Pressure Dependent Product Formation in the Photochemically Initiated Allyl + Allyl Reaction

Abstract
:1. Introduction
2. Results and Discussion
Product Branching


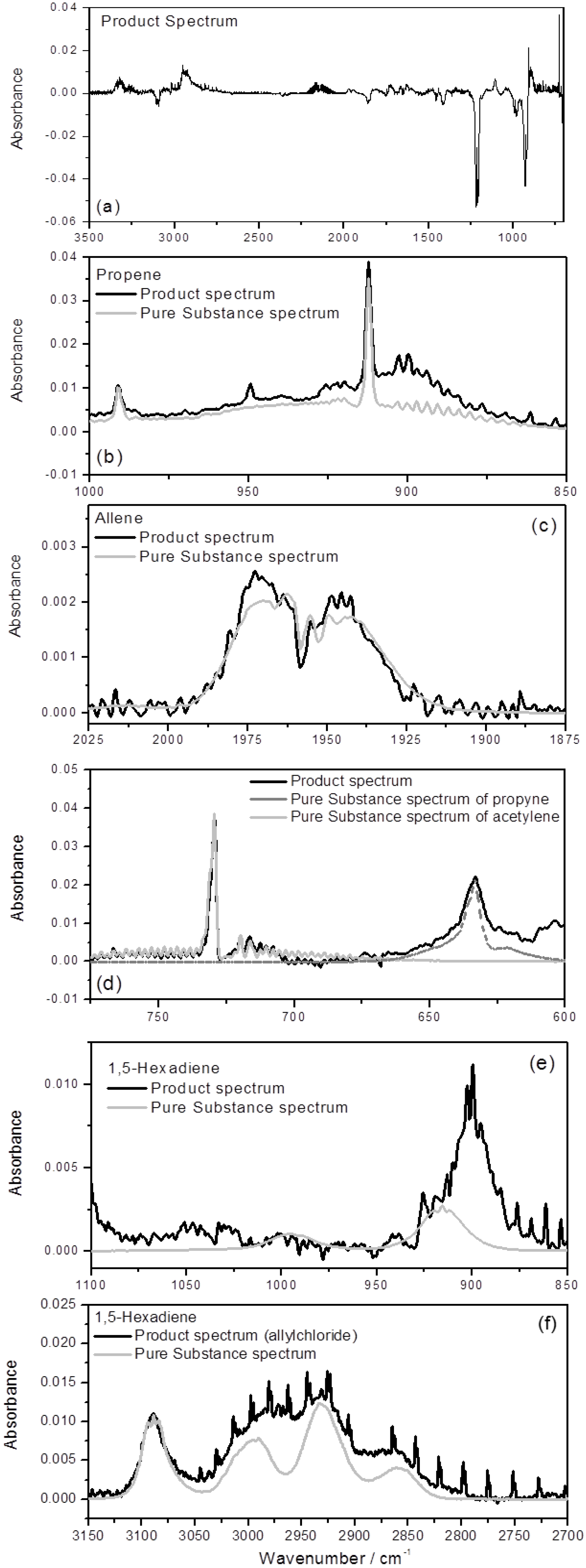{kind=link}
{kind=link}
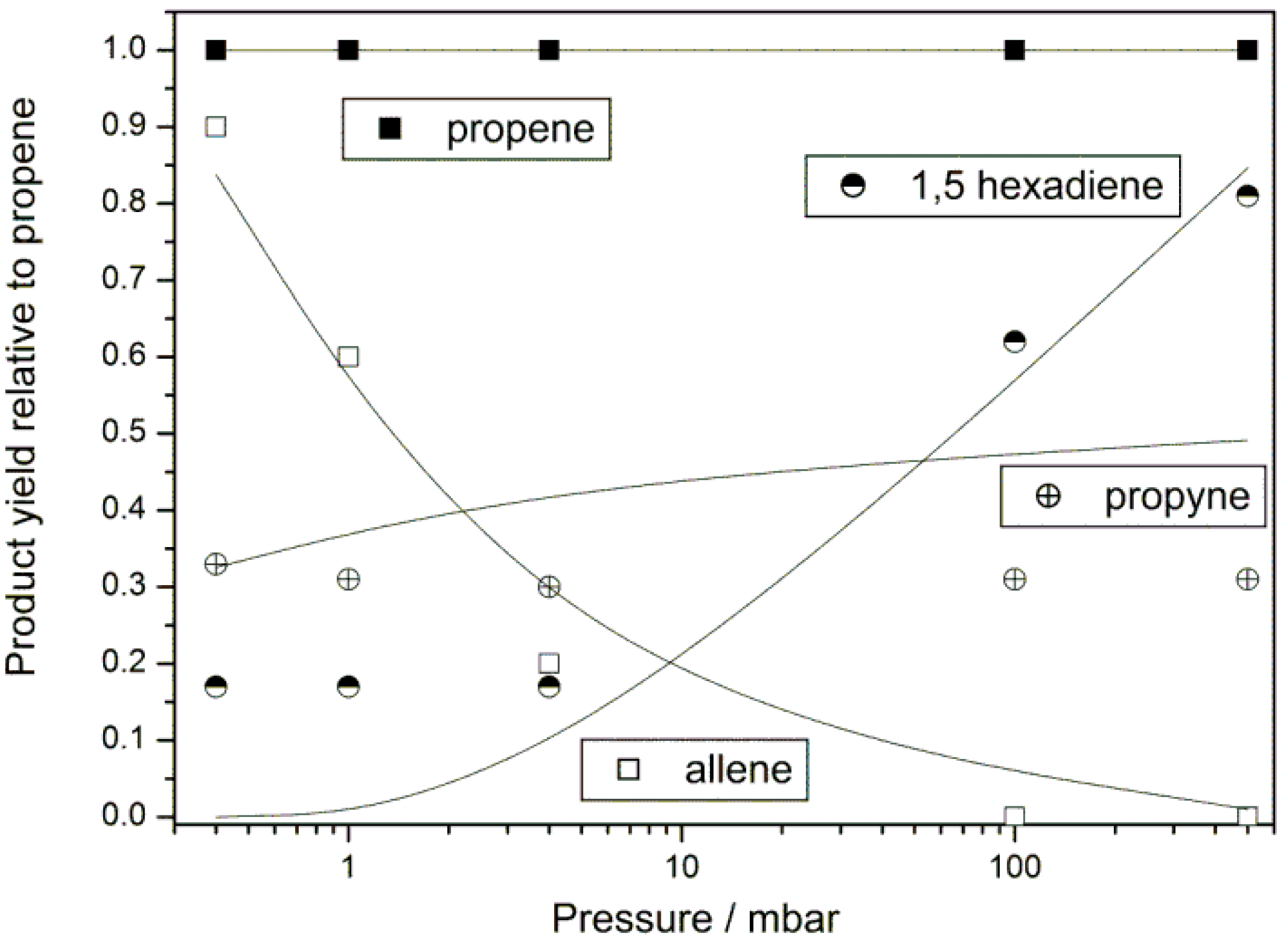{kind=link}
| Precursor Product | C3H5Br | C3H5Cl | 1,5-hexadiene |
|---|---|---|---|
| Propene | 30 ± 5% | 7 ± 2% | 80 ± 10% |
| Propyne | 10 ± 3% | 3 ± 1% | 24 ± 5% |
| Allene | 6 ± 2% | 6 ± l% | 25 ± 5% |
| Acetylene | 15 ± 3% | 4 ± 1% | 26 ± 4% |
| 1,5-Hexadiene | <5% | 14 ± 5% | — |


| No. | Reaction | A | n | Ea | Pressure [bar] or Ref. |
|---|---|---|---|---|---|
| 1 | C3H5*→ HCCCH3 + H | 2.0E02 | 0.00 | 0.00 | |
| 2 | C3H5 + H→C3H6 | 1.0E13 | 0.00 | 0.00 | |
| 3 | C6H10*→C3H5* + C3H5* | 1.0E05 | 0.00 | 0.00 | |
| 4 | C3H5* + C3H5*→C6H10* | 1.0E13 | 0.00 | 0.00 | |
| 5 | C3H5*→ C3H5 | 1.0E02 | 0.00 | 0.00 | 4.0E-4 |
| 4.0E02 | 0.00 | 0.00 | 4.0E-3 | ||
| 1.1E03 | 0.00 | 0.00 | 5.0E-1 | ||
| 6 | C3H5* + C3H5*→H + H + C6H8 | 2.0E11 | 0.00 | 0.00 | |
| 7 | C3H5 + C3H5→C6H10 | 1.6E13 | 0.00 | 0.00 | [26] |
| 8 | C3H5 + C3H5→H2CCCH2 + C3H6 | 1.5E11 | 0.00 | 0.00 | |
| 9 | C3H5* + C3H5*→H2CCCH2 + C3H6 | 1.5E12 | 0.00 | 0.00 | 4.0E-4 |
| 1.5E11 | 0.00 | 0.00 | 4.0E-3 | ||
| 1.0E06 | 0.00 | 0.00 | 5.0E-1 | ||
| 10 | C6H10 + H→C3H6 + C3H5 | 1.0E12 | 0.00 | 0.00 | 1/10 of [28] |
3. Experimental
Chemicals
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Warnatz, J.; Maas, U.; Dibble, R.W. Combustion; Springer: Heidelberg, Germany, 1997. [Google Scholar]
- Qin, Z.; Lissianski, V.; Yang, H.; Gardiner, W.C.; Davis, S.G.; Wang, H. Combustion chemistry of propane: A case study of detailed reaction mechanism optimization. Proc. Combust. Inst. 2000, 28, 1663–1669. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Mauß, F.; Zeuch, T. The Generation of a compact n-heptane toluene reaction mechanism using the Chemistry Guided Reduction (CGR) technique. Z. Phys. Chem. 2009, 222, 551–563. [Google Scholar] [CrossRef]
- Krasnopolsky, V.A. A photochemical model of Titan’s atmosphere and ionosphere. Icarus 2009, 201, 226–256. [Google Scholar] [CrossRef]
