Template-Based de Novo Design for Type II Kinase Inhibitors and Its Extended Application to Acetylcholinesterase Inhibitors

Abstract
:1. Introduction
2. Procedure

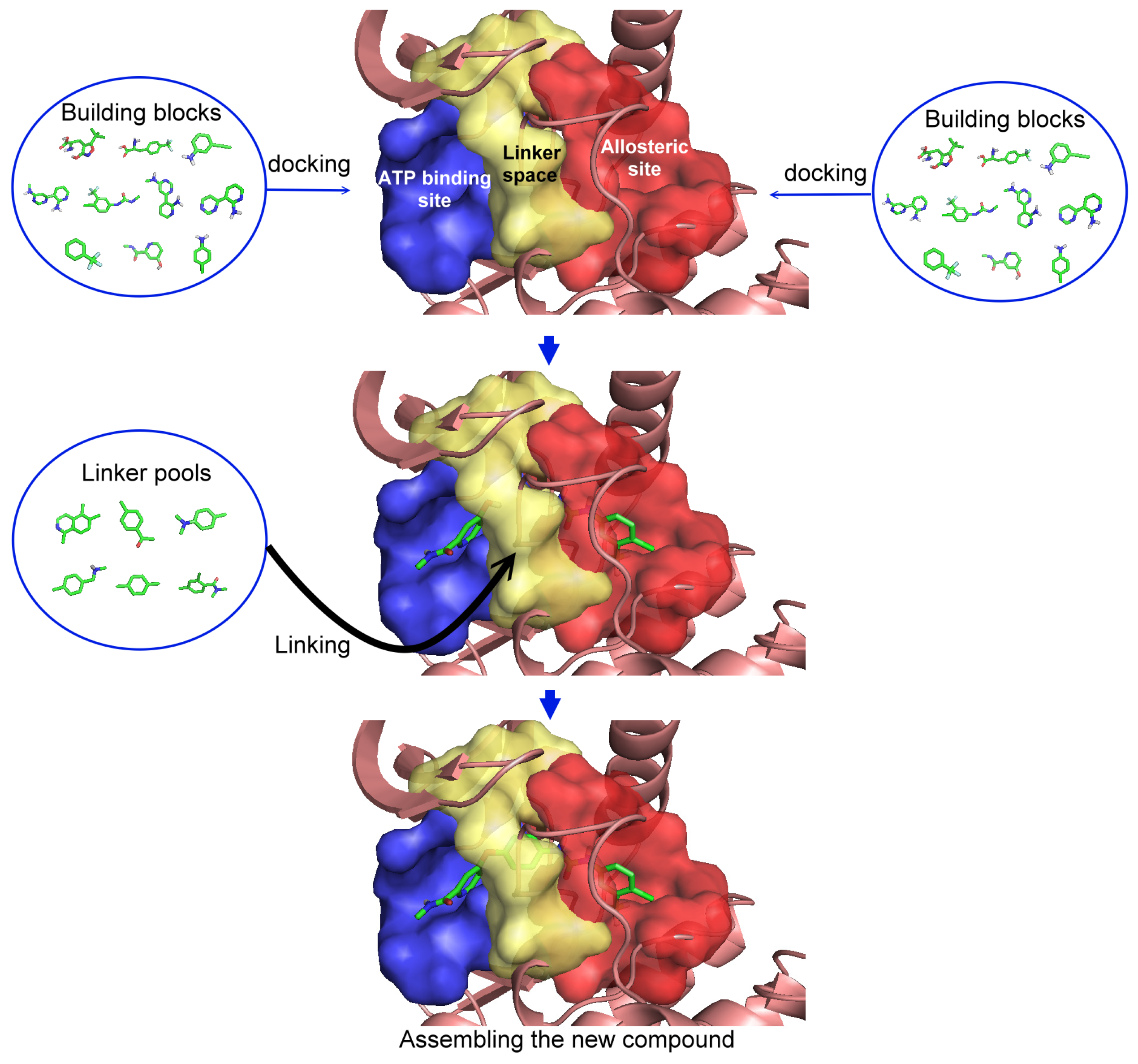
2.1. Step 1: Indicate the Space (Regions) for Structural Substitution

2.2. Step 2: Evaluate Each Building Block

2.3. Step 3: Assemble to Form New Structures
2.4. Step 4: Apply Drug-Likeness Filters
2.5. Step 5: Prioritize the New Structures
3. Results and Discussion
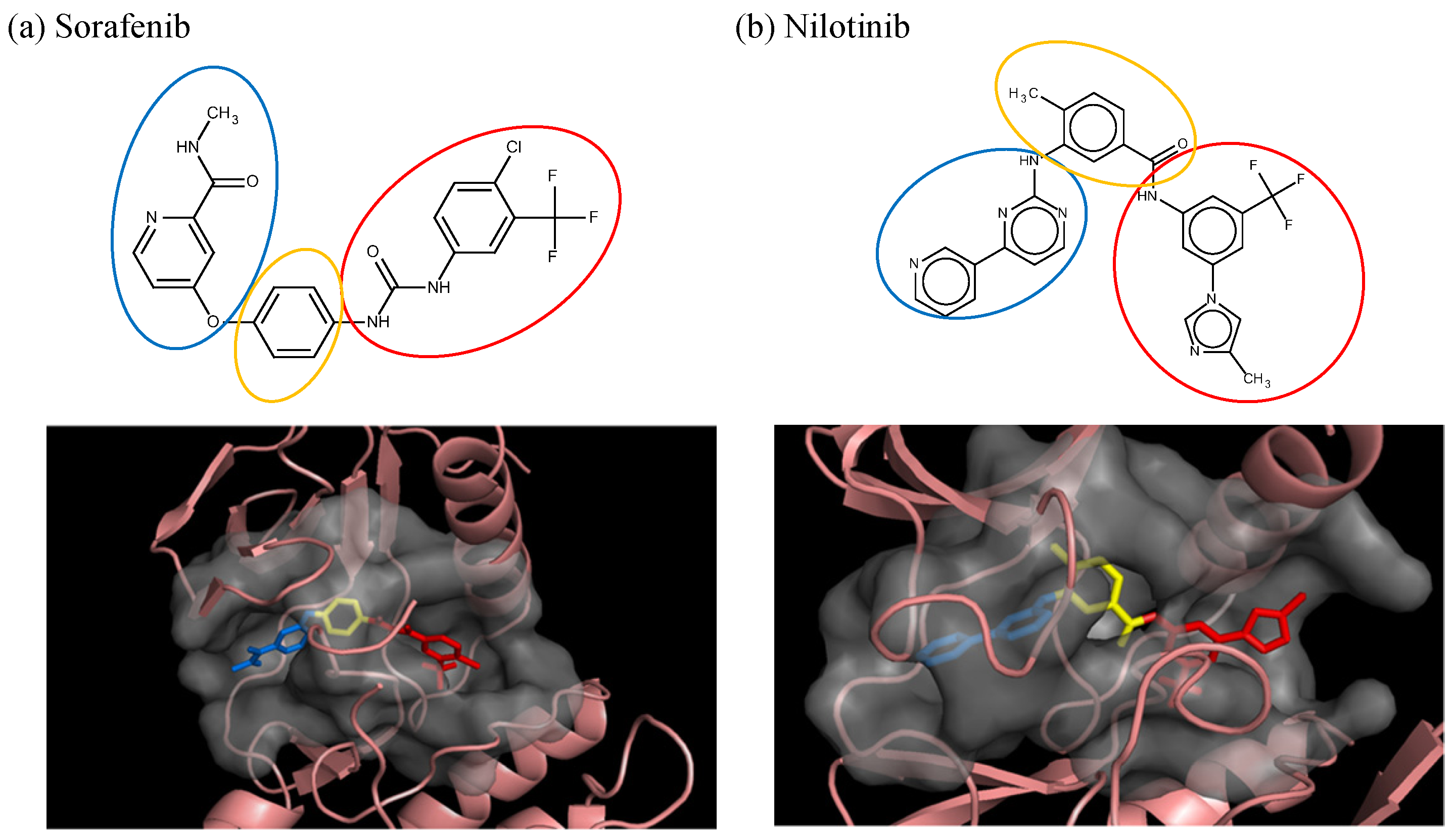
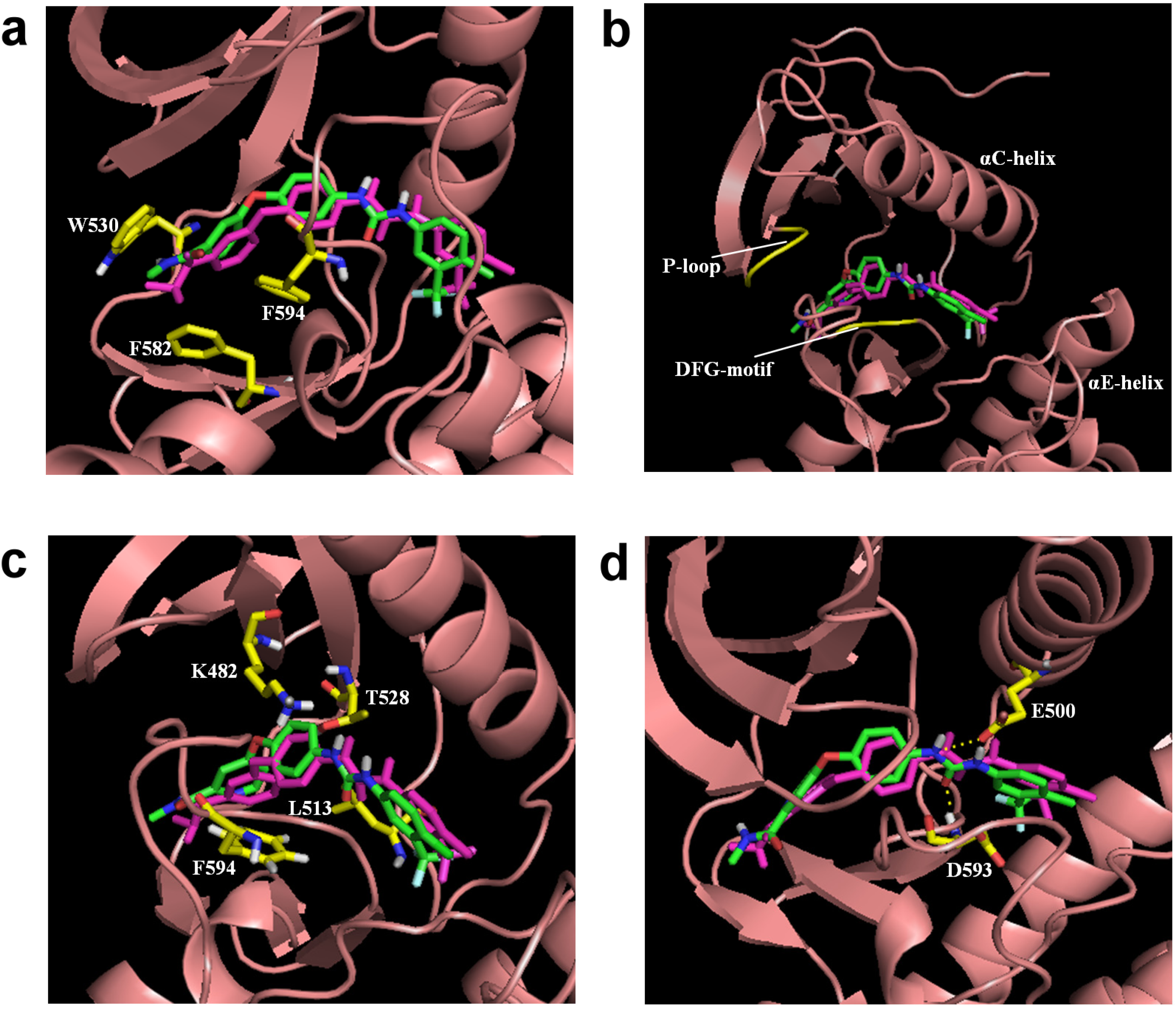
3.1. Sorafenib Reassembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | GE score of fragment 1 | GE score of fragment 2 | Docking energy of fragment 1 (kcal/mol) | Docking energy of fragment 2 (kcal/mol) |
|---|---|---|---|---|
| Sorafenib | 0.46 | 0.40 | −5.1 | −6.1 |
| Nilotinib | 0.60 | 0.39 | −7.8 | −6.6 |

3.2. Nilotinib Reassembly

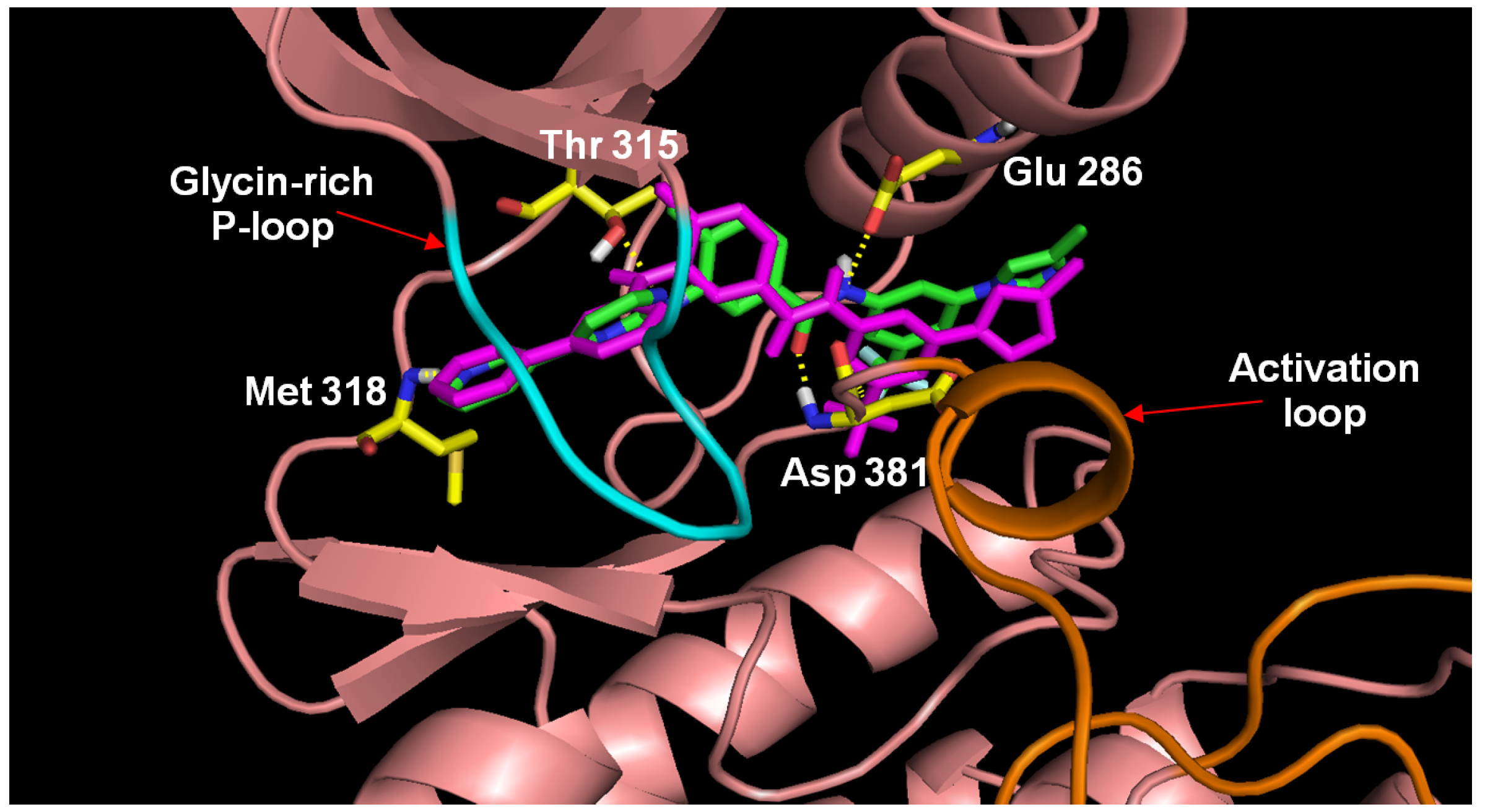
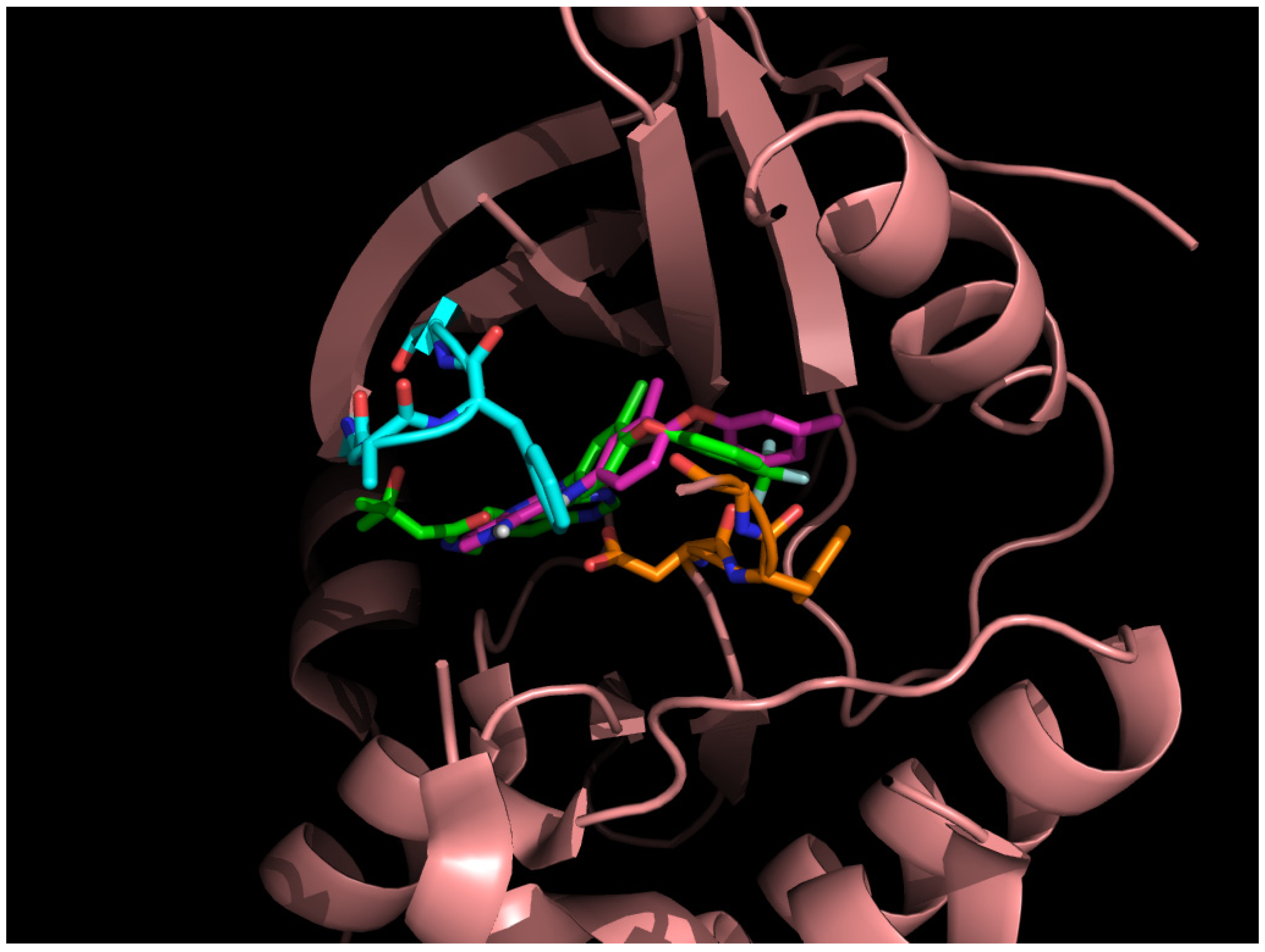
3.3. Optimization on the Series of Aminoisoquinoline Derivatives
| Rank | B-RAF, IC50 (nM) | Structure | Binding Energy (kCal/mol) | Sum of GE Score | Docking Energy (KCal/mol) |
|---|---|---|---|---|---|
| 1 | 1.6 |  1 | −33.0 | 1.09 | −12.4 |
| 2 | 17 |  12a | −27.2 | 1 | −11.4 |
| 3 | 56 |  13 | −27.0 | 0.95 | −11.5 |
| 4 | 18 |  15 | −26.7 | 1 | −11.3 |
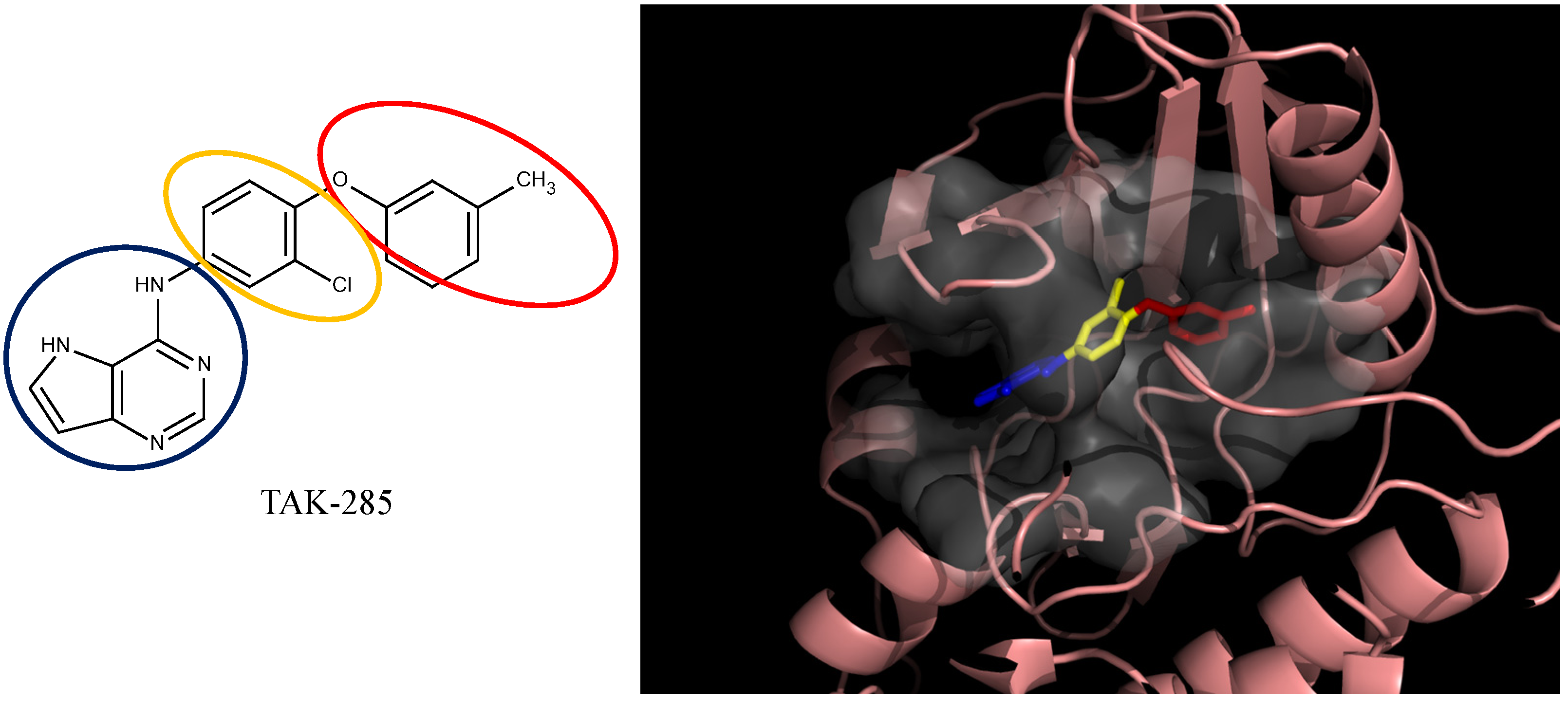
3.4. Optimization on the Series of TAK-285 Analogues Inhibiting HER2 Kinase
| Rank | HER2, IC50(nM) | Structure | Binding Energy (kCal/mol) |
|---|---|---|---|
| 1 | 20 |  6c | −21.1 |
