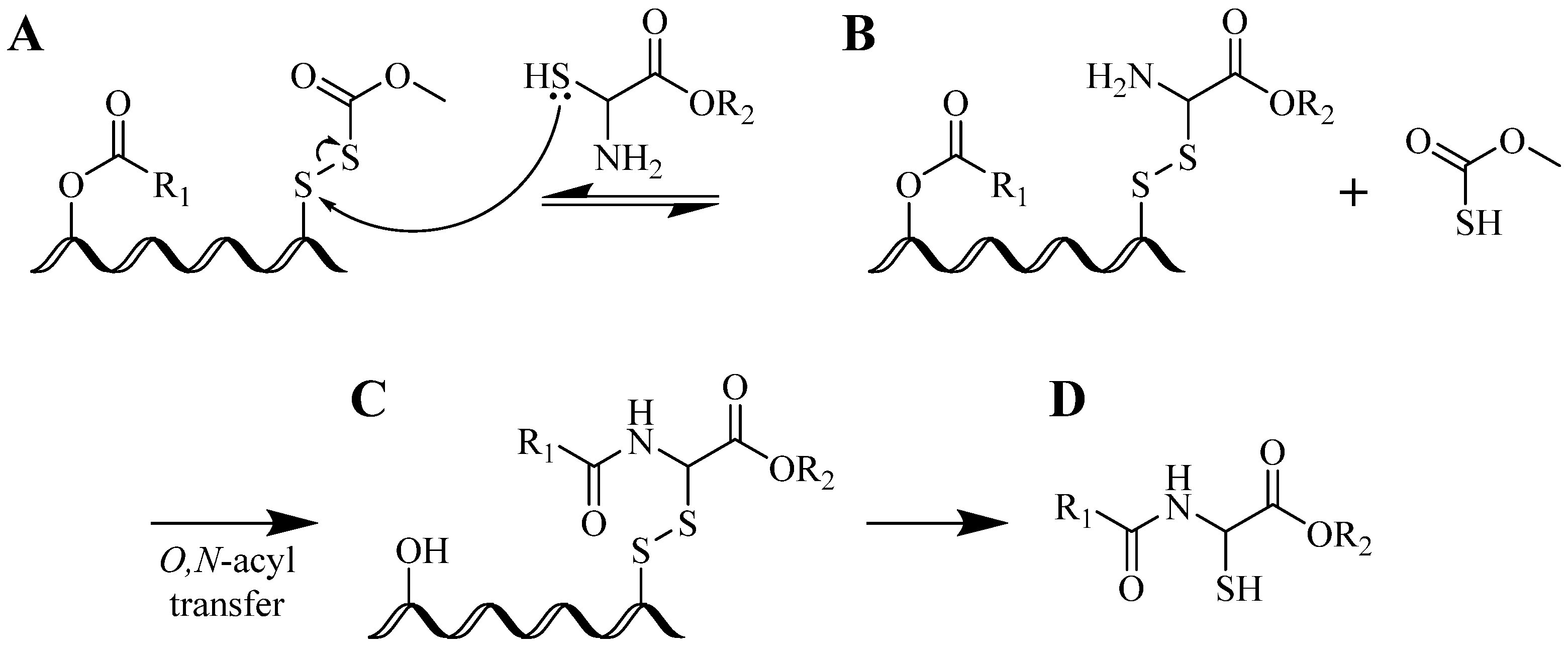

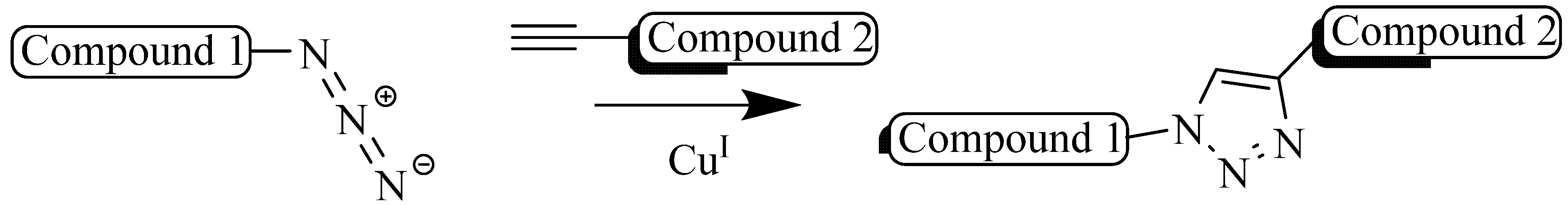
2. Copper (I) Catalyzed Alkyne-azide 1,3-Dipolarcycloaddition (CuAAC)
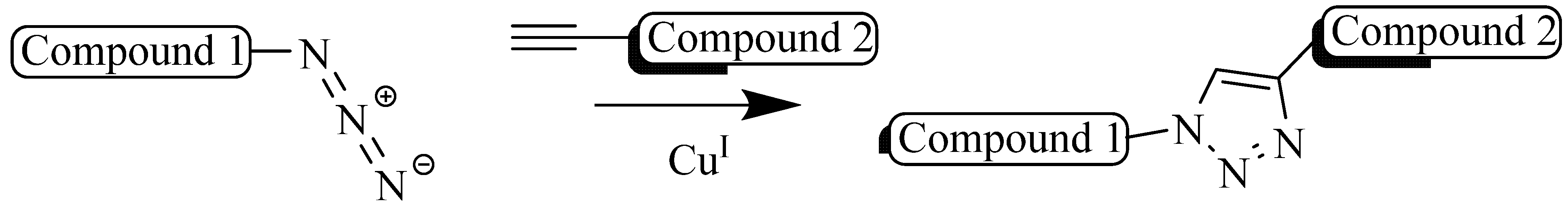
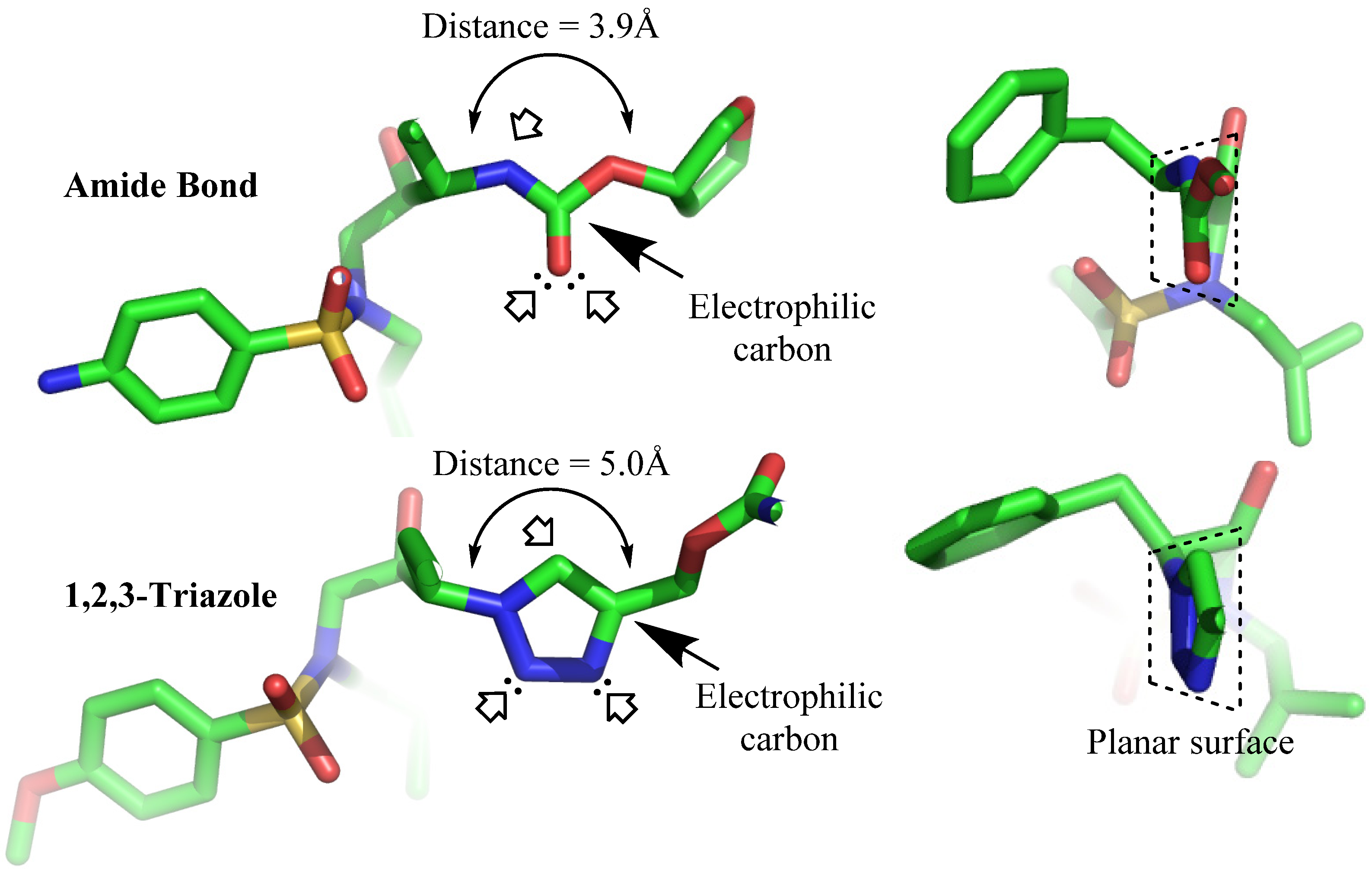
3. Structural Studies of Amide Bond and 1,4-Disubstituted Triazole
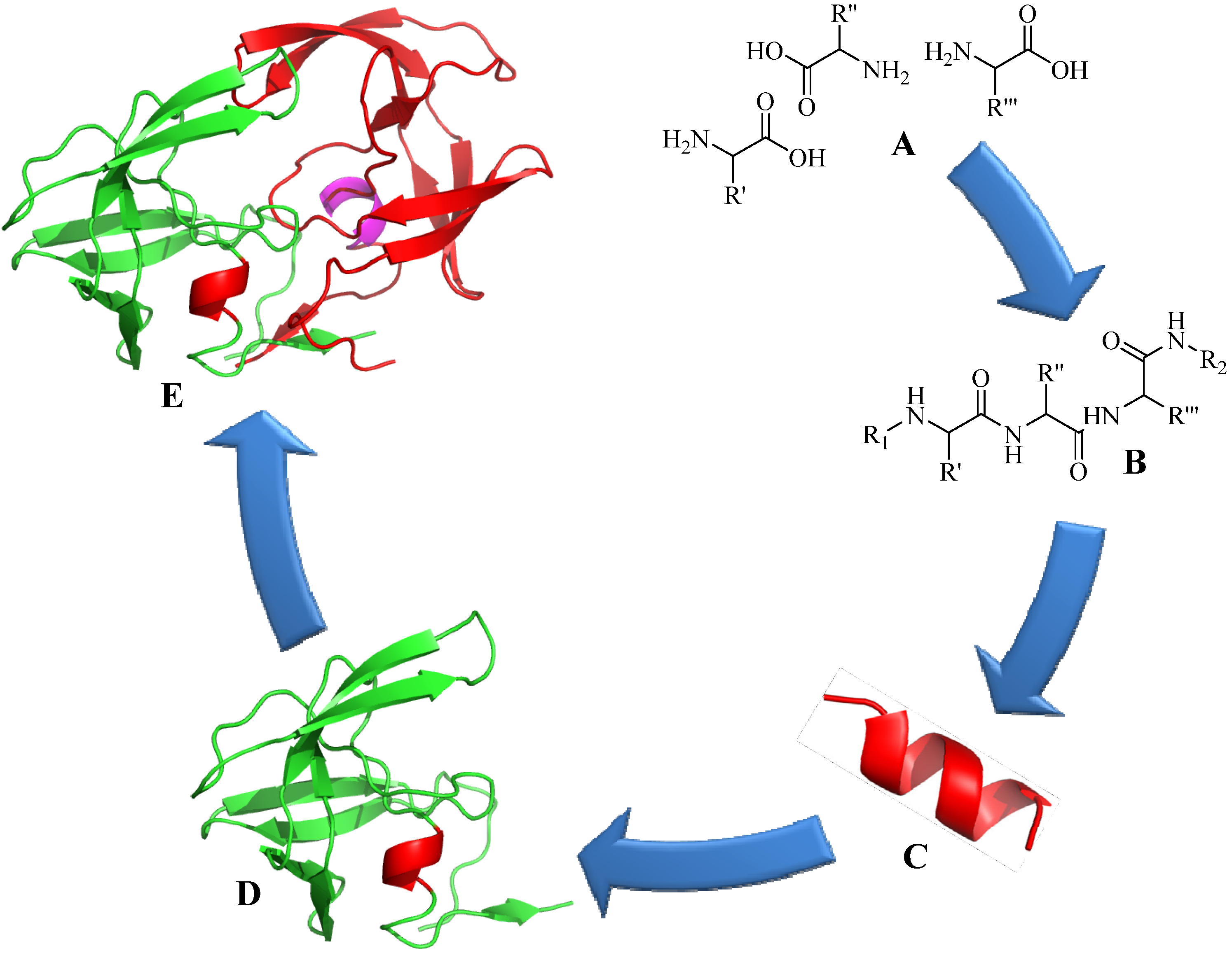


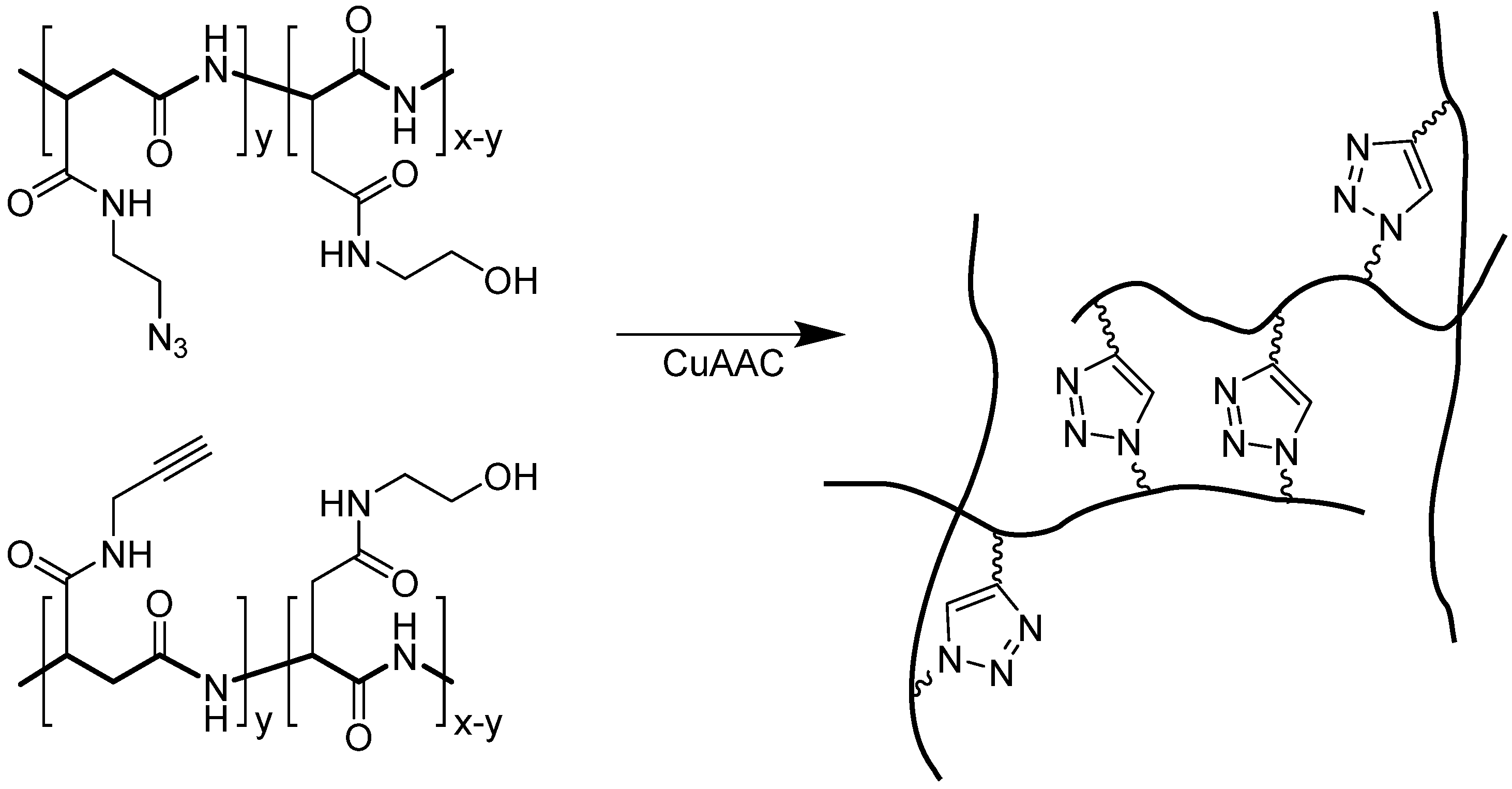
4. Application of CuAAC in Peptide Modifications
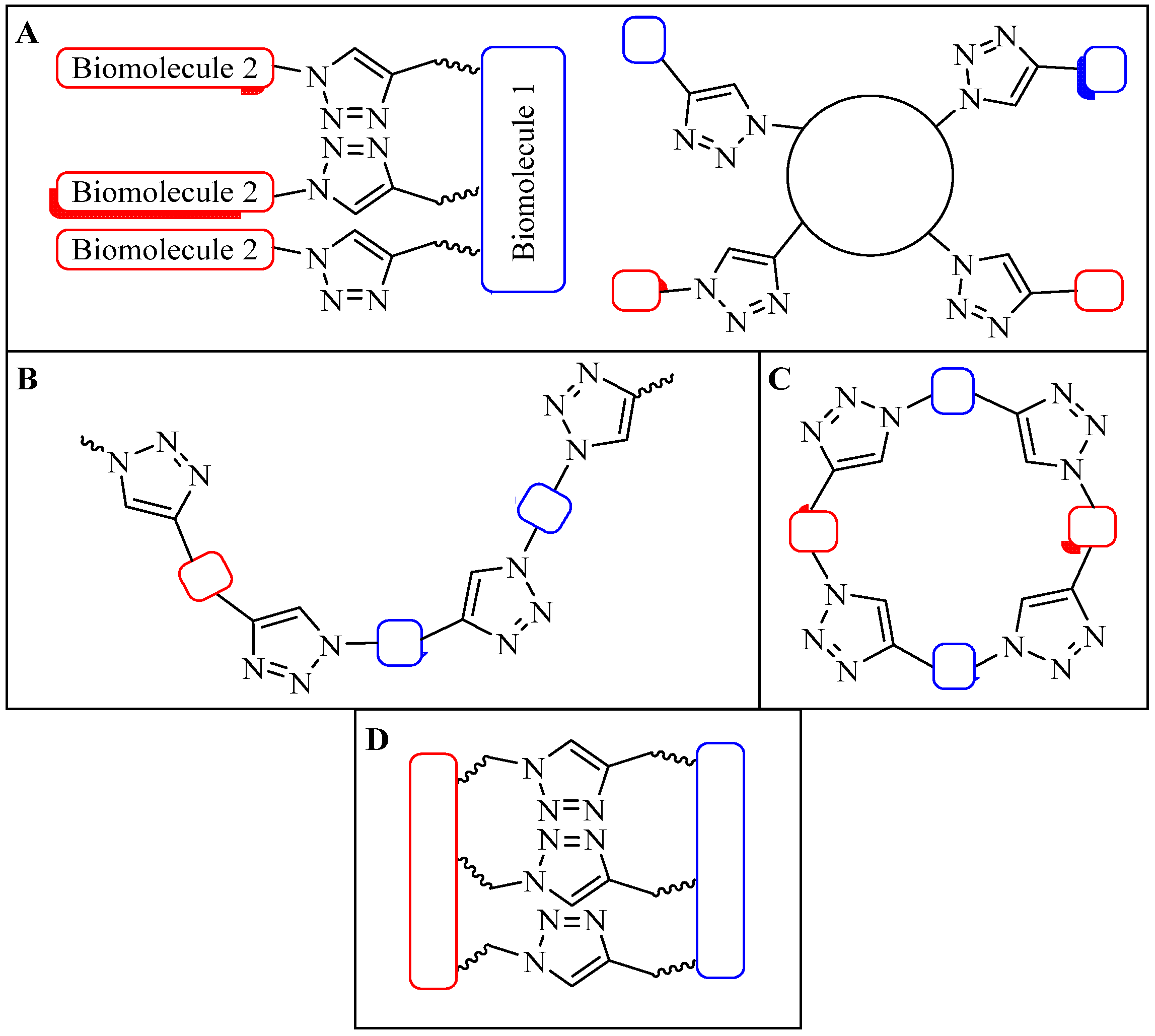
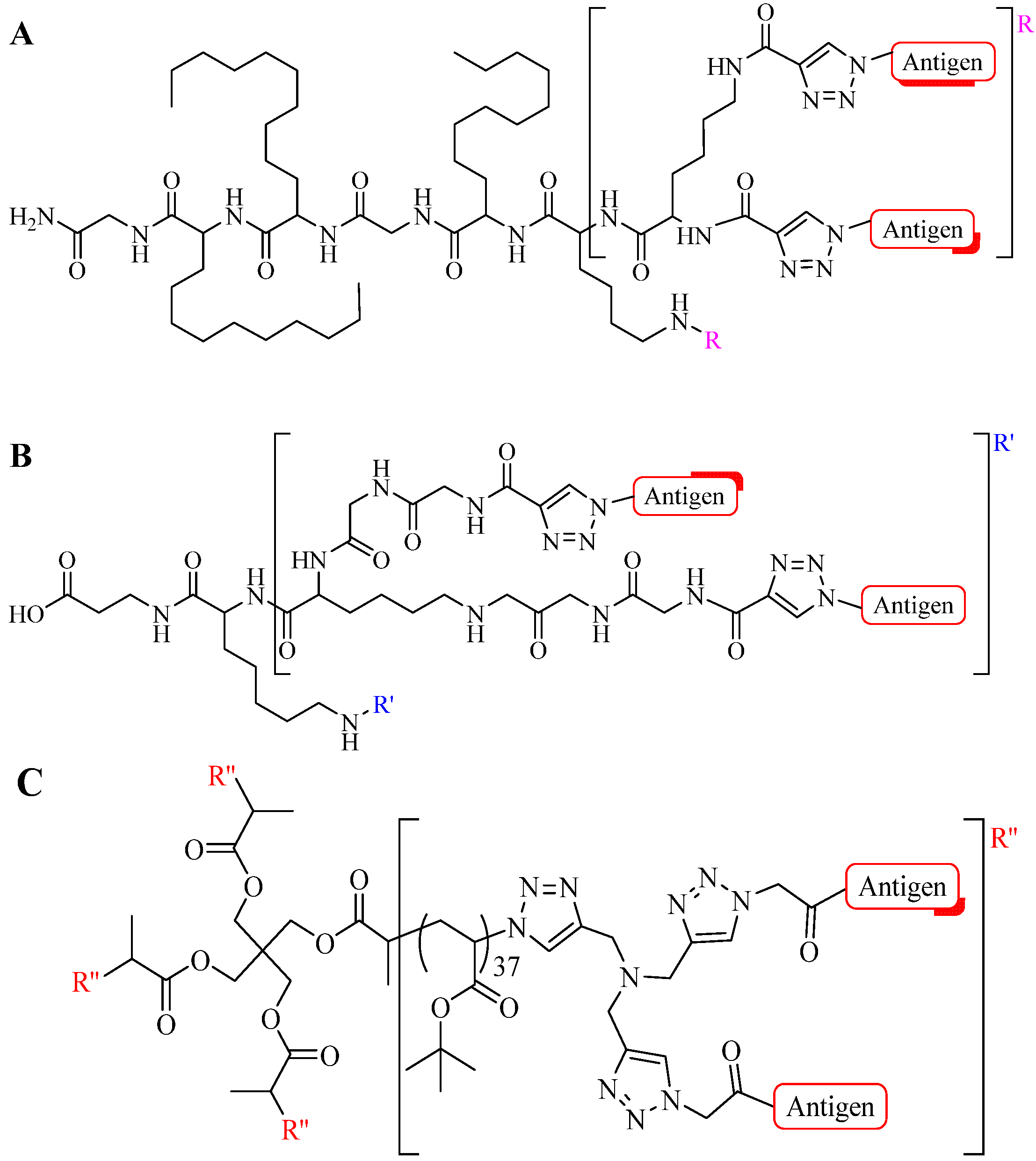
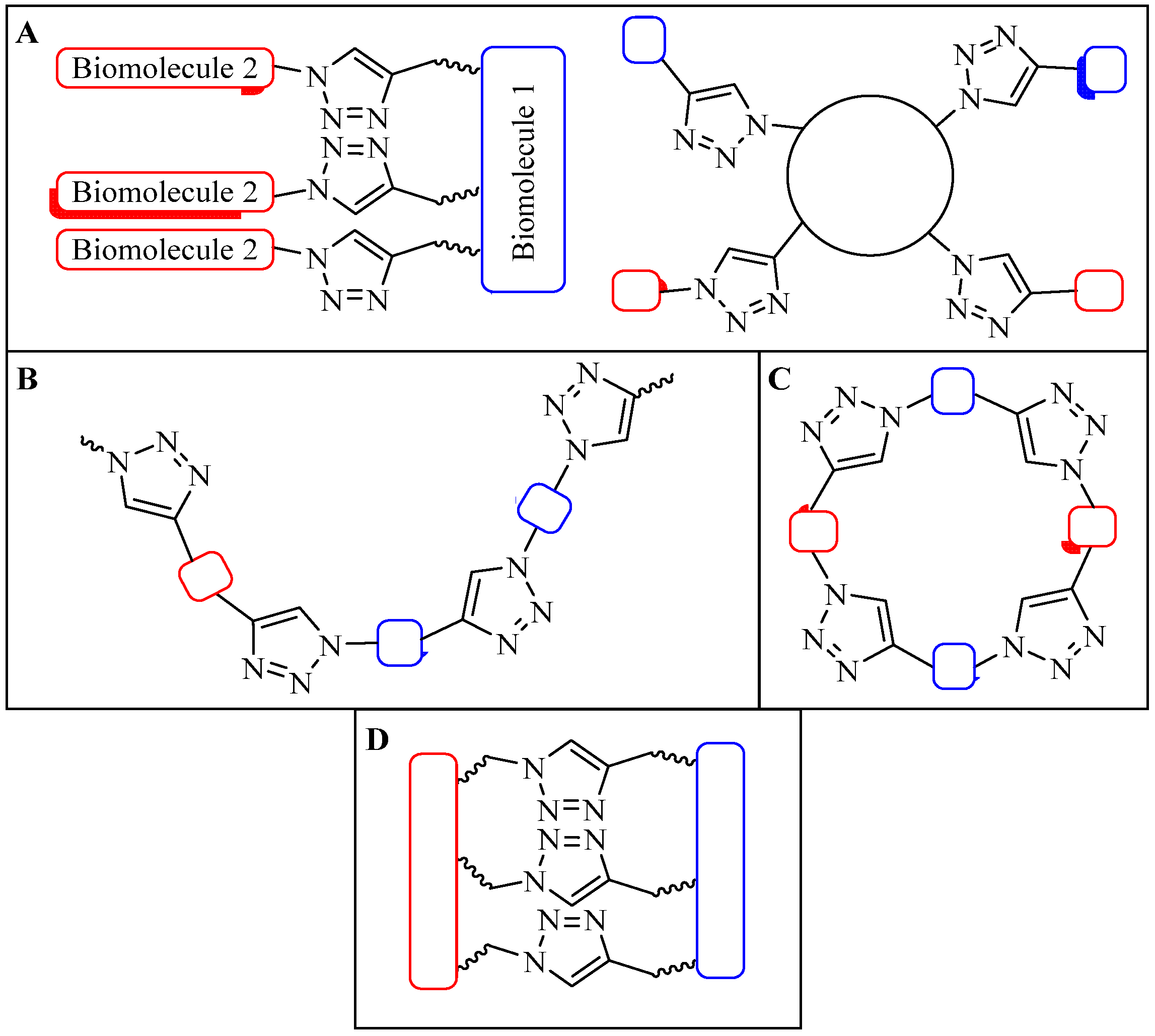