- Cathonnet, M.; Boettner, J.C.; James, H. Experimental study and numerical modeling of high temperature oxidation of propane and n-butane. Proc. Combust. Inst. 1981, 18, 903–913. [Google Scholar]
- Davis, S.G.; Law, C.K.; Wang, H. Propene pyrolysis and oxidation kinetics in a flow reactor and laminar flames. Combust. Flame 1999, 119, 375–399. [Google Scholar] [CrossRef]
- Simmie, J.M. Detailed chemical kinetic models for the combustion of hydrocarbon fuels. Prog. Energy Combust. Sci. 2003, 29, 599–634. [Google Scholar] [CrossRef]
- Atakan, B.; Hartlieb, A.T.; Brand, J.; Kohse-Höinghaus, K. An experimental investigation of premixed fuel-rich low pressure propene/oxygen/argon flames by laser spectroscopy and molecular-beam mass spectrometry. Proc. Comb. Inst. 1998, 27, 435–444. [Google Scholar]
- Hoyermann, K.; Mauß, F.; Zeuch, T. A detailed chemical reaction mechanism for the oxidation of hydrocarbons and its application to the analysis of benzene formation in fuel-rich premixed laminar acetylene and propene flames. Phys. Chem. Chem. Phys. 2004, 6, 3824–3835. [Google Scholar] [CrossRef]
- Hoyermann, K.; Nacke, F.; Nothdurft, J.; Olzmann, M.; Wehmeyer, J.; Zeuch, T. The reaction of allyl radicals with oxygen atoms - rate coefficient and product branching. Proc. Comb. Inst. 2009, 32, 157–164. [Google Scholar] [CrossRef]
- Hébrard, E.; Dobrijevic, M.; Loison, J.C.; Bergeat, A.; Hickson, K.M.; Caralp, F. Photochemistry of C3Hp hydrocarbons in Titan’s stratosphere revisited. Astron. Astrophys. 2013, 552, A132. [Google Scholar] [CrossRef]
- Zeuch, T.; Moréac, G.; Ahmed, S.S.; Mauß, F. A comprehensive skeletal mechanism for the oxidation of n-heptane generated by chemistry-guided reduction. Combust. Flame 2008, 155, 651–674. [Google Scholar] [CrossRef]
- Wang, H. Formation of nascent soot and other condensed-phase materials in flames. Proc. Comb. Inst. 2011, 33, 41–67. [Google Scholar] [CrossRef]
- Imanaka, H.; Khare, B.N.; Elsila, J.E.; Bakes, E.L.O.; McKay, C.P.; Cruikshank, D.P.; Sugita, S.; Matsui, T.; Zare, R.N. Laboratory experiments of titan tholin formed in cold plasma at various pressures: Implications for nitrogen-containing polycyclic aromatic compounds in Titan haze. Icarus 2004, 168, 344–366. [Google Scholar] [CrossRef]