| 2 | 4.1 |  10e | −21.0 |
| 3 | 4.6 |  6m | −20.9 |
| 4 | 26 |  6n | −19.4 |
| 5 | 8.3 |  6e | −19.2 |
| 6 | 17 |  8l | −17.0 |
| 7 | 12 |  10j | −16.7 |
| 8 | 3.3 |  8e | −15.3 |


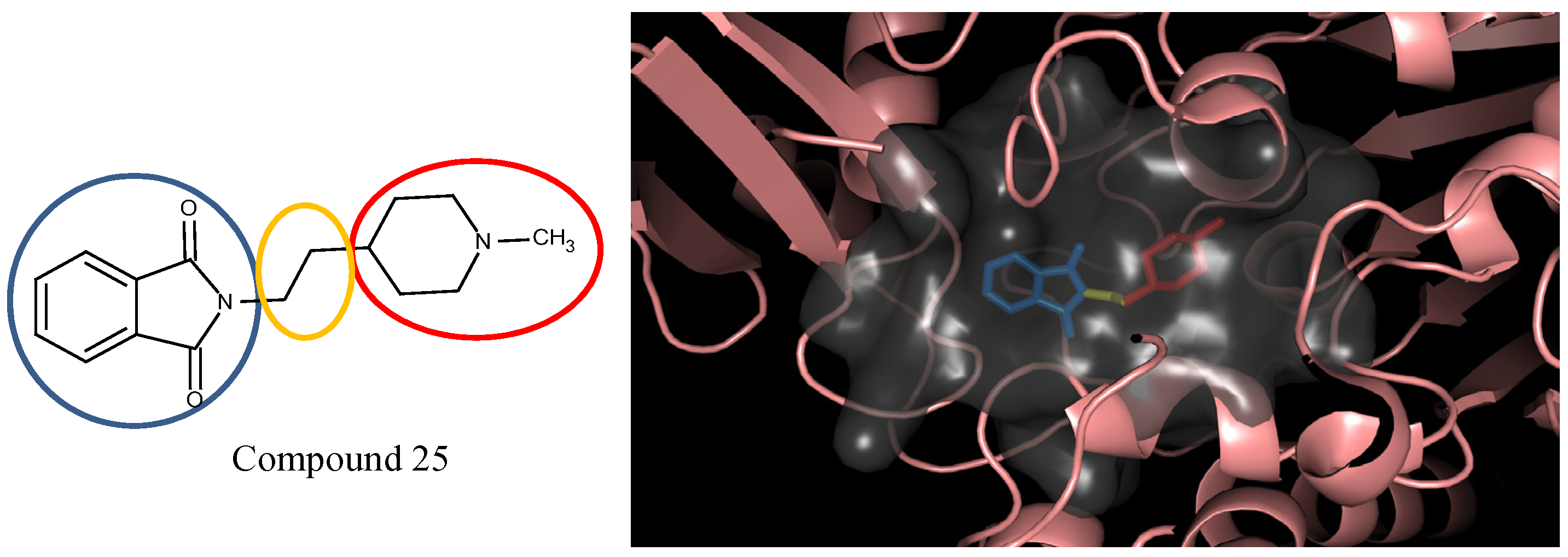
3.5. Optimization on the Series of E2020 Analogues
| Rank | AChE, IC50 (nM) | Structure | Binding Energy (kCal/mol) |
|---|---|---|---|
| 1 | 0.9 |  25 | −10.23 |
| 3 | 7.7 |  18B | −9.65 |
| 2 | 4.2 |  26 | −9.47 |
| 4 | 4400 |  22 | −7.73 |


4. Experimental
4.1. Datasets
4.2. Molecular Dynamics Simulation
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dey, F.; Caflisch, A. Fragment-based de novo ligand design by multiobjective evolutionary optimization. J. Chem. Inf. Model. 2008, 48, 679–690. [Google Scholar] [CrossRef]
- Schneider, G.; Lee, M.L.; Stahl, M.; Schneider, P. De novo design of molecular architectures by evolutionary assembly of drug-derived building blocks. J. Comput. Aided Mol. Des. 2000, 14, 487–494. [Google Scholar] [CrossRef]
- Pegg, S.C.; Haresco, J.J.; Kuntz, I.D. A genetic algorithm for structure-based de novo design. J. Comput. Aided Mol. Des. 2001, 15, 911–933. [Google Scholar] [CrossRef]
- Liu, Q.; Masek, B.; Smith, K.; Smith, J. Tagged fragment method for evolutionary structure-based de novo lead generation and optimization. J. Med. Chem. 2007, 50, 5392–5402. [Google Scholar] [CrossRef]
- Wang, R.; Gao, Y.; Lai, L. LigBuilder: A multi-purpose program for structure-based drug design. J. Mol. Model. 2000, 6, 498–516. [Google Scholar] [CrossRef]