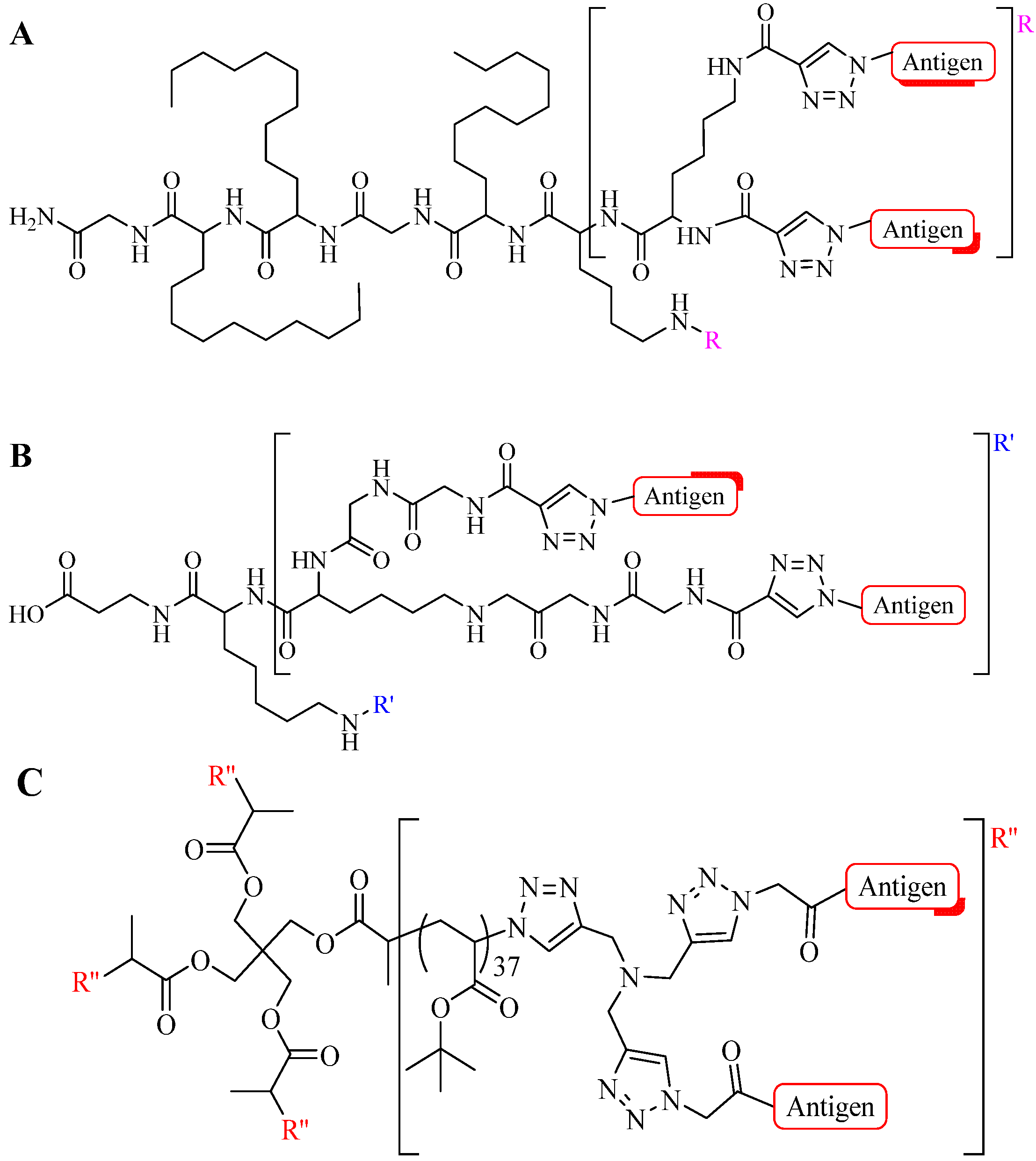
4.1. Intermolecular Linker
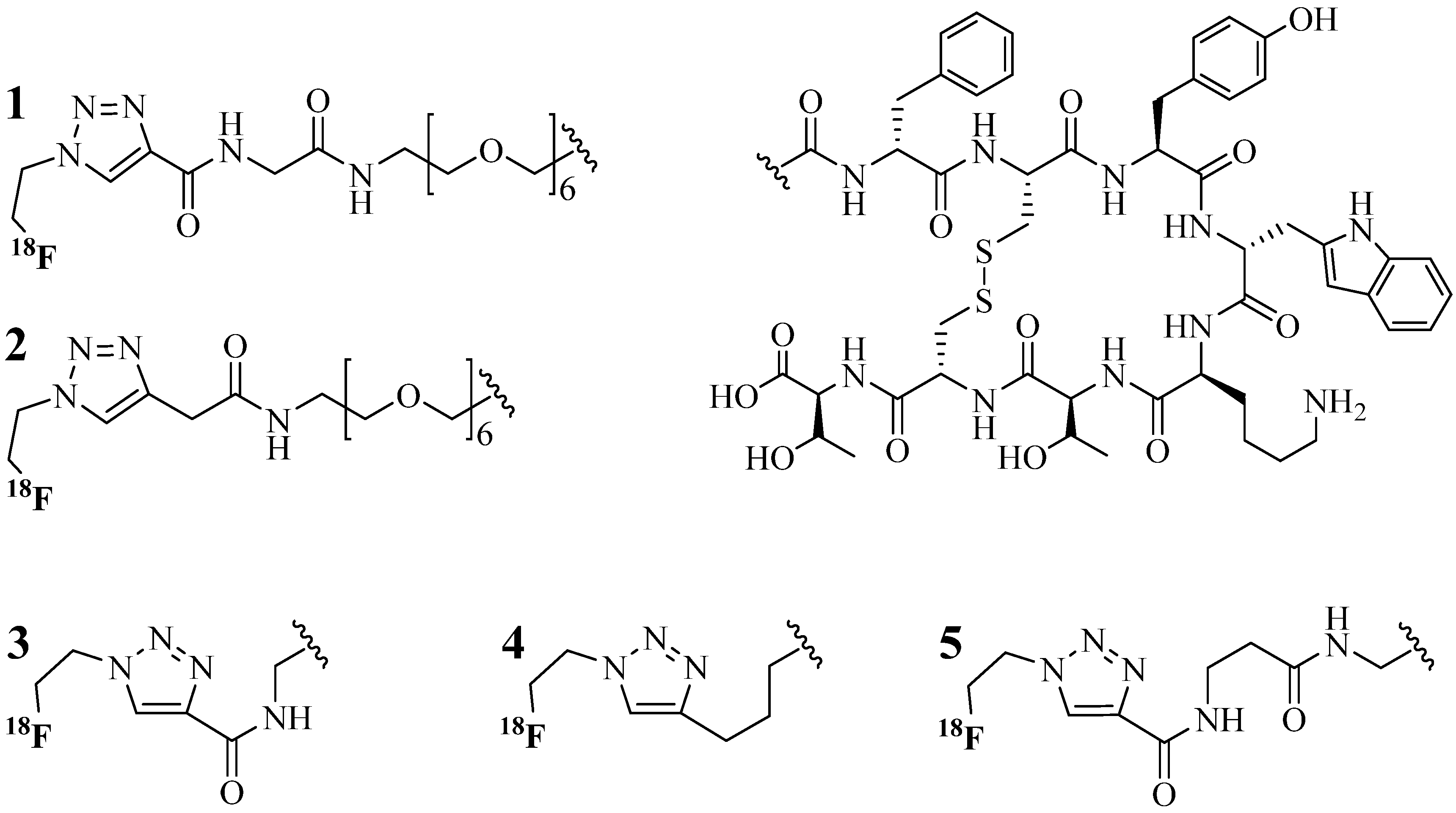
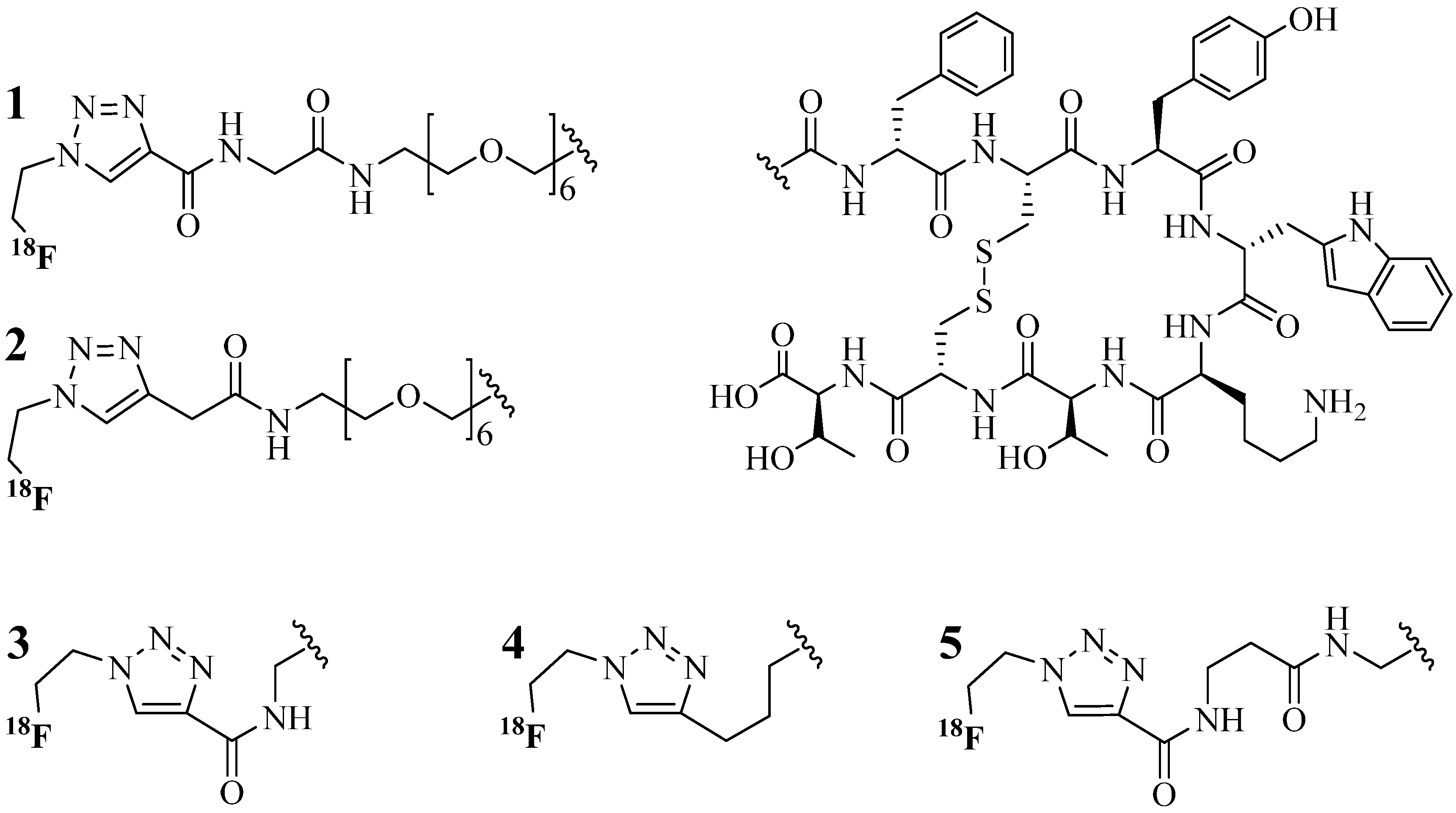
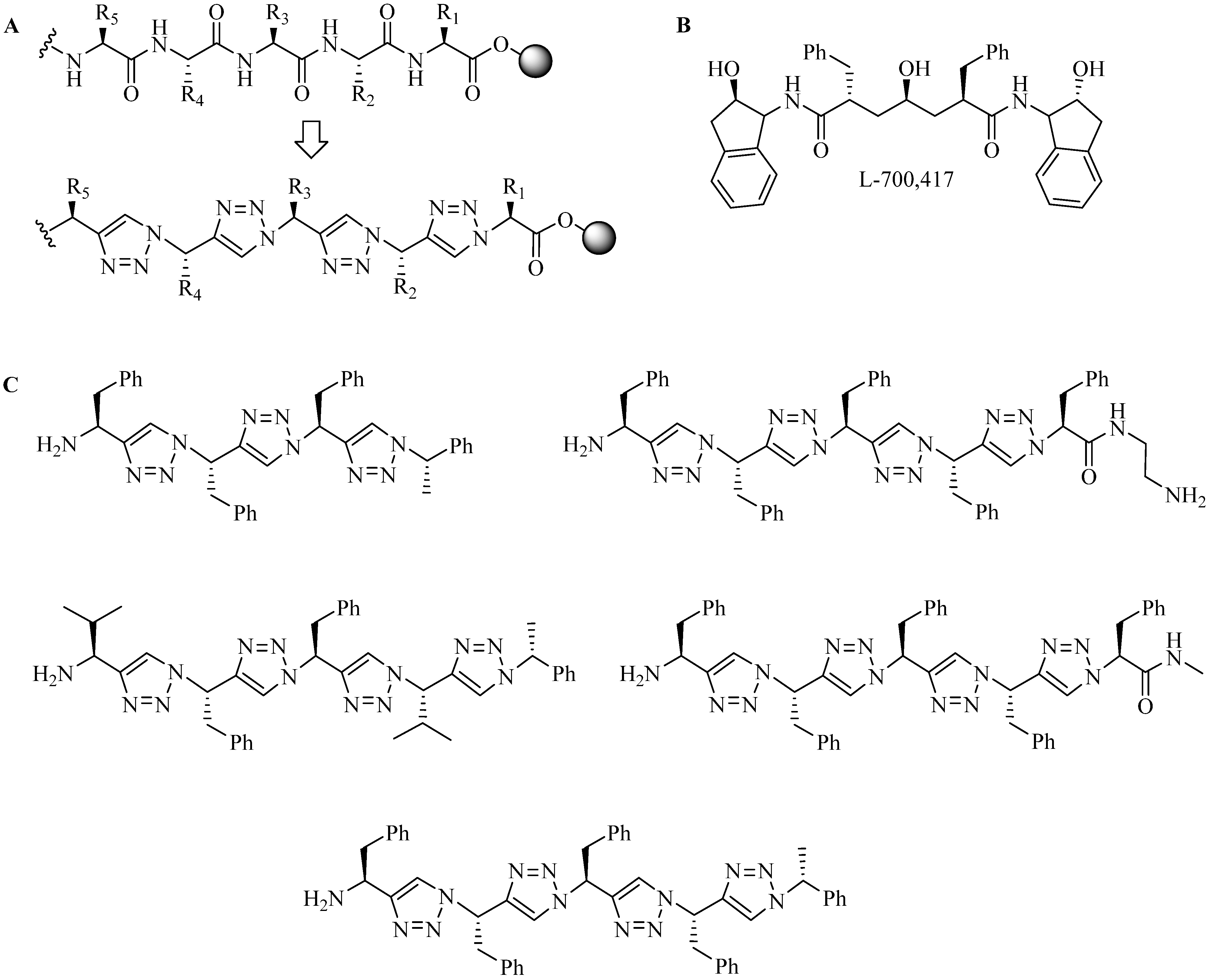
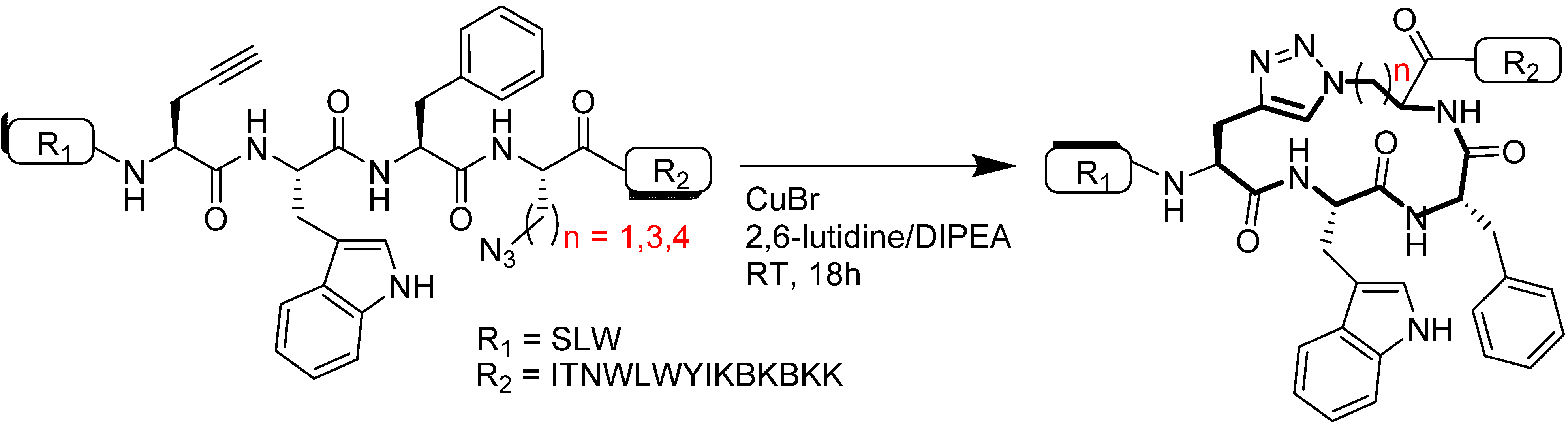
4.1.1. Single-Site Intermolecular Linker
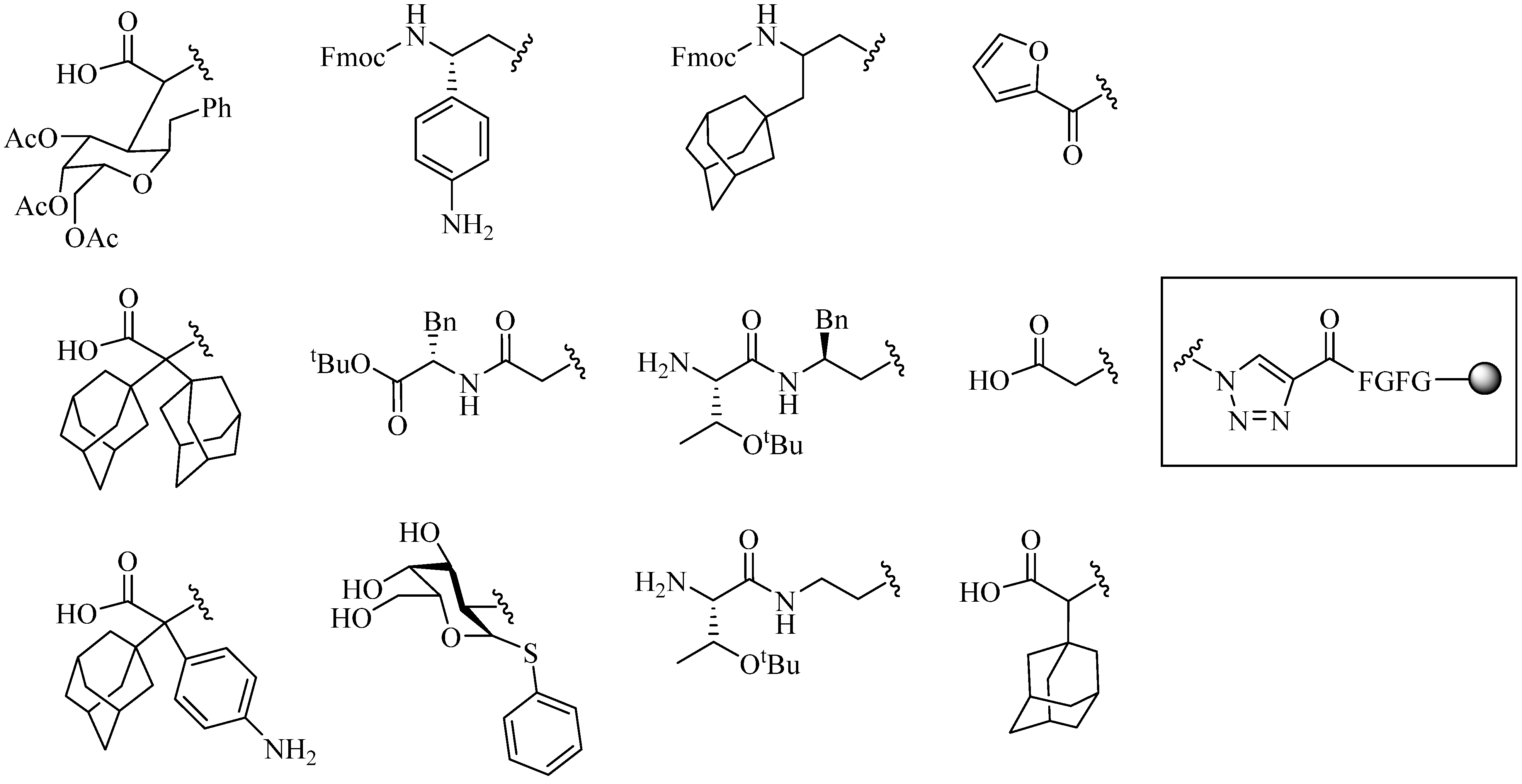
- (1)
- Short half-life (low stability) of 18F—thus the synthesis needed to be completed within a short time;
- (2)
- Consideration of the safety of the operator working on the high gamma energy 18F;