- Wolf, J.L.; Richters, S.; Pecher, J.; Zeuch, T. Pressure dependent mechanistic branching in the formation pathways of secondary organic aerosol from cyclic-alkene gas-phase ozonolysis. Phys. Chem. Chem. Phys. 2011, 13, 10952–10964. [Google Scholar] [CrossRef]
- Leonori, F.; Balucani, N.; Capozza, G.; Segoloni, E.; Stranges, D.; Casavecchia, P. Crossed beam studies of radical–radical reactions: O(3P) + C3H5 (allyl). Phys. Chem. Chem. Phys. 2007, 9, 1307–1311. [Google Scholar] [CrossRef]
- FitzPatrick, B.L.; Lau, K.-C.; Butler, L.J.; Lee, S.-H.; Lin, J.J.-M. Investigation of the O + allyl addition/elimination reaction pathways from the OCH2CHCH2 radical intermediate. J. Chem. Phys. 2008, 129, 084301. [Google Scholar] [CrossRef]
- Hoyermann, K.; Maarfeld, S.; Nacke, F.; Nothdurft, J.; Olzmann, M.; Wehmeyer, J.; Welz, O.; Zeuch, T. Rate coefficients for cycloalkyl + O reactions and product branching in the decomposition of chemically activated cycloalkoxy radicals: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2010, 12, 8954–8968. [Google Scholar]
- Hoyermann, K.; Olzmann, M.; Welz, O.; Zeuch, T. The reaction of iso-propyl radicals with oxygen atoms: Rate coefficient, product branching, and relevance for combustion modeling. Proc. Comb. Inst. 2011, 33, 283–291. [Google Scholar] [CrossRef]
- Ismail, H.; Abel, P.R.; Green, W.H.; Fahr, A.; Jusinski, L.E.; Knepp, A.M.; Zádor, J.; Meloni, G.; Selby, T.M.; Osborn, D.L.; et al. Temperature-Dependent kinetics of the vinyl radical (C2H3) self-reaction. J. Phys. Chem. A 2009, 113, 1278–1286. [Google Scholar] [CrossRef]
- Tulloch, J.M.; Macpherson, M.T.; Morgan, C.A.; Pilling, M.J. Flash photolysis studies of free-radical reactions: C3H5 + C3H5 (293–691 K) and C3H5 + NO (295–400 K). J. Phys. Chem. 1982, 86, 3812–3819. [Google Scholar] [CrossRef]
- Boyd, A.A.; Nozière, B.; Lesclaux, R. Kinetics and thermochemistry of the reversible combination reactions of the allyl and benzyl radicals with NO. J. Phys. Chem. 1995, 99, 10815–10823. [Google Scholar]