- Gillet, V.; Johnson, A.P.; Mata, P.; Sike, S.; Williams, P. SPROUT: A program for structure generation. J. Comput. Aided Mol. Des. 1993, 7, 127–153. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C. Multiple highly diverse structures complementary to enzyme binding sites: Results of extensive application of a de novo design method incorporating combinatorial growth. J. Am. Chem. Soc. 1994, 116, 5560–5571. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Murcko, M.A. CONCERTS: Dynamic connection of fragments as an approach to de novo ligand design. J. Med. Chem. 1996, 39, 1651–1663. [Google Scholar] [CrossRef]
- Holland, J.H. Adaptation in Natural and Artificial Systems; University of Michigan Press: Ann Arbor, MI, USA, 1975. [Google Scholar]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Vinkers, H.M.; de Jonge, M.R.; Daeyaert, F.F.; Heeres, J.; Koymans, L.M.; van Lenthe, J.H.; Lewi, P.J.; Timmerman, H.; van Aken, K.; Janssen, P.A. SYNOPSIS: Synthesize and optimize system in silico. J. Med. Chem. 2003, 46, 2765–2773. [Google Scholar] [CrossRef]
- Lewell, X.Q.; Judd, D.B.; Watson, S.P.; Hann, M.M. RECAP-retrosynthetic combinatorial analysis procedure: A powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J. Chem. Inf. Model. 1998, 38, 511–522. [Google Scholar] [CrossRef]
- Cramer, R.D.; Soltanshahi, F.; Jilek, R.; Campbell, B. AllChem: Generating and searching 1020 synthetically accessible structures. J. Comput. Aided Mol. Des. 2007, 21, 341–350. [Google Scholar] [CrossRef]