- (3)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R′ | R1 | R2 | R3 | R4 | R5 | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | -CF3 | -CH3 | -CH3 | -CH3 |  |  | 56 |
| 2 | -CF3 |  | -H | -H |  |  | 40 |
| 3 | -CF3 | | -H | -H | Bn | -CF3 | 45 |
| 4 | -CF3 | -H | -CH3 | -CH3 | Bn | -CF3 | 45 |
| 5 | -C(O)OCH3 | i-Pr | -H | -H | Bn | -CF3 | 65 |
| 6 | -C(O)OCH3 | i-Pr | -H | -H | Bn | | 55 |
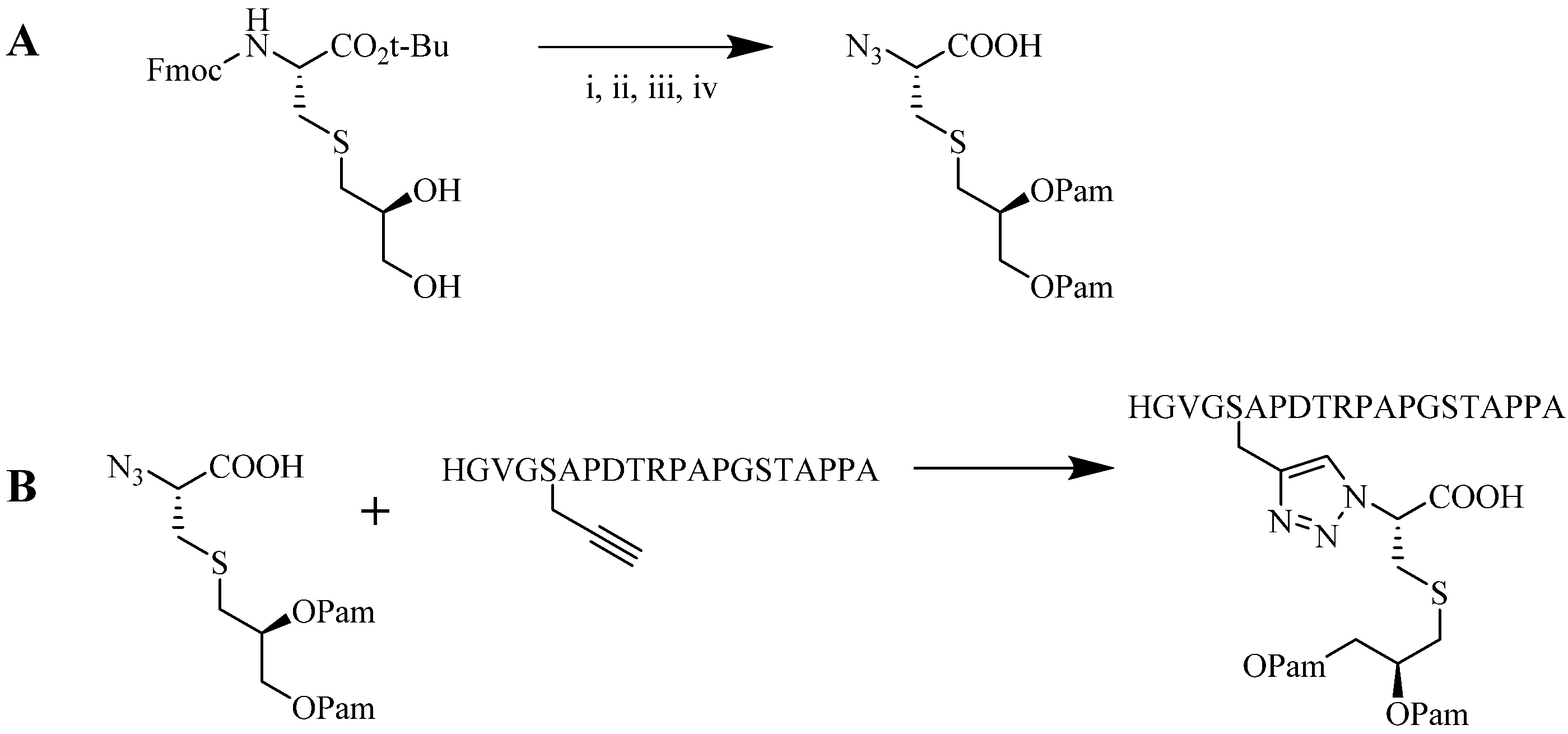
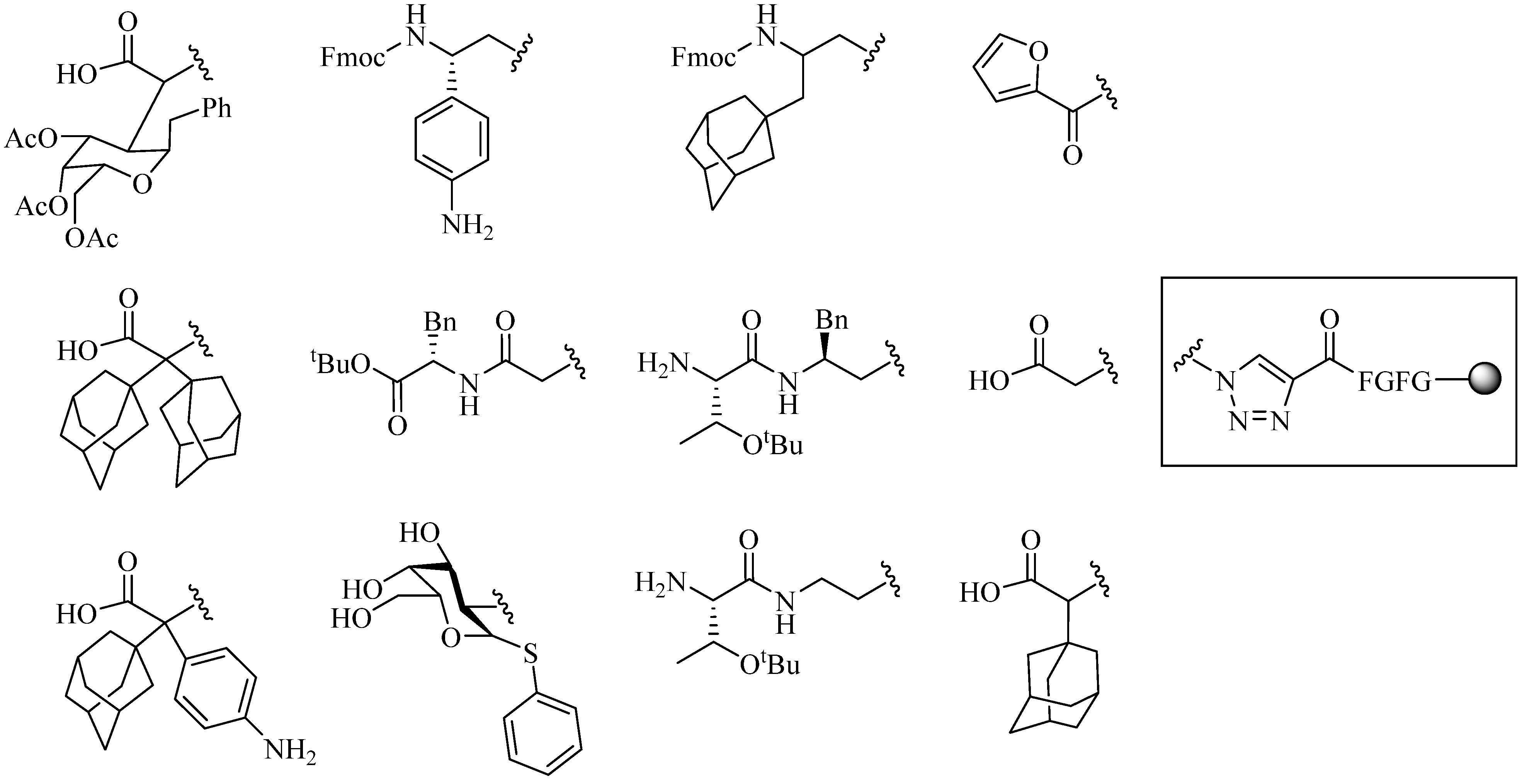
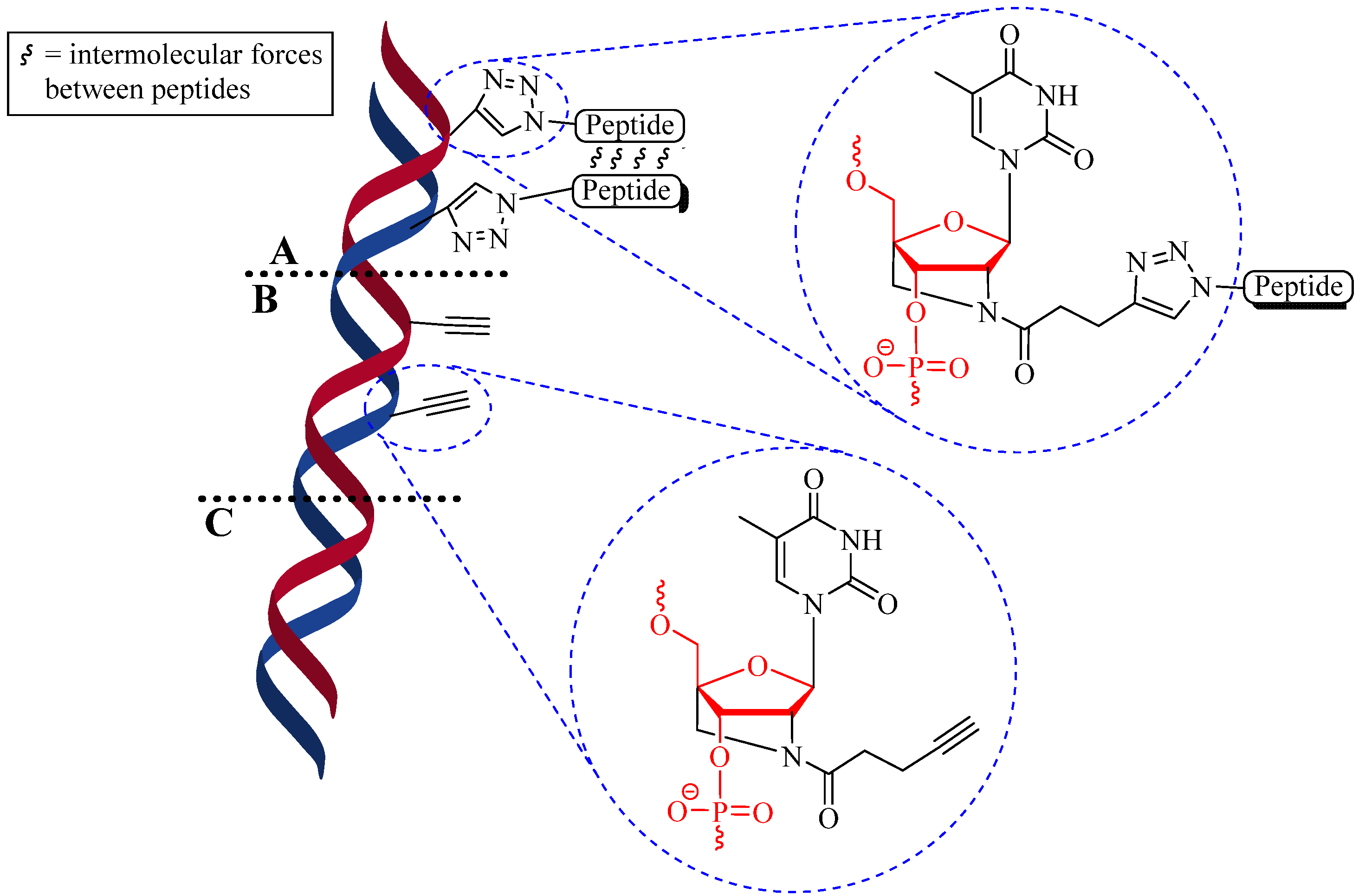
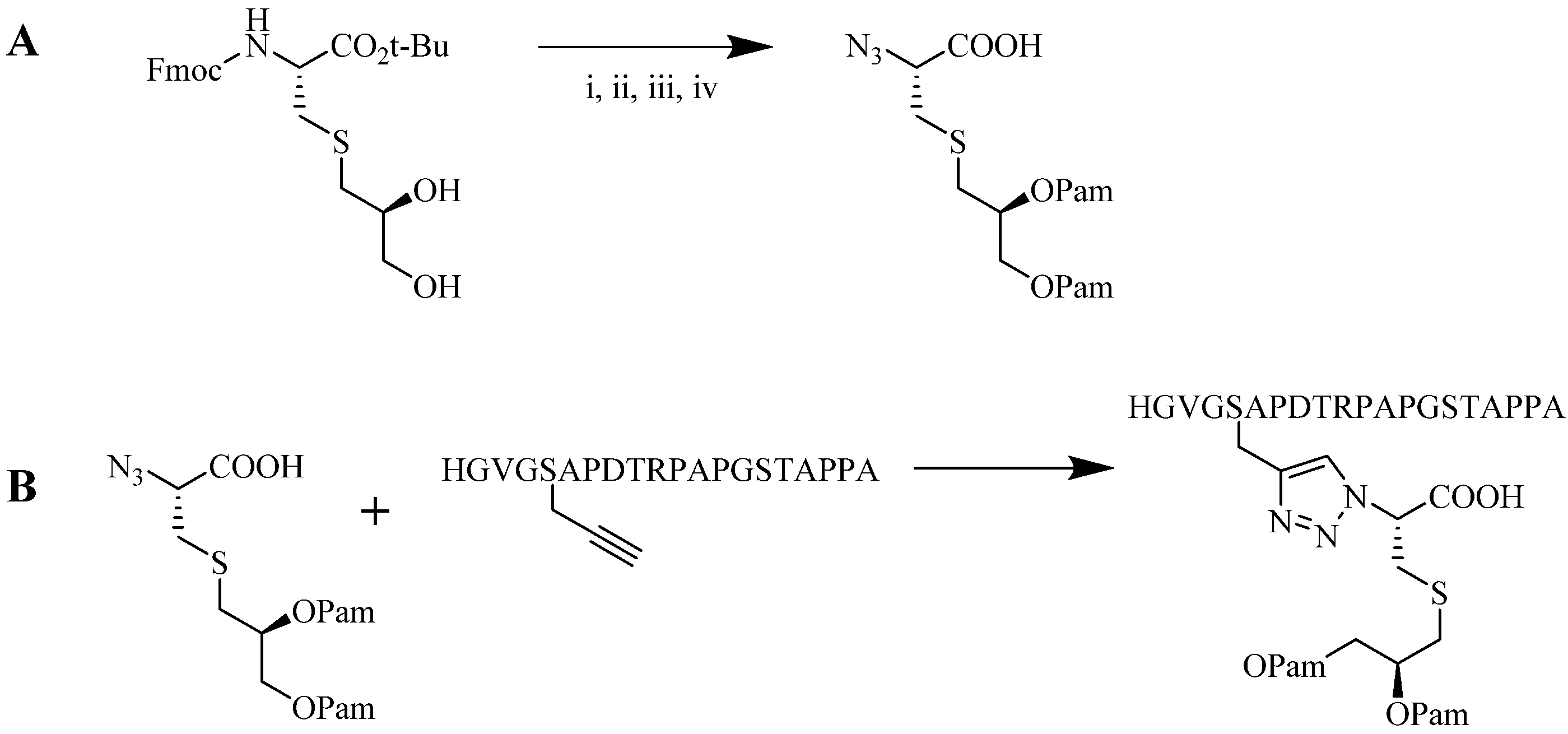
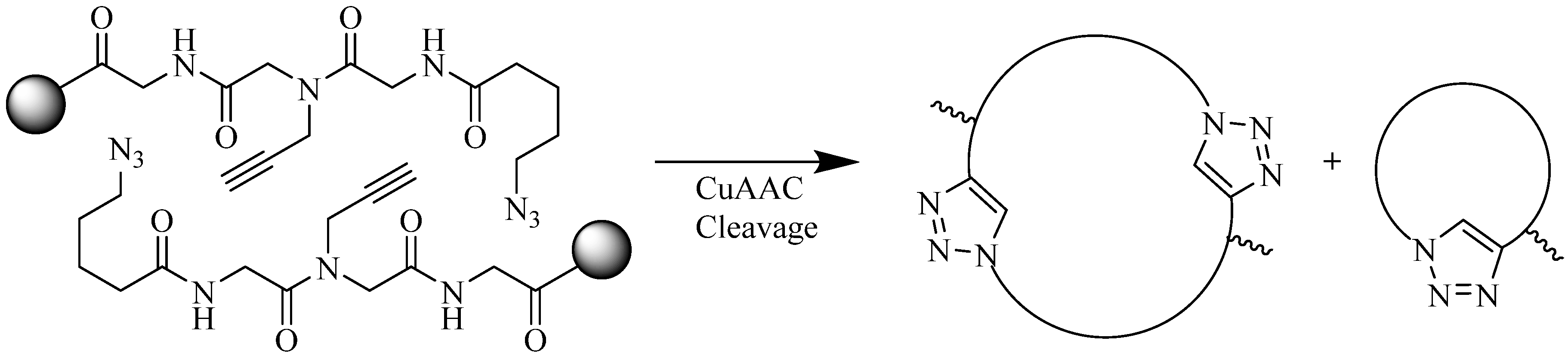
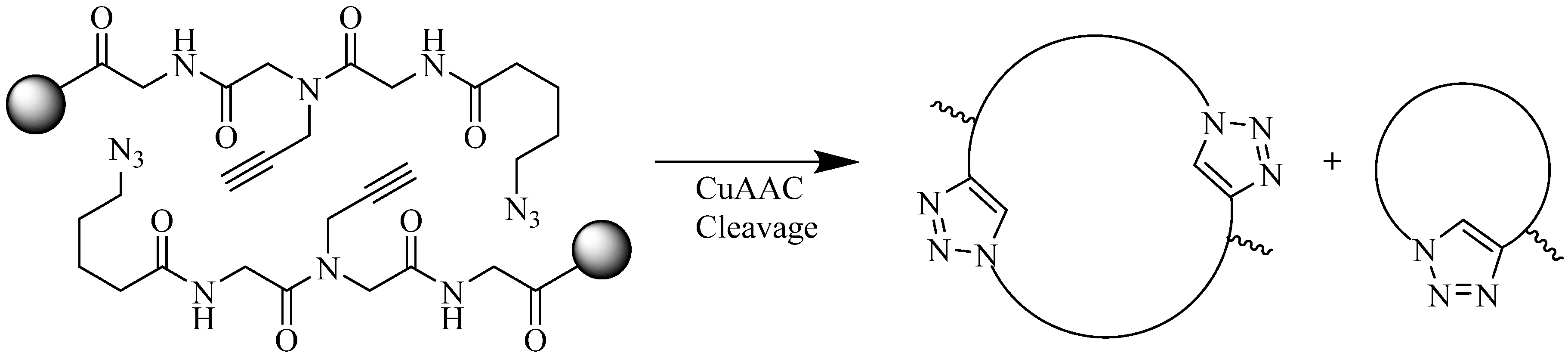
4.1.2. Multiple-Sites Intermolecular Conjugates


- (a)
- The distance between the active groups (azide or alkyne) and the resin—as the distance increased, the yield of bicyclic product decreased;
- (b)
- The distance between the active groups—a minimum of six amino acids promotes cyclodimerization;
- (c)
- Solvent composition ratio—affects resin swelling and interstrand hydrogen bonding thus affecting the dimerization, and;
- (d)
- The structural homolog of the peptide (α, β, γ etc.)—α and β homologs readily form the cyclodimer while γ homologs do not [89].