- Matsugi, A.; Suma, K.; Miyoshi, A. Kinetics and Mechanisms of the Allyl plus Allyl and Allyl plus Propargyl Recombination Reactions. J. Phys. Chem. A 2011, 115, 7610–7624. [Google Scholar] [CrossRef]
- Georgievskii, Y.; Miller, J.A.; Klippenstein, S.J. Association rate constants for reactions between resonance-stabilized radicals: C3H3 + C3H3, C3H3 + C3H5, and C3H5 + C3H5. Phys. Chem. Chem. Phys. 2007, 9, 4259–4268. [Google Scholar] [CrossRef]
- James, D.G.L.; Kambanis, S.M. Pyrolysis of diallyl oxalate vapour. Part 1. Kinetics of the unsensitized decomposition. Trans. Faraday Soc. 1969, 65, 1350–1356. [Google Scholar] [CrossRef]
- Selby, T.M.; Meloni, G.; Goulay, F.; Leone, S.R.; Fahr, A.; Taatjes, C.A.; Osborn, D.L. Synchrotron photoionization mass spectrometry measurements of kinetics and product formation in the allyl radical (H2CCHCH2) self-reaction. J. Phys. Chem. A 2008, 112, 9366–9373. [Google Scholar] [CrossRef]
- Lynch, P.T.; Annesley, C.J.; Aul, C.J.; Yang, X.; Tranter, R.S. Recombination of allyl radicals in the high temperature fall-off regime. J. Phys. Chem. A 2003, 117, 4750–4761. [Google Scholar]
- Fridlyand, A.; Lynch, P.T.; Tranter, R.S.; Brezinsky, K. Single pulse shock tube study of allyl radical recombination. J. Phys. Chem. A 2013, 117, 4762–4776. [Google Scholar] [CrossRef]
- Zhang, H.R.; Eddings, E.G.; Sarofim, A.F.; Westbrook, C.K. Fuel dependence of benzene pathways. Proc. Comb. Inst. 2009, 32, 377–385. [Google Scholar] [CrossRef]
- Simon, F.G.; Heydtmann, H. Bestimmung der Geschwindigkeitskonstanten der Reaktion von1CH2 mit Fluor- und Chlorethen. Ber. Bunsenges. Phys. Chem. 1986, 90, 543–545. [Google Scholar] [CrossRef]
- Taatjes, C.A.; Hansen, N.; Osborn, D.L.; Kohse-Höinghaus, K.; Cool, T.A.; Westmoreland, P.R. “Imaging” combustion chemistry via multiplexed synchrotron-photoionization mass spectrometry. Phys. Chem. Chem. Phys. 2008, 10, 20–34. [Google Scholar] [CrossRef]
- Wallington, T.J.; Skewes, L.M.; Siegl, W.O. Kinetics of the gas phase reaction of chlorine atoms with a series of alkenes, alkynes and aromatic species at 295 K. J. Photochem. Photobiol. 1988, 45, 167–175. [Google Scholar] [CrossRef]