- Hecht, D.; Fogel, G.B. A novel in silico approach to drug discovery via computational intelligence. J. Chem. Inf. Model. 2009, 49, 1105–1121. [Google Scholar] [CrossRef]
- Douguet, D.; Munier-Lehmann, H.; Labesse, G.; Pochet, S. LEA3D: A computer-aided ligand design for structure-based drug design. J. Med. Chem. 2005, 48, 2457–2468. [Google Scholar] [CrossRef]
- Burns, J.A.; Whitesides, G.M. Feed-forward neural networks in chemistry: Mathematical systems for classification and pattern recognition. Chem. Rev. 1993, 93, 2583–2601. [Google Scholar] [CrossRef]
- Kosko, B.; Isaka, S. Fuzzy logic. Sci. Am. 1993, 269, 62–67. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Rees, D.C. Group efficiency: A guideline for hits-to-leads chemistry. ChemMedChem 2008, 3, 1179–1180. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Chen, K.; Sharp, K.A.; Kollman, P.A. The maximal affinity of ligands. Proc. Natl. Acad. Sci. USA 1999, 96, 9997–10002. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discovering Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Navia, M.A.; Chaturvedi, P.R. Design principles for orally bioavailable drugs. Drug Discovering Today 1996, 1, 179–189. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Saxty, G.; Woodhead, S.J.; Berdini, V.; Davies, T.G.; Verdonk, M.L.; Wyatt, P.G.; Boyle, R.G.; Barford, D.; Downham, R.; Garrett, M.D.; et al. Identification of inhibitors of protein kinase B using fragment-based lead discovery. J. Med. Chem. 2007, 50, 2293–2296. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Tounge, B.A.; Bembenek, S.D. Ligand binding efficiency: Trends, Physical basis, and implications. J. Med. Chem. 2008, 51, 2432–2438. [Google Scholar] [CrossRef]