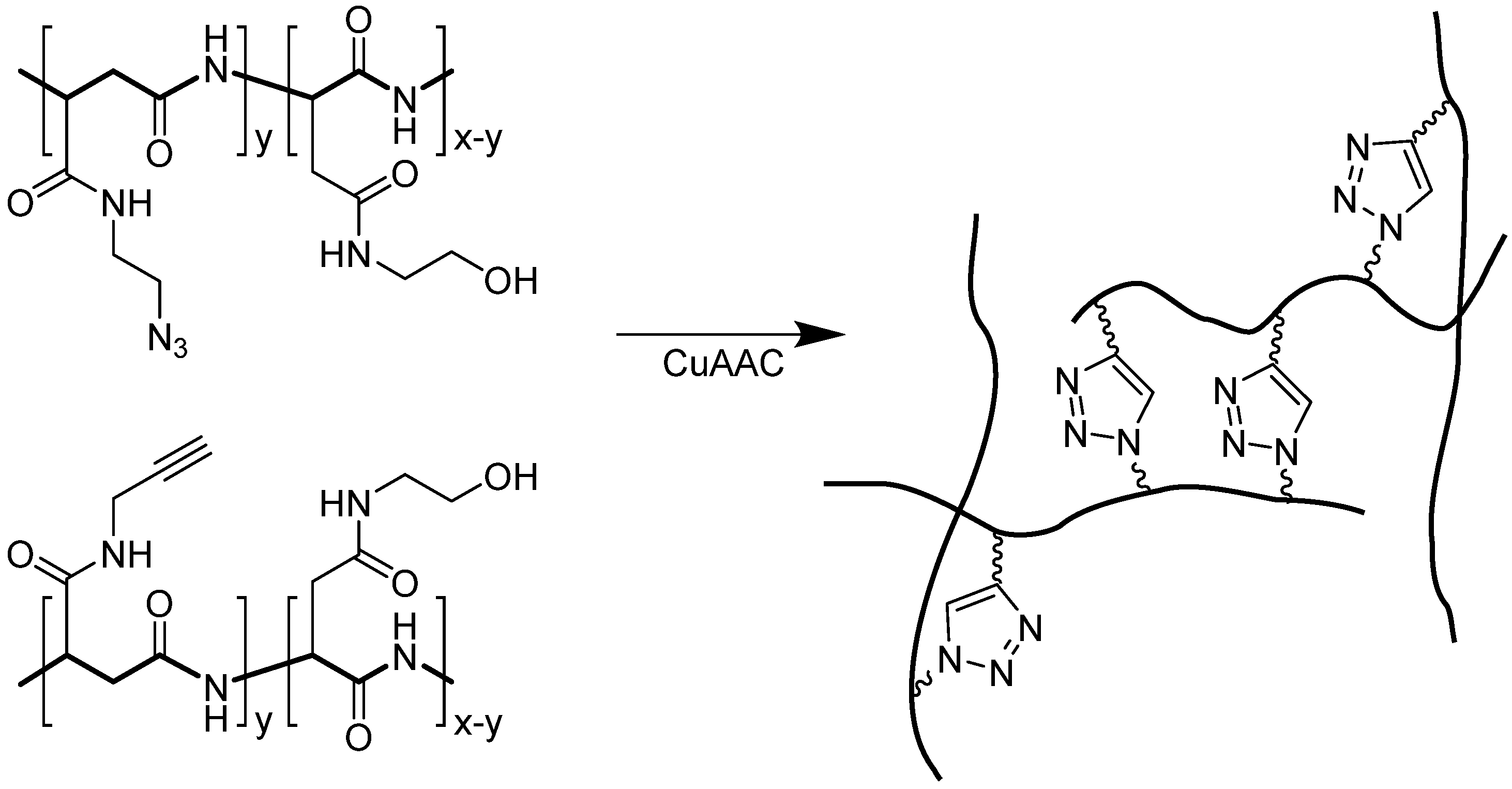
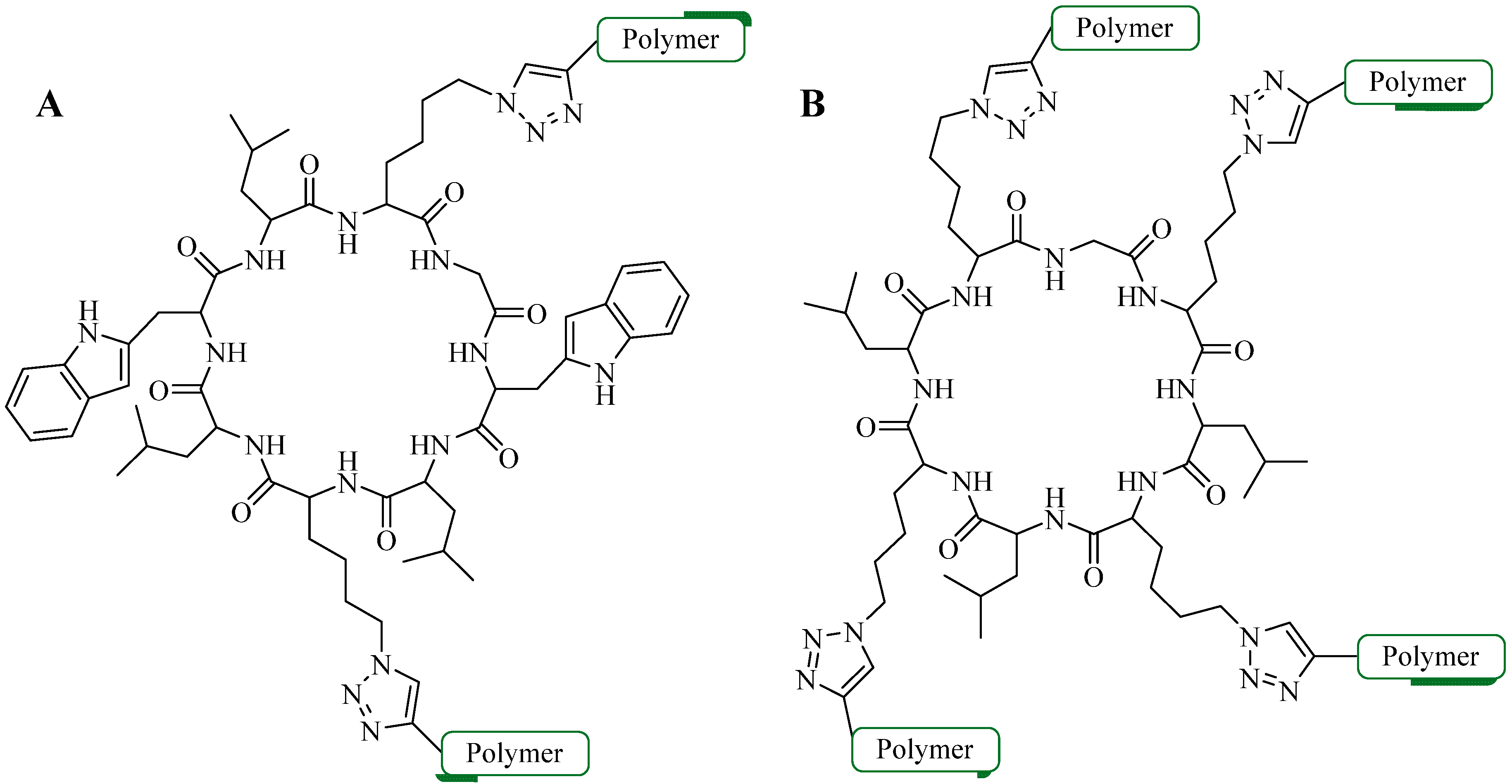
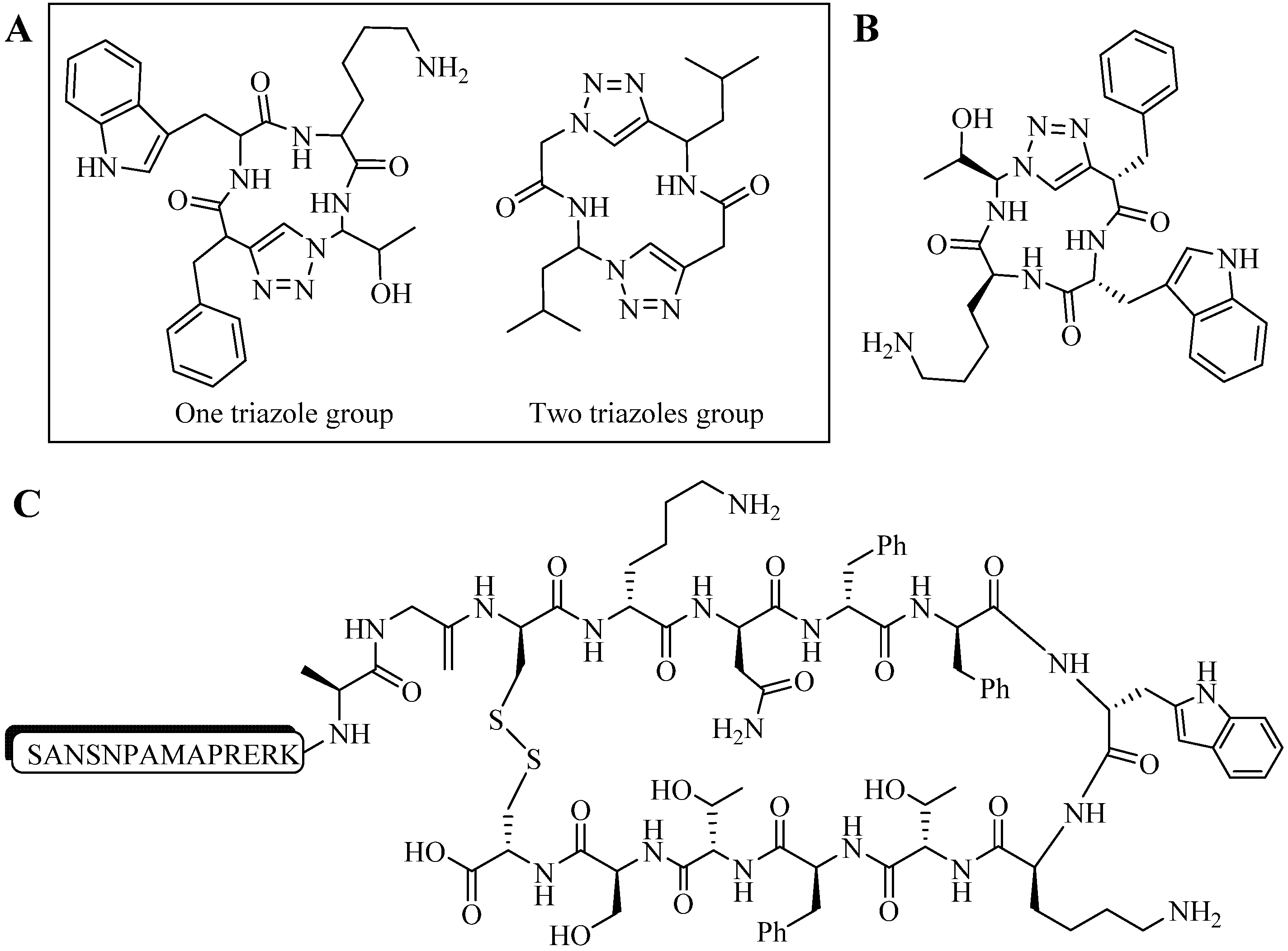
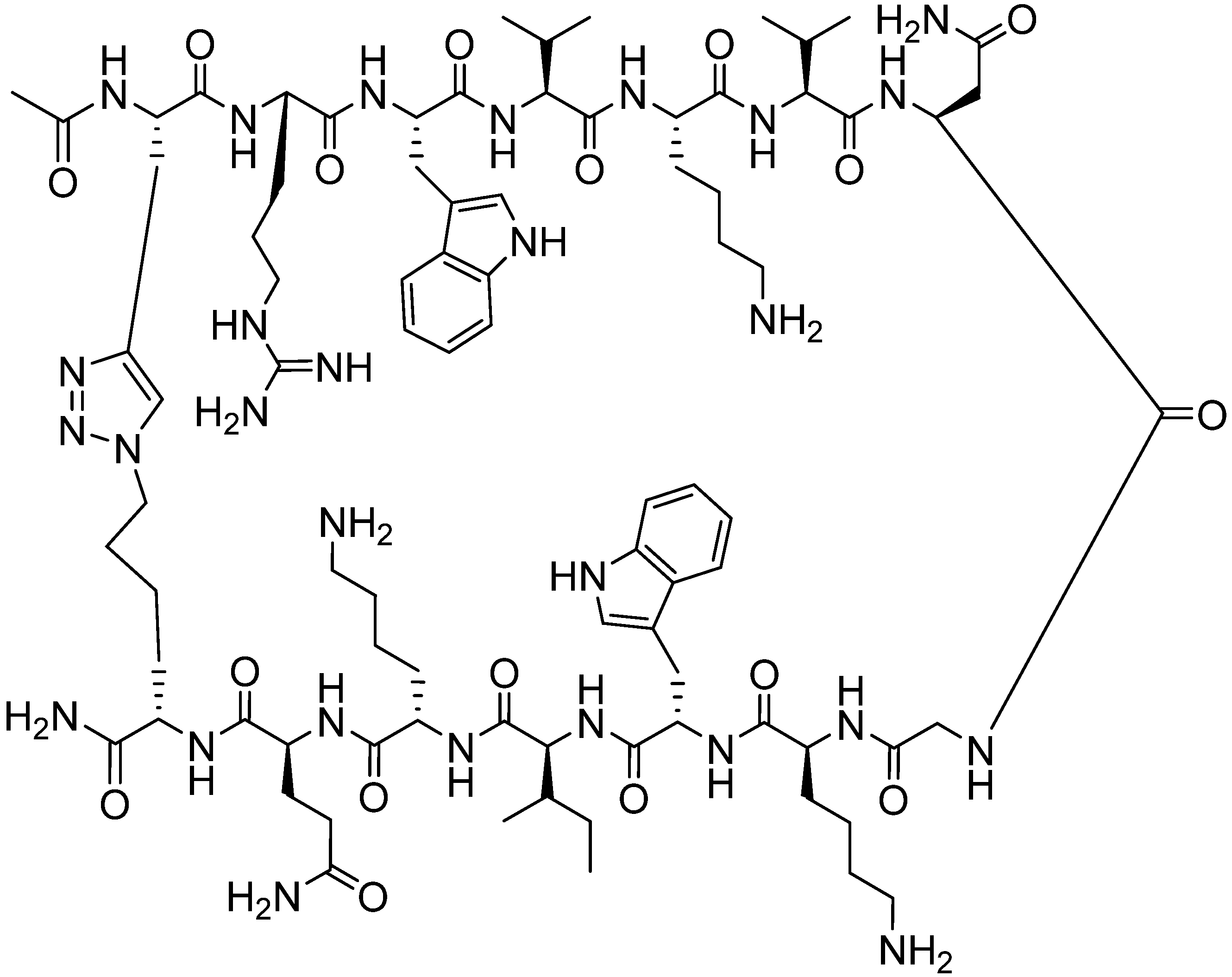
4.2. Intramolecular Triazoles Linker
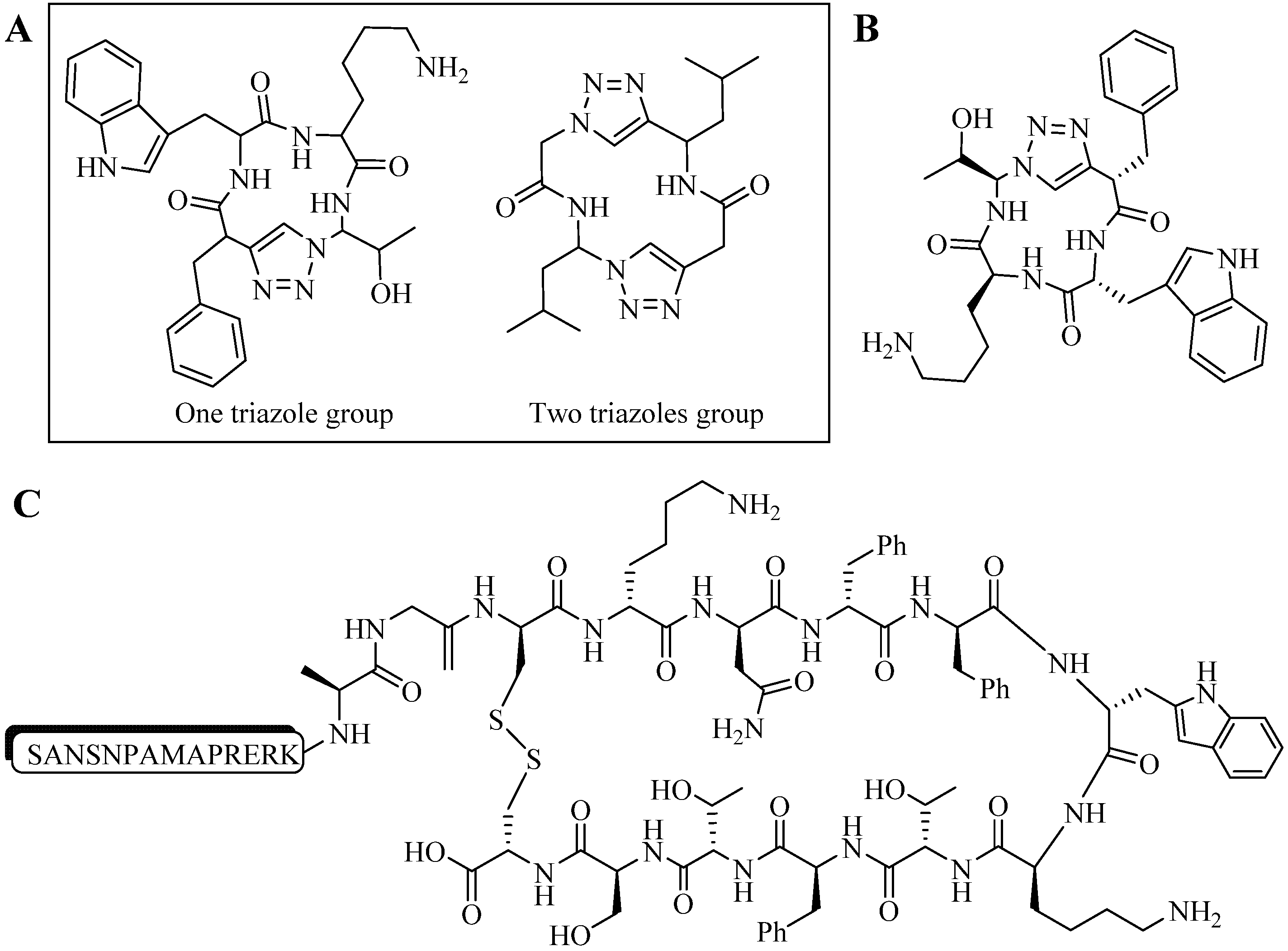
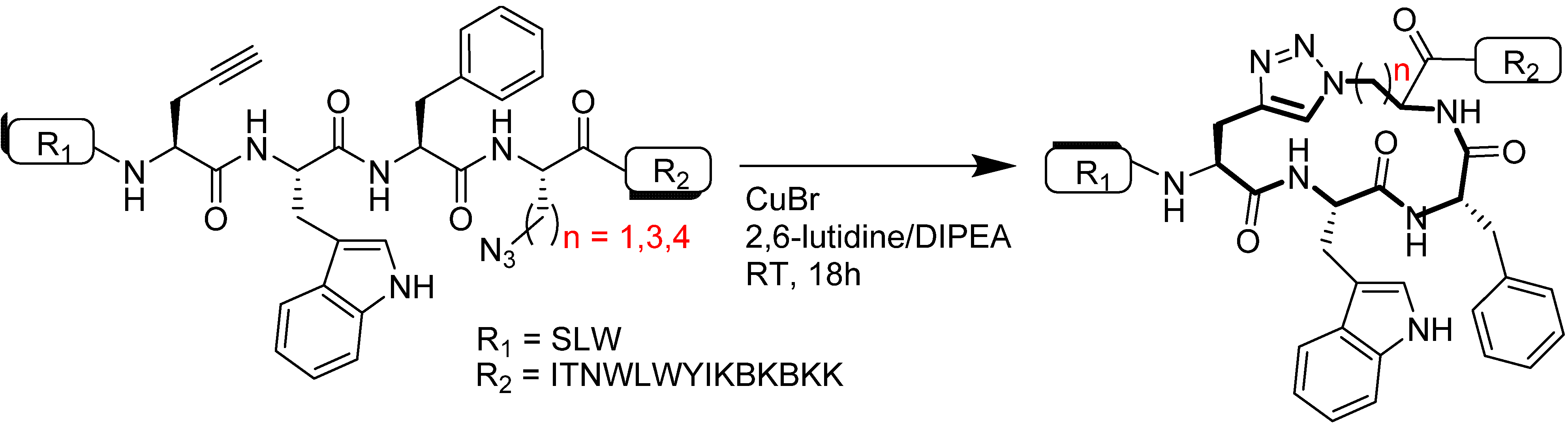
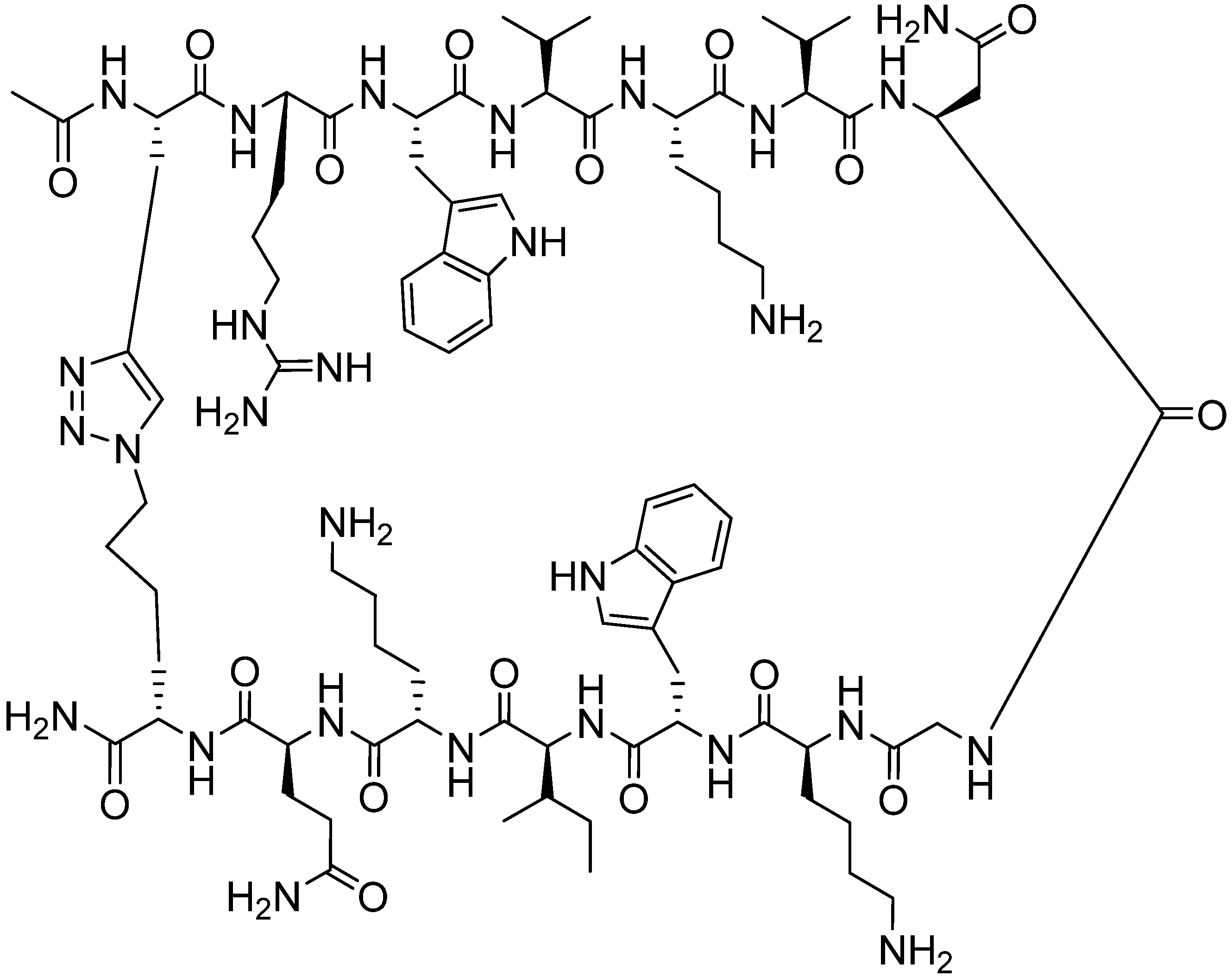
4.2.1. Side Chain Stapling/Macrocyclization
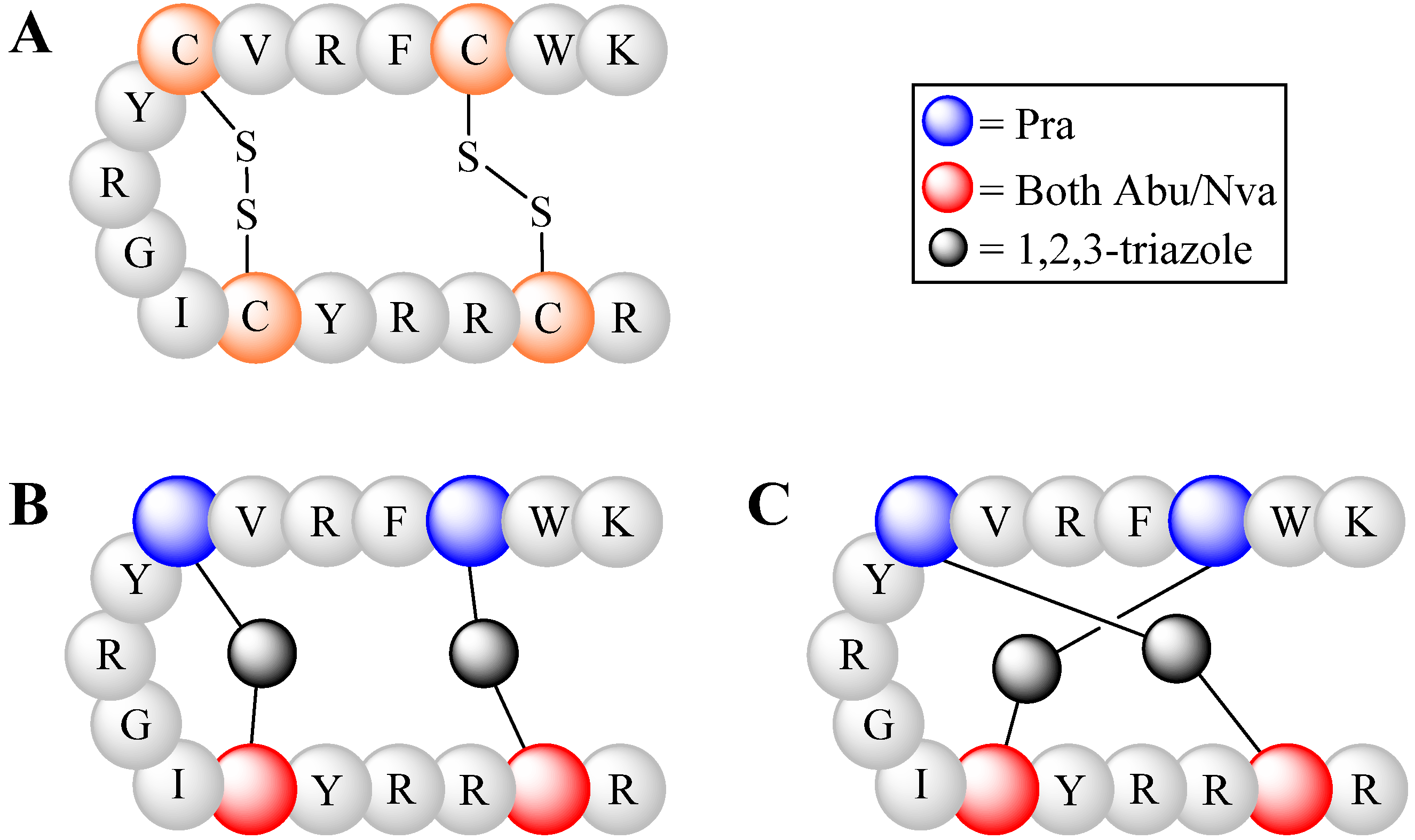
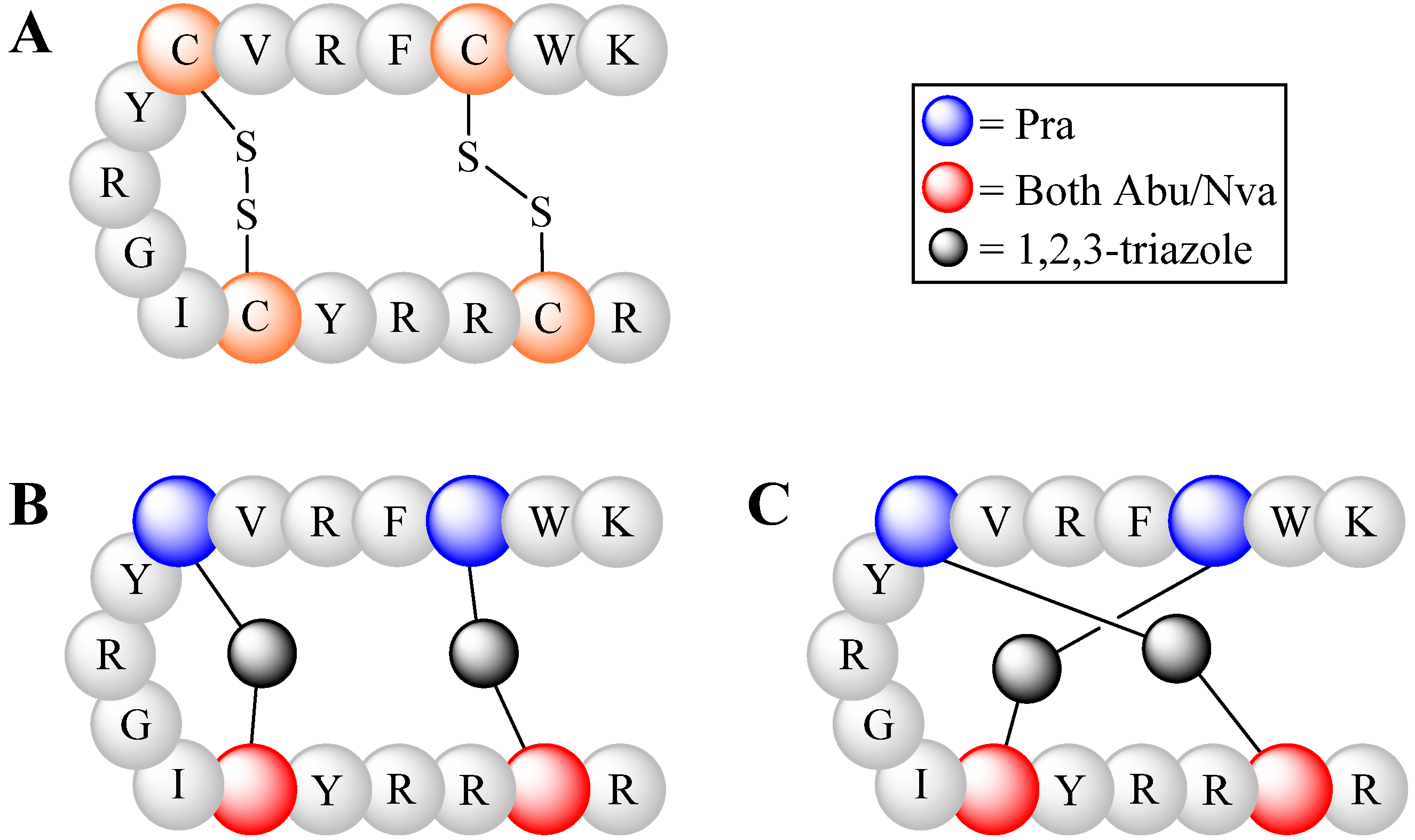
4.2.2. Triazoles as Disulfide Bridge Substitutions
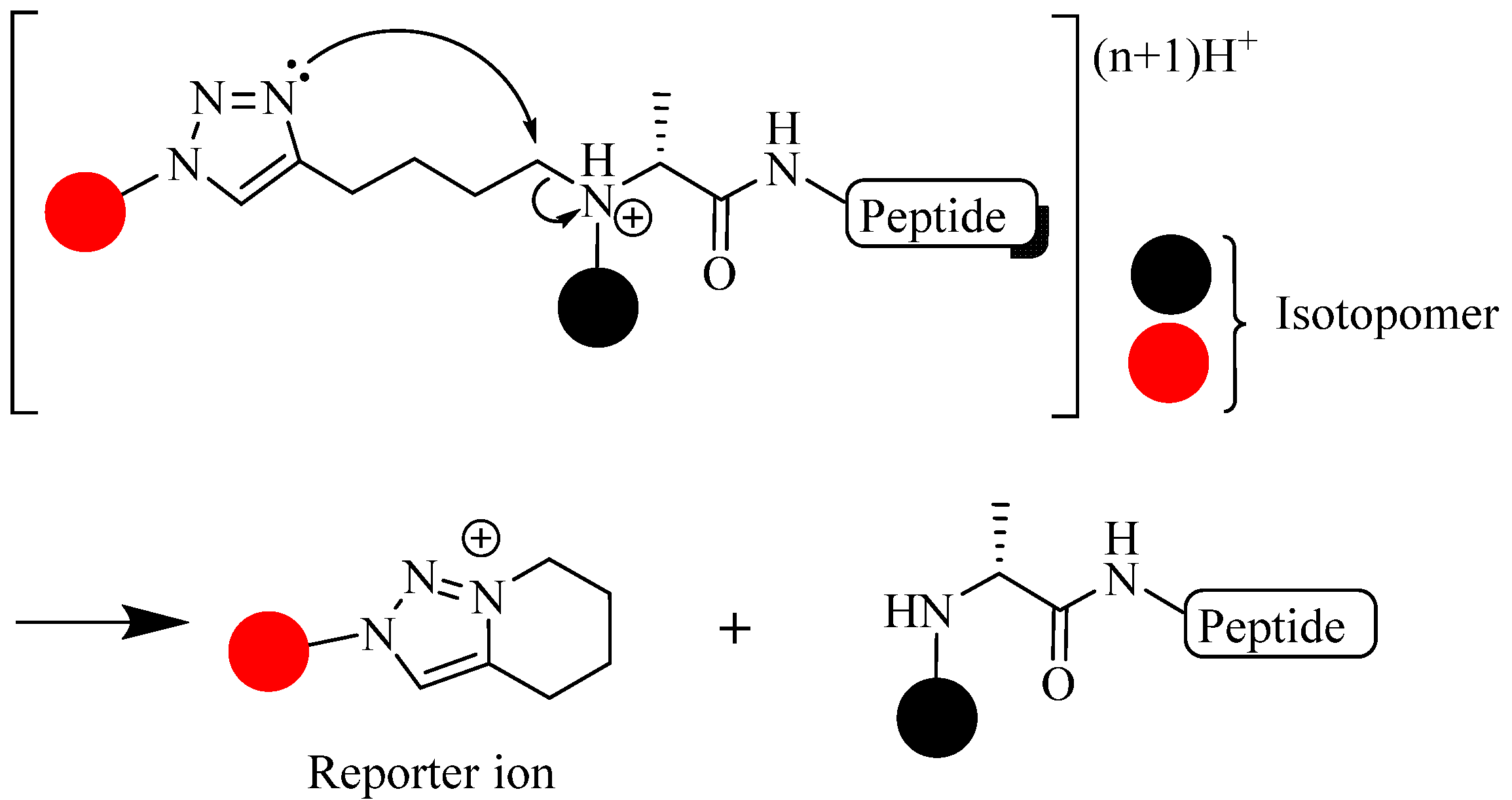
4.3. Other Applications
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Campbell, N.A.; Reece, J.B. Biology, 6th ed.; Pearson Education, Inc.: San Francisco, CA, USA, 2002; p. 1245. [Google Scholar]
- Curtius, T. Ueber einige neue hippursaureanalog constituierte synthetisch dargestellte aminosauren. J. Prakt. Chem. 1882, 26, 145–208. [Google Scholar] [CrossRef]
- Fisher, E.; Fourneau, E. Ueber einige derivate des glykokolls. Ber. Dtsch. Chem. Ges. 1901, 34, 2868–2879. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase peptide synthesis.1. Synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Vigneaud, V.D.; Ressler, C.; Swan, J.M.; Roberts, C.W.; Katsoyannis, P.G. The synthesis of oxytocin. J. Am. Chem. Soc. 1954, 76, 3115–3121. [Google Scholar]
- Kent, S.B. Total chemical synthesis of proteins. Chem. Soc. Rev. 2009, 38, 338–351. [Google Scholar] [CrossRef]
- Hofmann, K.; Yajima, H.; Yanaibara, N.; Liu, T.; Lande, S. Studies on polypeptides. XIII. The synthesis of a tricosapeptide posessing essentially the full biological activity of natural ACTH1–3. J. Am. Chem. Soc. 1961, 83, 486–487. [Google Scholar]
- Kimura, T.; Takai, M.; Masui, Y.; Morikawa, T.; Sakakibara, S. Strategy for the synthesis of large peptides—an application to the total synthesis of human parathyroid-hormone [hpth(1–84)]. Biopolymers 1981, 20, 1823–1832. [Google Scholar] [CrossRef]
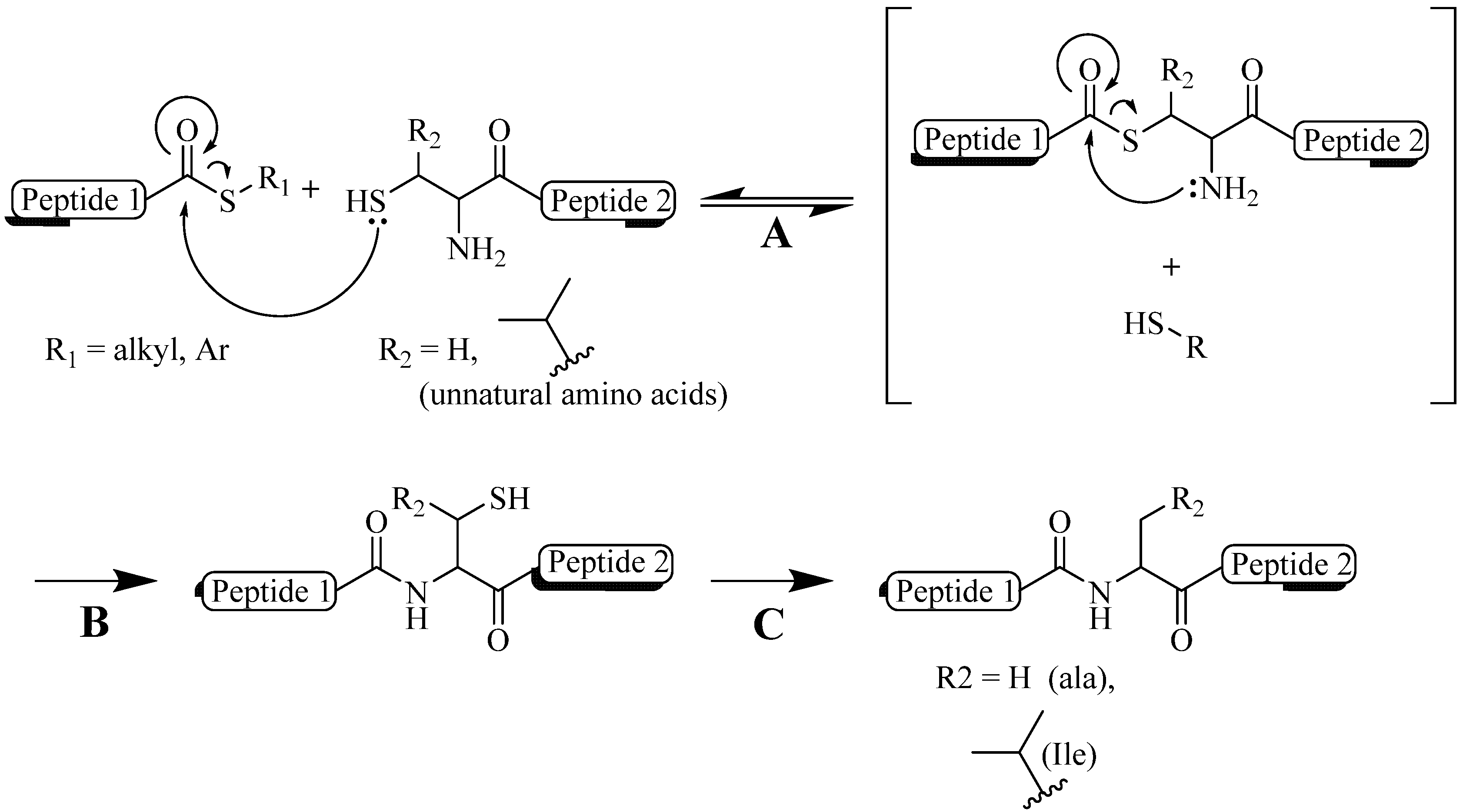
- Kemp, D.S.; Leung, S.L.; Kerkman, D.J. Models that demonstrate peptide-bond formation by prior thiol capture.1. Capture by disulfide formation. Tetrahedron Lett. 1981, 22, 181–184. [Google Scholar]
- Wieland, T.; Bokelmann, E.; Bauer, L.; Lang, H.U.; Lau, H. Über peptidsynthesen. 8. Bildung von s-haltigen peptiden durch intramolekulare wanderung von aminoacylresten. Justus Liebigs Ann. Chem. 1953, 583, 129–149. [Google Scholar] [CrossRef]
- Chen, J.; Wan, Q.; Yuan, Y.; Zhu, J.L.; Danishefsky, S.J. Native chemical ligation at valine: A contribution to peptide and glycopeptide synthesis. Angew. Chem. Int. Ed. Engl. 2008, 47, 8521–8524. [Google Scholar] [CrossRef]
- Tan, Z.; Shang, S.; Danishefsky, S.J. Insights into the finer issues of native chemical ligation: An approach to cascade ligations. Angew. Chem. Int. Ed. Engl. 2010, 49, 9500–9503. [Google Scholar] [CrossRef]
- Moyle, P.M.; Hari, Y.; Huang, N.; Olive, C.; Good, M.F.; Toth, I. A technique for the synthesis of highly-pure, mono-epitopic, multi-valent lipid core peptide vaccines. Tetrahedron Lett. 2007, 48, 4965–4967. [Google Scholar] [CrossRef]
- Fujita, Y.; Moyle, P.M.; Hieu, S.; Simerska, P.; Toth, I. Investigation toward multi-epitope vaccine candidates using native chemical ligation. Biopolymers 2008, 90, 624–632. [Google Scholar] [CrossRef]
- Chandrudu, S.; Simerska, P.; Toth, I. Chemical methods for peptide and protein production. Molecules 2013, 18, 4373–4388. [Google Scholar] [CrossRef]
- Raibaut, L.; Ollivier, N.; Melnyk, O. Sequential native peptide ligation strategies for total chemical protein synthesis. Chem. Soc. Rev. 2012, 41, 7001–7015. [Google Scholar] [CrossRef]
- Van Berkel, S.S.; van Eldijk, M.B.; van Hest, J.C.M. Staudinger ligation as a method for bioconjugation. Angew. Chem. Int. Ed. Engl. 2011, 50, 8806–8827. [Google Scholar] [CrossRef]
- De Araujo, A.D.; Palomo, J.M.; Cramer, J.; Seitz, O.; Alexandrov, K.; Waldmann, H. Diels-alder ligation of peptides and proteins. Chemistry 2006, 12, 6095–6109. [Google Scholar] [CrossRef]
- Debets, M.F.; van Berkel, S.S.; Dommerholt, J.; Dirks, A.T.; Rutjes, F.P.; van Delft, F.L. Bioconjugation with strained alkenes and alkynes. Acc. Chem. Res. 2011, 44, 805–815. [Google Scholar] [CrossRef]
- Michael, A. Ueber die einwirkung von diazobenzolimid auf acetylendicarbonsäuremethylester. J. Prakt. Chem. 1983, 48, 94–95. [Google Scholar] [CrossRef]
- Huisgen, R. Centenary lecture - 1,3-dipolar cycloadditions. Proc. Chem. Soc. Lond. 1961, 357–396. [Google Scholar]
- Labbe, G. Are azidocumulenes accessible. Bull. Soc. Chim. Belg. 1984, 93, 579–592. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(i)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(i)-catalyzed synthesis of azoles. Dft study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Meldal, M. Polymer “clicking” by cuaac reactions. Macromol. Rapid Commun. 2008, 29, 1016–1051. [Google Scholar] [CrossRef]
- Jones, G.O.; Ess, D.H.; Houk, K.N. Activation energies and reaction energetics for 1,3-dipolar cycloadditions of hydrazoic acid with c-c and c-n multiple bonds from high-accuracy and density functional quantum mechanical calculations. Helv. Chim. Acta 2005, 88, 1702–1710. [Google Scholar] [CrossRef]
- Golas, P.L.; Matyjaszewski, K. Marrying click chemistry with polymerization: Expanding the scope of polymeric materials. Chem. Soc. Rev. 2010, 39, 1338–1354. [Google Scholar] [CrossRef]
- Astakhova, I.K.; Hansen, L.H.; Vester, B.; Wengel, J. Peptide-lna oligonucleotide conjugates. Org. Biomol. Chem. 2013, 11, 4240–4249. [Google Scholar] [CrossRef] [Green Version]