- Wallington, T.J.; Andino, J.M.; Ball, J.C.; Japar, S.M. Fourier transform infrared studies of the reaction of Cl atoms with PAN, PPN, CH3OOH, HCOOH, CH3COCH3 and CH3COC2H5 at 295 ± 2 K. J. Atmos. Chem. 1990, 10, 301–313. [Google Scholar] [CrossRef]
- Grosjean, D.; Williams, E.L., II. Environmental persistence of organic compounds estimated from structure-reactivity and linear free-energy relationships. Unsaturated aliphatics. Atmos. Environ. Part A 1992, 26, 1395–1405. [Google Scholar] [CrossRef]
- Wakamatsu, H.; Hidaka, Y. Shock-Tube and modeling study of chloroethane pyrolysis and oxidation. Int. J. Chem. Kinet. 2008, 40, 320–339. [Google Scholar] [CrossRef]
- Wallington, J.; Siegl, W.O.; Skewes, L.M.; Japar, S.M. A relative rate study of the reaction of bromine atoms with a variety of organic compounds at 295 K. Int. J. Chem. Kinet. 1989, 21, 1069–1076. [Google Scholar] [CrossRef]
- Barnes, I.; Bastian, V.; Becker, K.H.; Overath, R.; Tong, Z. Rate constants for the reactions of Br atoms with a series of alkanes, alkenes, and alkynes in the presence of O2. Int. J. Chem. Kinet. 1989, 21, 499–517. [Google Scholar] [CrossRef]
- Bierbach, A.; Barnes, I.; Becker, K.H. Rate coefficients for the gas-phase reactions of bromine radicals with a series of alkenes, dienes, and aromatic hydrocarbons at 298 ± 2K. Int. J. Chem. Kinet. 1996, 28, 565–577. [Google Scholar] [CrossRef]
- Bedjanian, Y.; Poulet, G.; le Bras, G. Low-Pressure Study of the Reactions of Br Atoms with Alkenes. 1. Reaction with Propene. J. Phys. Chem. A 1998, 102, 5867–5875. [Google Scholar] [CrossRef]
- McMillen, D.F.; Golden, D.M. Hydrocarbon bond dissociation energies. Ann. Rev. Phys. Chem. 1982, 33, 493–532. [Google Scholar] [CrossRef]
- Mueller, J.A.; Miller, J.L.; Butler, L.J.; Qi, F.; Sorkhabi, O.; Suits, A.G. Dependence of the H + Allene/H + Propyne product branching from the unimolecular dissociation of 2-Propenyl radicals. J. Phys. Chem. A 2000, 104, 11261–11264. [Google Scholar] [CrossRef]
- Knyazev, V.D.; Stoliarov, S.I.; Slagle, I.R. Kinetics of the reaction of vinyl radicals with acetylene. Proc. Combust. Inst. 1996, 26, 513–519. [Google Scholar]