- Perola, E. An analysis of the binding efficiencies of drugs and their leads in successful drug discovery programs. J. Med. Chem. 2010, 53, 2986–2997. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Bembenek, S.D.; Tounge, B.A. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007, 17, 4258–4261. [Google Scholar] [CrossRef]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Clark, J.W.; Eder, J.P.; Ryan, D.; Lathia, C.; Lenz, H.J. Safety and pharmacokinetics of the dual action raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43–9006, in patients with advanced, refractory solid tumors. Clin. Cancer Res. 2005, 11, 5472–5480. [Google Scholar] [CrossRef]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Bruggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef]
- Smith, A.L.; DeMorin, F.F.; Paras, N.A.; Huang, Q.; Petkus, J.K.; Doherty, E.M.; Nixey, T.; Kim, J.L.; Whittington, D.A.; Epstein, L.F.; et al. Selective inhibitors of the mutant b-raf pathway: Discovery of a potent and orally bioavailable aminoisoquinoline. J. Med. Chem. 2009, 52, 6189–6192. [Google Scholar]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H.; et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar]
- Kawakami, Y.; Inoue, A.; Kawai, T.; Wakita, M.; Sugimoto, H.; Hopfinger, A.J. The rationale for E2020 as a potent acetylcholinesterase inhibitor. Bioorg. Med. Chem. 1996, 4, 1429–1446. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Hajduk, P.J. Fragment-based drug design: How big is too big? J. Med. Chem. 2006, 49, 6972–6976. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar]