- Ahmad Fuaad, A.A.; Jia, Z.; Hartas, J.; Ziora, Z.M.; Lin, I.C.; Moyle, P.M.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Skwarczynski, M.; et al. Polymer-peptide hybrids as a highly immunogenic single-dose nanovaccine. Nanomedicine (Lond.) 2013, in press. [Google Scholar]
- Hu, X.; Yan, L.; Xiao, H.; Li, H.; Jing, X. Application of microwave-assisted click chemistry in the preparation of functionalized copolymers for drug conjugation. J. Appl. Polym. Sci. 2012, 127, 3365–3373. [Google Scholar]
- Barge, A.; Tagliapietra, S.; Binello, A.; Cravotto, G. Click chemistry under microwave or ultrasound irradiation. Curr. Org. Chem. 2011, 15, 189–203. [Google Scholar] [CrossRef]
- Jang, H.J.; Fafarman, A.; Holub, J.M.; Kirshenbaum, K. Click to fit: Versatile polyvalent display on a peptidomimetic scaffold. Org. Lett. 2005, 7, 1951–1954. [Google Scholar] [CrossRef]
- Li, Z.G.; Bittman, R. Synthesis and spectral properties of cholesterol- and fty720-containing boron dipyrromethene dyes. J. Org. Chem. 2007, 72, 8376–8382. [Google Scholar] [CrossRef]
- Molander, G.A.; Ham, J. Synthesis of functionalized organotrifluoroborates via the 1,3-dipolar cycloaddition of azides. Org. Lett. 2006, 8, 2767–2770. [Google Scholar] [CrossRef]
- Meldal, M.; Tornoe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Kennedy, D.C.; McKay, C.S.; Legault, M.C.B.; Danielson, D.C.; Blake, J.A.; Pegoraro, A.F.; Stolow, A.; Mester, Z.; Pezacki, J.P. Cellular consequences of copper complexes used to catalyze bioorthogonal click reactions. J. Am. Chem. Soc. 2011, 133, 17993–18001. [Google Scholar] [CrossRef]
- Van Hest, J.C.M.; van Delft, F.L. Protein modification by strain-promoted alkyne-azide cycloaddition. ChemBioChem 2011, 12, 1309–1312. [Google Scholar] [CrossRef]
- Uttamapinant, C.; Tangpeerachaikul, A.; Grecian, S.; Clarke, S.; Singh, U.; Slade, P.; Gee, K.R.; Ting, A.Y. Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew. Chem. Int. Ed. Engl. 2012, 51, 5852–5856. [Google Scholar] [CrossRef]
- Besanceney-Webler, C.; Jiang, H.; Zheng, T.Q.; Feng, L.; del Amo, D.S.; Wang, W.; Klivansky, L.M.; Marlow, F.L.; Liu, Y.; Wu, P. Increasing the efficacy of bioorthogonal click reactions for bioconjugation: A comparative study. Angew. Chem. Int. Ed. Engl. 2011, 50, 8051–8056. [Google Scholar]
- Skwarczynski, M.; Zaman, M.; Urbani, C.N.; Lin, I.C.; Jia, Z.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Polyacrylate dendrimer nanoparticles: A self-adjuvanting vaccine delivery system. Angew. Chem. Int. Ed. Engl. 2010, 49, 5742–5745. [Google Scholar]
- Skwarczynski, M.; Ahmad Fuaad, A.A.H.; Rustanti, L.; Ziora, Z.M.; Aqil, M.; Batzloff, M.R.; Good, M.F.; Toth, I. Group a streptococcus vaccine candidates based on the conserved conformational epitope from m protein. Drug Deliv. Lett. 2011, 1, 2–8. [Google Scholar]
- Liu, T.Y.; Hussein, W.M.; Jia, Z.; Ziora, Z.M.; McMillan, N.A.; Monteiro, M.J.; Toth, I.; Skwarczynski, M. Self-adjuvanting polymer-peptide conjugates as therapeutic vaccine candidates against cervical cancer. Biomacromolecules 2013, 14, 2798–2806. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, J.E.; Kelly, J.A.; Veinot, J.G.C. Iron/iron oxide nanoparticle sequestration of catalytic metal impurities from aqueous media and organic reaction products. Langmuir 2007, 23, 9543–9545. [Google Scholar] [CrossRef]
- Nikiforovich, G.V.; Marshall, G.R. Conformation-function relationships in lhrh analogs.1. Conformations of lhrh peptide backbone. Int. J. Pept. Protein Res. 1993, 42, 171–180. [Google Scholar] [CrossRef]
- Nikiforovich, G.V.; Marshall, G.R. Conformation-function relationships in lhrh analogs.2. Conformations of lhrh peptide agonists and antagonists. Int. J. Pept. Protein Res. 1993, 42, 181–193. [Google Scholar] [CrossRef]
- Brandt, E.R.; Sriprakash, K.S.; Hobb, R.I.; Hayman, W.A.; Zeng, W.G.; Batzloff, M.R.; Jackson, D.C.; Good, M.F. New multi-determinant strategy for a group a streptococcal vaccine designed for the australian aboriginal population. Nat. Med. 2000, 6, 455–459. [Google Scholar] [CrossRef]
- Choudhary, A.; Raines, R.T. An evaluation of peptide-bond isosteres. ChemBioChem 2011, 12, 1801–1807. [Google Scholar] [CrossRef]
- Baginski, M.; Piela, L.; Skolnick, J. The ethylene group as a peptide-bond mimicking unit—a theoretical conformational-analysis. J. Comput. Chem. 1993, 14, 471–477. [Google Scholar] [CrossRef]
- Jenkins, C.L.; Vasbinder, M.M.; Miller, S.J.; Raines, R.T. Peptide bond isosteres: Ester or (e)-alkene in the backbone of the collagen triple helix. Org. Lett. 2005, 7, 2619–2622. [Google Scholar] [CrossRef]
- Horne, W.S.; Yadav, M.K.; Stout, C.D.; Ghadiri, M.R. Heterocyclic peptide backbone modifications in an alpha-helical coiled coil. J. Am. Chem. Soc. 2004, 126, 15366–15367. [Google Scholar]
- Brik, A.; Alexandratos, J.; Lin, Y.C.; Elder, J.H.; Olson, A.J.; Wlodawer, A.; Goodsell, D.S.; Wong, C.H. 1,2,3-triazole as a peptide surrogate in the rapid synthesis of hiv-1 protease inhibitors. ChemBioChem 2005, 6, 1167–1169. [Google Scholar] [CrossRef]
- Hou, J.L.; Liu, X.F.; Shen, J.; Zhao, G.L.; Wang, P.G. The impact of click chemistry in medicinal chemistry. Expert Opin. Drug Dis. 2012, 7, 489–501. [Google Scholar] [CrossRef]
- Li, X.C. Click to join peptides/proteins together. Chem. Asian J. 2011, 6, 2606–2616. [Google Scholar]
- Jacobsen, O.; Maekawa, H.; Ge, N.H.; Gorbitz, C.H.; Rongved, P.; Ottersen, O.P.; Amiry-Moghaddam, M.; Klaveness, J. Stapling of a 3(10)-helix with click chemistry. J. Org. Chem. 2011, 76, 1228–1238. [Google Scholar] [CrossRef]