- Somnitz, H.; Ufer, T.; Zellner, R. Acetone photolysis at 248 nm revisited: Pressure dependence of the CO and CO2 quantum yields. Phys. Chem. Chem. Phys. 2009, 11, 8522–8531. [Google Scholar] [CrossRef]
- Imanaka, H.; Smith, M.A. Formation of nitrogenated organic aerosols in the Titan upper atmosphere. Proc. Natl. Acad. Sci. USA 2010, 107, 12423–12428. [Google Scholar] [CrossRef]
- Sagan, C.; Khare, B.N. Tholins: Organic chemistry of interstellar grains and gas. Nature 1979, 277, 102–107. [Google Scholar] [CrossRef]
- Raulin, F.; Brassé, C.; Poch, O.; Coll, P. Prebiotic-like chemistry on Titan. Chem. Soc. Rev. 2012, 41, 5380–5393. [Google Scholar] [CrossRef]
- Hörst, S.M.; Yelle, R.V.; Buch, A.; Carrasco, N.; Cernogora, G.; Dutuit, O.; Quirico, E.; Sciamma-O’Brien, E.; Smith, M.A.; Somogyi, Á.; et al. Formation of amino acids and nucleotide bases in a Titan atmosphere simulation experiment. Astrobiology 2012, 12, 809–817. [Google Scholar] [CrossRef]
- Hoyermann, K.; Nothdurft, J.; Olzmann, M.; Wehmeyer, J.; Zeuch, T. Formation and decomposition of chemically activated cyclopentoxy radicals from the c-C5H9 + O reaction. J. Phys. Chem. A 2006, 110, 3165–3173. [Google Scholar] [CrossRef]
- Glowacki, D.R.; Lockhart, J.; Blitz, M.A.; Klippenstein, S.J.; Pilling, M.J.; Robertson, S.H.; Seakins, P.W. Interception of excited vibrational quantum states by O2 in atmospheric association reactions. Science 2012, 337, 1066–1069. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Seidel, L.; Hoyermann, K.; Mauß, F.; Nothdurft, J.; Zeuch, T. Pressure Dependent Product Formation in the Photochemically Initiated Allyl + Allyl Reaction. Molecules 2013, 18, 13608-13622. https://doi.org/10.3390/molecules181113608
Seidel L, Hoyermann K, Mauß F, Nothdurft J, Zeuch T. Pressure Dependent Product Formation in the Photochemically Initiated Allyl + Allyl Reaction. Molecules. 2013; 18(11):13608-13622. https://doi.org/10.3390/molecules181113608
Chicago/Turabian StyleSeidel, Lars, Karlheinz Hoyermann, Fabian Mauß, Jörg Nothdurft, and Thomas Zeuch. 2013. "Pressure Dependent Product Formation in the Photochemically Initiated Allyl + Allyl Reaction" Molecules 18, no. 11: 13608-13622. https://doi.org/10.3390/molecules181113608