- Alavi, A.; Hood, J.D.; Frausto, R.; Stupack, D.G.; Cheresh, D.A. Role of raf in vascular protection from distinct apoptotic stimuli. Science 2003, 301, 94–96. [Google Scholar]
- Golemovic, M.; Verstovsek, S.; Giles, F.; Cortes, J.; Manshouri, T.; Manley, P.W.; Mestan, J.; Dugan, M.; Alland, L.; Griffin, J.D.; et al. AMN107, a novel aminopyrimidine inhibitor of BCR-ABL, has in vitro activity against imatinib-resistant chronic myeloid leukemia. Clin. Cancer Res. 2005, 11, 4941–4947. [Google Scholar]
- Bartram, C.R.; de Klein, A.; Hagemeijer, A.; van, A.T.; Geurts, V.K.A.; Bootsma, D.; Grosveld, G.; Ferguson-Smith, M.A.; Davies, T.; Stone, M.; et al. Translocation of c-Abl oncogene correlates with the presence of a philadelphia chromosome in chronic myelocytic leukaemia. Nature 1983, 306, 277–280. [Google Scholar]
- Chan, L.C.; Karhi, K.K.; Rayter, S.I.; Heisterkamp, N.; Eridani, S.; Powle, R.; Lawler, S.D.; Groffen, J.; Foulkes, J.G.; Greaves, M.F.; et al. A Novel abl protein expressed in philadelphia chromosome positive acute lymphoblastic leukaemia. Nature 1987, 325, 635–637. [Google Scholar]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; de Klein, A.; Bartram, C.; Grosveld, G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984, 36, 93–99. [Google Scholar]
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990, 247, 1079–1082. [Google Scholar]