- Chen, Z.G.; Li, Y.; Chen, E.; Hall, D.L.; Darke, P.L.; Culberson, C.; Shafer, J.A.; Kuo, L.C. Crystal-structure at 1.9-angstrom resolution of human-immunodeficiency-virus (hiv)-ii protease complexed with l-735,524, an orally bioavailable inhibitor of the hiv proteases. J. Biol. Chem. 1994, 269, 26344–26348. [Google Scholar]
- Saric, T.; Graef, C.I.; Goldberg, A.L. Pathway for degradation of peptides generated by proteasomes: A key role for thimet oligopeptidase and other metallopeptidases. J. Biol. Chem. 2004, 279, 46723–46732. [Google Scholar] [CrossRef]
- Kim, E.E.; Baker, C.T.; Dwyer, M.D.; Murcko, M.A.; Rao, B.G.; Tung, R.D.; Navia, M.A. Crystal-structure of hiv-1 protease in complex with vx-478, a potent and orally bioavailable inhibitor of the enzyme. J. Am. Chem. Soc. 1995, 117, 1181–1182. [Google Scholar]
- Bejot, R.; Goggi, J.; Moonshi, S.S.; Robins, E.G. A practical synthesis of [f-18]ftrgd: An angiogenesis biomarker for pet. J. Label. Compd. Radiopharm. 2013, 56, 42–49. [Google Scholar] [CrossRef]
- Daumar, P.; Wanger-Baumann, C.A.; Pillarsetty, N.; Fabrizio, L.; Carlin, S.D.; Andreev, O.A.; Reshetnyak, Y.K.; Lewis, J.S. Efficient (18)f-labeling of large 37-amino-acid phlip peptide analogues and their biological evaluation. Bioconjug. Chem. 2012, 23, 1557–1566. [Google Scholar] [CrossRef]
- Gill, H.S.; Marik, J. Preparation of f-18-labeled peptides using the copper(i)-catalyzed azide-alkyne 1,3-dipolar cycloaddition. Nat. Protoc. 2011, 6, 1718–1725. [Google Scholar]
- Iddon, L.; Leyton, J.; Indrevoll, B.; Glaser, M.; Robins, E.G.; George, A.J.T.; Cuthbertson, A.; Luthra, S.K.; Aboagye, E.O. Synthesis and in vitro evaluation of [f-18]fluoroethyl triazole labelled [tyr(3)]octreotate analogues using click chemistry. Bioorg. Med. Chem. Lett. 2011, 21, 3122–3127. [Google Scholar] [CrossRef]
- Li, J.B.; Shi, L.L.; Jia, L.N.; Jiang, D.W.; Zhou, W.; Hu, W.Q.; Qi, Y.J.; Zhang, L. Radiolabeling of rgd peptide and preliminary biological evaluation in mice bearing u87mg tumors. Bioorg. Med. Chem. 2012, 20, 3850–3855. [Google Scholar] [CrossRef]
- Reubi, J.C. New specific radioligand for one subpopulation of brain somatostatin receptors. Life Sci. 1985, 36, 1829–1836. [Google Scholar] [CrossRef]
- Olberg, D.E.; Hjelstuen, O.K. Labeling strategies of peptides with f-18 for positron emission tomography. Curr. Top. Med. Chem. 2010, 10, 1669–1679. [Google Scholar] [CrossRef]
- Li, X.G.; Autio, A.; Ahtinen, H.; Helariutta, K.; Liljenback, H.; Jalkanen, S.; Roivainen, A.; Airaksinen, A.J. Translating the concept of peptide labeling with 5-deoxy-5-[f-18] fluororibose into preclinical practice: F-18-labeling of siglec-9 peptide for pet imaging of inflammation. Chem. Commun. 2013, 49, 3682–3684. [Google Scholar] [CrossRef]
- Dijkgraaf, I.; Terry, S.Y.A.; McBride, W.J.; Goldenberg, D.M.; Laverman, P.; Franssen, G.M.; Oyen, W.J.G.; Boerman, O.C. Imaging integrin alpha-v-beta-3 expression in tumors with an 18f-labeled dimeric rgd peptide. Contrast Media Mol. Imaging 2013, 8, 238–245. [Google Scholar] [CrossRef]
- Mamat, C.; Ramenda, T.; Wuest, F.R. Recent applications of click chemistry for the synthesis of radiotracers for molecular imaging. Mini Rev. Org. Chem. 2009, 6, 21–34. [Google Scholar] [CrossRef]
- Golas, P.L.; Tsarevsky, N.V.; Matyjaszewski, K. Structure-reactivity correlation in “click” chemistry: Substituent effect on azide reactivity. Macromol. Rapid Commun. 2008, 29, 1167–1171. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (cuaac) and beyond: New reactivity of copper(i) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- Nahrwold, M.; Weiss, C.; Bogner, T.; Mertink, F.; Conradi, J.; Sammet, B.; Palmisano, R.; Gracia, S.R.; Preusse, T.; Sewald, N. Conjugates of modified cryptophycins and rgd-peptides enter target cells by endocytosis. J. Med. Chem. 2013, 56, 1853–1864. [Google Scholar] [CrossRef]
- Sokolova, N.V.; Vorobyeva, D.V.; Osipov, S.N.; Vasilyeva, T.P.; Nenajdenko, V.G. Synthesis of alpha-trifluoromethyl-alpha-hydroxy acid-peptide conjugates via click chemistry. Synth.—Stuttg. 2012, 44, 130–136. [Google Scholar] [CrossRef]
- Sokolova, N.V.; Nenajdenko, V.G.; Sokolov, V.B.; Serebryakova, O.G.; Makhaeva, G.F. Synthesis and testing of trifluoromethyl-containing phosphonate-peptide conjugates as inhibitors of serine hydrolases. Bioorg. Med. Chem. Lett. 2011, 21, 7216–7218. [Google Scholar] [CrossRef]
- Hussein, W.M.; Liu, T.Y.; Toth, I.; Skwarczynski, M. Microwave-assisted synthesis of difficult sequence-containing peptides using the isopeptide method. Org. Biomol. Chem. 2013, 11, 2370–2376. [Google Scholar]
- Dehn, S.; Castelletto, V.; Hamley, I.W.; Perrier, S. Altering peptide fibrillization by polymer conjugation. Biomacromolecules 2012, 13, 2739–2747. [Google Scholar]
- Yeung, H.; Lee, D.J.; Williams, G.M.; Harris, P.W.R.; Dunbar, R.P.; Brimble, M.A. A method for the generation of pam(2)cys-based lipopeptide mimics via cuaac click chemistry. Synlett 2012, 11, 1617–1620. [Google Scholar]
- Fujita, Y.; Taguchi, H. Current status of multiple antigen-presenting peptide vaccine systems: Application of organic and inorganic nanoparticles. Chem. Cent. J. 2011, 5. [Google Scholar] [CrossRef]
- Gupta, K.; Singh, S.; Gupta, K.; Khan, N.; Sehgal, D.; Haridas, V.; Roy, R.P. A bioorthogonal chemoenzymatic strategy for defined protein dendrimer assembly. ChemBioChem 2012, 13, 2489–2494. [Google Scholar]
- Skwarczynski, M.; Kamaruzaman, K.A.; Srinivasan, S.; Zaman, M.; Lin, I.C.; Batzloff, M.R.; Good, M.F.; Toth, I. M-protein-derived conformational peptide epitope vaccine candidate against group a streptococcus. Curr. Drug Deliv. 2013, 10, 39–45. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Parhiz, B.H.; Soltani, F.; Srinivasan, S.; Kamaruzaman, K.A.; Lin, I.C.; Toth, I. Lipid peptide core nanoparticles as multivalent vaccine candidates against streptococcus pyogenes. Aust. J. Chem. 2012, 65, 35–39. [Google Scholar]
- Zaman, M.; Skwarczynski, M.; Malcolm, J.M.; Urbani, C.N.; Jia, Z.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Self-adjuvanting polyacrylic nanoparticulate delivery system for group a streptococcus (gas) vaccine. Nanomedicine 2011, 7, 168–173. [Google Scholar] [CrossRef]
- Moen, A.; Nicholson, D.G. Reduction of copper(ii) with subsequent disproportionation of copper(i) during the hydrothermal syntheses of microporous silicoaluminium phosphates sapo-5 and sapo-11. J. Chem. Soc. Faraday Trans. 1995, 91, 3529–3535. [Google Scholar] [CrossRef]
- Poon, C.K.; Chapman, R.; Jolliffe, K.A.; Perrier, S. Pushing the limits of copper mediated azide-alkyne cycloaddition (cuaac) to conjugate polymeric chains to cyclic peptides. Polym. Chem. UK 2012, 3, 1820–1826. [Google Scholar] [CrossRef]
- Jochim, A.L.; Miller, S.E.; Angelo, N.G.; Arora, P.S. Evaluation of triazolamers as active site inhibitors of hiv-1 protease. Bioorg. Med. Chem. Lett. 2009, 19, 6023–6026. [Google Scholar] [CrossRef]
- Angelo, N.G.; Arora, P.S. Solution- and solid-phase synthesis of triazole oligomers that display protein-like functionality. J. Org. Chem. 2007, 72, 7963–7967. [Google Scholar] [CrossRef]
- Angelo, N.G.; Arora, P.S. Nonpeptidic foldamers from amino acids: Synthesis and characterization of 1,3-substituted triazole oligomers. J. Am. Chem. Soc. 2005, 127, 17134–17135. [Google Scholar]
- Beierle, J.M.; Horne, W.S.; van Maarseveen, J.H.; Waser, B.; Reubi, J.C.; Ghadiri, M.R. Conformationally homogeneous heterocyclic pseudotetrapeptides as three-dimensional scaffolds for rational drug design: Receptor-selective somatostatin analogues. Angew. Chem. Int. Ed. Engl. 2009, 48, 4725–4729. [Google Scholar]
- Jagasia, R.; Holub, J.M.; Bollinger, M.; Kirshenbaum, K.; Finn, M.G. Peptide cyclization and cyclodimerization by cu-i-mediated azide-alkyne cycloaddition. J. Org. Chem. 2009, 74, 2964–2974. [Google Scholar]
- Ingale, S.; Dawson, P.E. On resin side-chain cyclization of complex peptides using cuaac. Org. Lett. 2011, 13, 2822–2825. [Google Scholar] [CrossRef]
- Huynh, N.T.; Jeon, Y.S.; Zrinyi, M.; Kim, J.H. Preparation of click’ hydrogels from polyaspartamide derivatives. Polym. Int. 2013, 62, 266–272. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Dougall, A.M.; Khoshnejad, M.; Chandrudu, S.; Pearson, M.S.; Loukas, A.; Toth, I. Peptide-based subunit vaccine against hookworm infection. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Zhong, W.; Skwarczynski, M.; Toth, I. Lipid core peptide system for gene, drug, and vaccine delivery. Aust. J. Chem. 2009, 62, 956–967. [Google Scholar]
- Schievano, E.; Pagano, K.; Mammi, S.; Peggion, E. Conformational studies of aib-rich peptides containing lactam-bridged side chains: Evidence of 3(10)-helix formation. Biopolymers 2005, 80, 294–302. [Google Scholar] [CrossRef]
- Ousaka, N.; Sato, T.; Kuroda, R. Intramolecular crosslinking of an optically inactive 3(10)-helical peptide: Stabilization of structure and helix sense. J. Am. Chem. Soc. 2008, 130, 463–465. [Google Scholar] [CrossRef]
- Scrima, M.; Le Chevalier-Isaad, A.; Rovero, P.; Papini, A.M.; Chorev, M.; D'Ursi, A.M. Cu-i-catalyzed azide-alkyne intramolecular i-to-(i+4) side-chain-to-side-chain cyclization promotes the formation of helix-like secondary structures. Eur. J. Org. Chem. 2010, 2010, 446–457. [Google Scholar]
- Park, J.H.; Waters, M.L. Positional effects of click cyclization on beta-hairpin structure, stability, and function. Org.Biomol. Chem. 2013, 11, 69–77. [Google Scholar] [CrossRef]
- Butterfield, S.M.; Sweeney, M.M.; Waters, M.L. The recognition of nucleotides with model ss-hairpin receptors: Investigation of critical contacts and nucleotide selectivity. J. Org. Chem. 2005, 70, 1105–1114. [Google Scholar] [CrossRef]
- Holland-Nell, K.; Meldal, M. Maintaining biological activity by using triazoles as disufide bond mimetics. Angew. Chem. Int. Ed. Engl. 2011, 50, 5204–5206. [Google Scholar] [CrossRef]
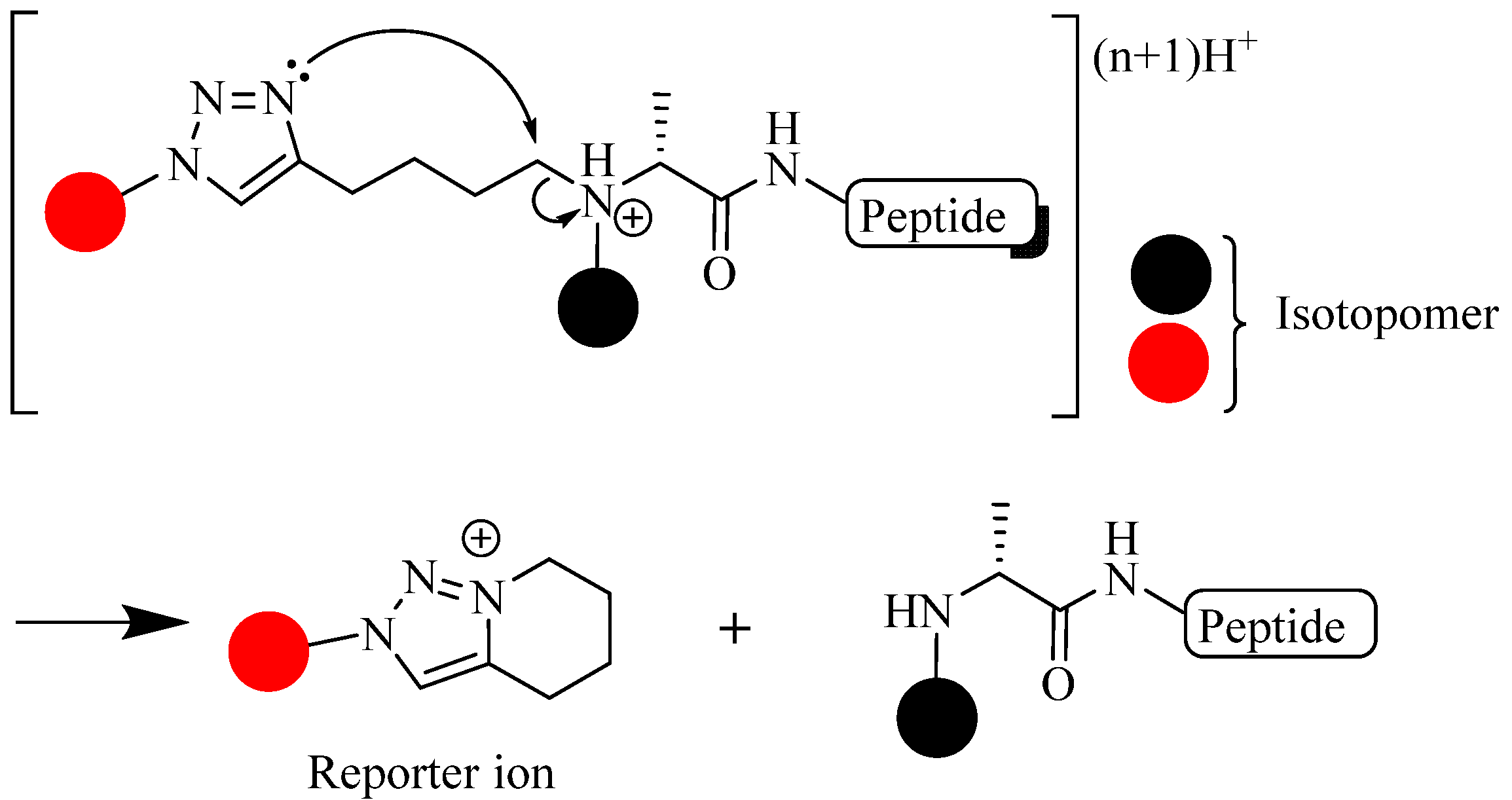
- Sohn, C.H.; Lee, J.E.; Sweredoski, M.J.; Graham, R.L.; Smith, G.T.; Hess, S.; Czerwieniec, G.; Loo, J.A.; Deshaies, R.J.; Beauchamp, J.L. Click chemistry facilitates formation of reporter ions and simplified synthesis of amine-reactive multiplexed isobaric tags for protein quantification. J. Am. Chem. Soc. 2012, 134, 2672–2680. [Google Scholar] [CrossRef]
- Chowdhury, S.M.; Du, X.X.; Tolic, N.; Wu, S.; Moore, R.J.; Mayer, M.U.; Smith, R.D.; Adkins, J.N. Identification of cross-linked peptides after click-based enrichment using sequential collision-induced dissociation and electron transfer dissociation tandem mass spectrometry. Anal. Chem. 2009, 81, 5524–5532. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ahmad Fuaad, A.A.H.; Azmi, F.; Skwarczynski, M.; Toth, I. Peptide Conjugation via CuAAC ‘Click’ Chemistry. Molecules 2013, 18, 13148-13174. https://doi.org/10.3390/molecules181113148
Ahmad Fuaad AAH, Azmi F, Skwarczynski M, Toth I. Peptide Conjugation via CuAAC ‘Click’ Chemistry. Molecules. 2013; 18(11):13148-13174. https://doi.org/10.3390/molecules181113148
Chicago/Turabian StyleAhmad Fuaad, Abdullah A. H., Fazren Azmi, Mariusz Skwarczynski, and Istvan Toth. 2013. "Peptide Conjugation via CuAAC ‘Click’ Chemistry" Molecules 18, no. 11: 13148-13174. https://doi.org/10.3390/molecules181113148