- Morphy, R. Selectively nonselective kinase inhibition: Striking the RIght balance. J. Med. Chem. 2009, 53, 1413–1437. [Google Scholar]
- Kantarjian, H.M.; Giles, F.; Gattermann, N.; Bhalla, K.; Alimena, G.; Palandri, F.; Ossenkoppele, G.J.; Nicolini, F.E.; O’Brien, S.G.; Litzow, M.; et al. Nilotinib (formerly AMN107), a highly selective bcr-abl tyrosine kinase inhibitor, is effective in patients with philadelphia chromosome–positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood 2007, 110, 3540–3546. [Google Scholar]
- Sekulic, A.; Haluska, P.J.; Miller, A.J.; Genebriera De Lamo, J.; Ejadi, S.; Pulido, J.S.; Salomao, D.R.; Thorland, E.C.; Vile, R.G.; Swanson, D.L.; et al. Malignant melanoma in the 21st century: The emerging molecular landscape. Mayo Clin. Proc. 2008, 83, 825–846. [Google Scholar]
- Soreq, H.; Seidman, S. Acetylcholinesterase—New roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar]
- Wild, R.; Pettit, T.; Burns, A. Cholinesterase inhibitors for dementia with lewy bodies. Cochrane Database Syst. Rev. 2003. [Google Scholar] [CrossRef]
- Kryger, G.; Harel, M.; Giles, K.; Toker, L.; Velan, B.; Lazar, A.; Kronman, C.; Barak, D.; Ariel, N.; Shafferman, A.; et al. Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr. D 2000, 56, 1385–1394. [Google Scholar]
- Molinspiration Cheminformatics. Collection of Substituents and Spacers Extracted from Bioactive Molecules. Available online: http://www.molinspiration.com/docu/fragments/index.html (accessed on 5 May, 2010).
- HyperChem(TM) Professional 7.51; Hypercube, Inc.: Gainesville, FL, USA, 2007.
- Polak, E. Computational Methods in Optimization; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Lindahl, E.; Hess, B.; Spoel, D.V.D. 0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration; D. Reidel Publishing Company: Dordrecht, The Netherlands, 1981. [Google Scholar]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aided Mol. Des. 1998, 12, 27–35. [Google Scholar]
- Cardozo, M.G.; Kawai, T.; Iimura, Y.; Sugimoto, H.; Yamanishi, Y.; Hopfinger, A.J. Conformational analyses and molecular-shape comparisons of a series of indanone-benzylpiperidine inhibitors of acetylcholinesterase. J. Med. Chem. 1992, 35, 590–601. [Google Scholar]
- Sample Availability: Not Available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Su, B.-H.; Huang, Y.-S.; Chang, C.-Y.; Tu, Y.-S.; Tseng, Y.J. Template-Based de Novo Design for Type II Kinase Inhibitors and Its Extended Application to Acetylcholinesterase Inhibitors. Molecules 2013, 18, 13487-13509. https://doi.org/10.3390/molecules181113487
Su B-H, Huang Y-S, Chang C-Y, Tu Y-S, Tseng YJ. Template-Based de Novo Design for Type II Kinase Inhibitors and Its Extended Application to Acetylcholinesterase Inhibitors. Molecules. 2013; 18(11):13487-13509. https://doi.org/10.3390/molecules181113487
Chicago/Turabian StyleSu, Bo-Han, Yi-Syuan Huang, Chia-Yun Chang, Yi-Shu Tu, and Yufeng J. Tseng. 2013. "Template-Based de Novo Design for Type II Kinase Inhibitors and Its Extended Application to Acetylcholinesterase Inhibitors" Molecules 18, no. 11: 13487-13509. https://doi.org/10.3390/molecules181113487