Deracemization of Axially Chiral Nicotinamides by Dynamic Salt Formation with Enantiopure Dibenzoyltartaric Acid (DBTA)

Abstract
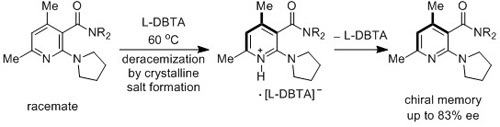
:
1. Introduction
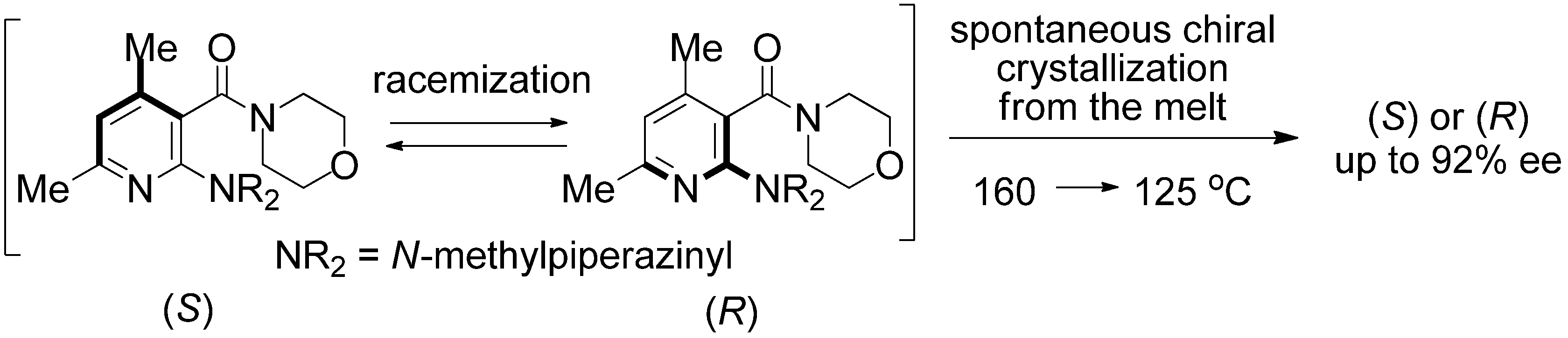
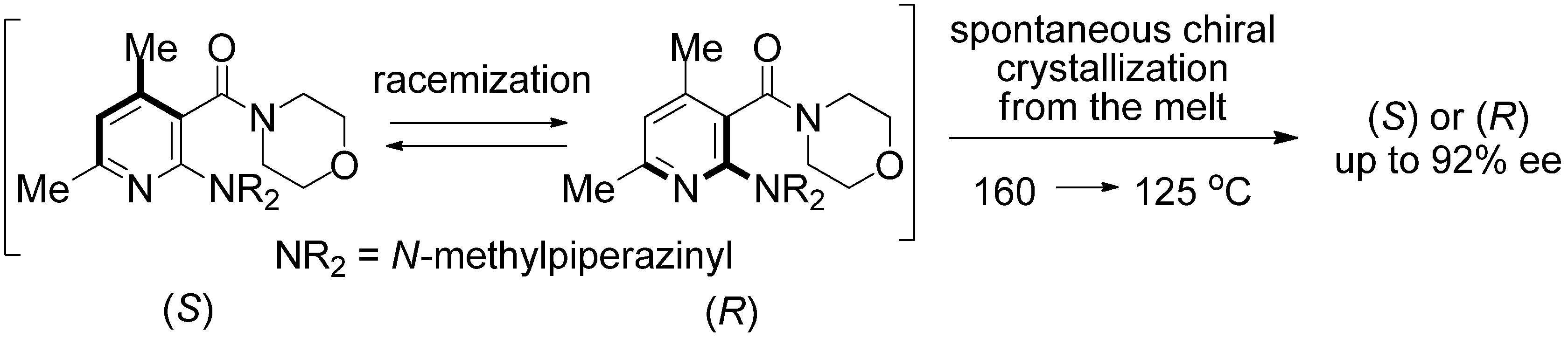
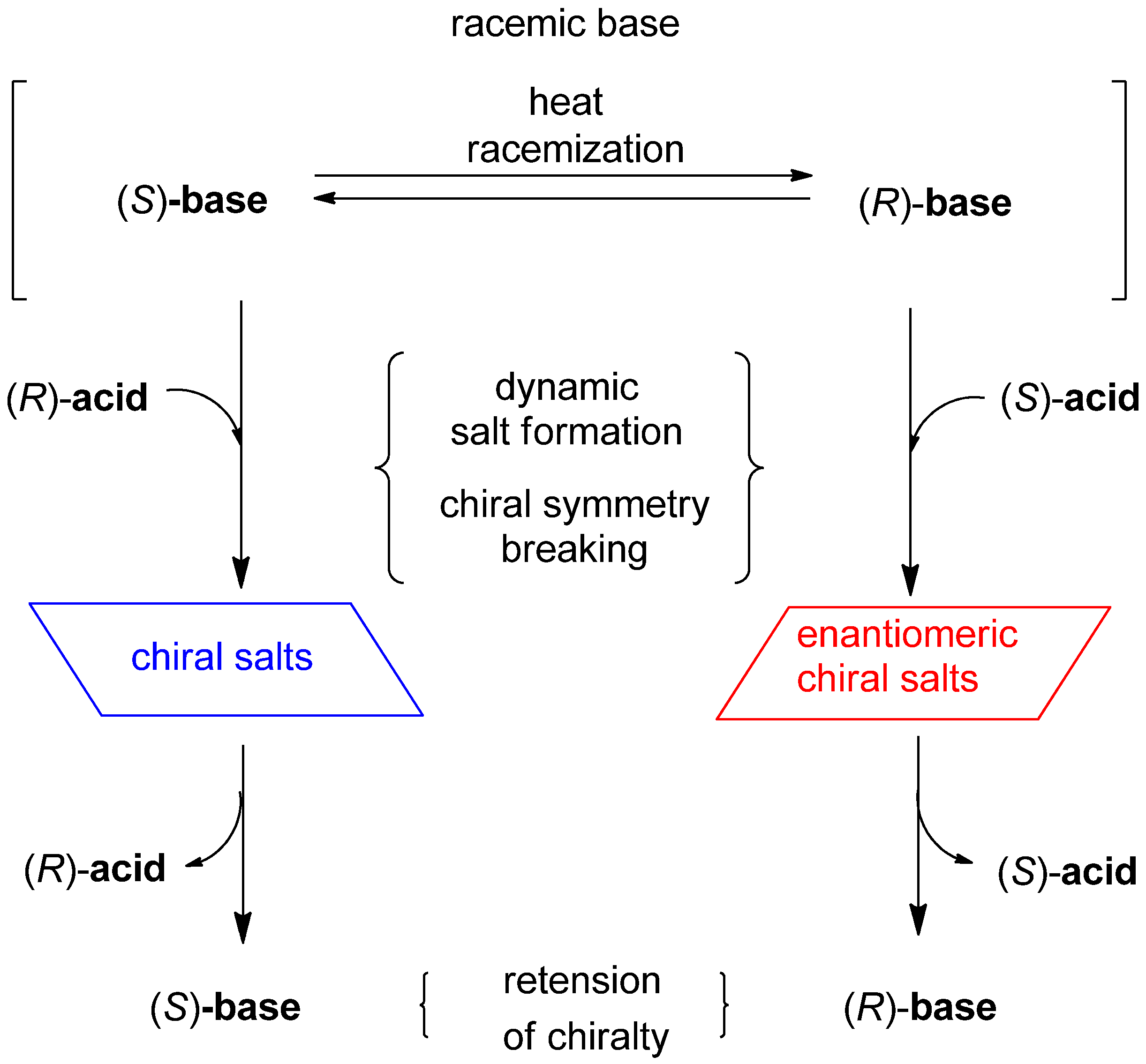
2. Results and Discussion
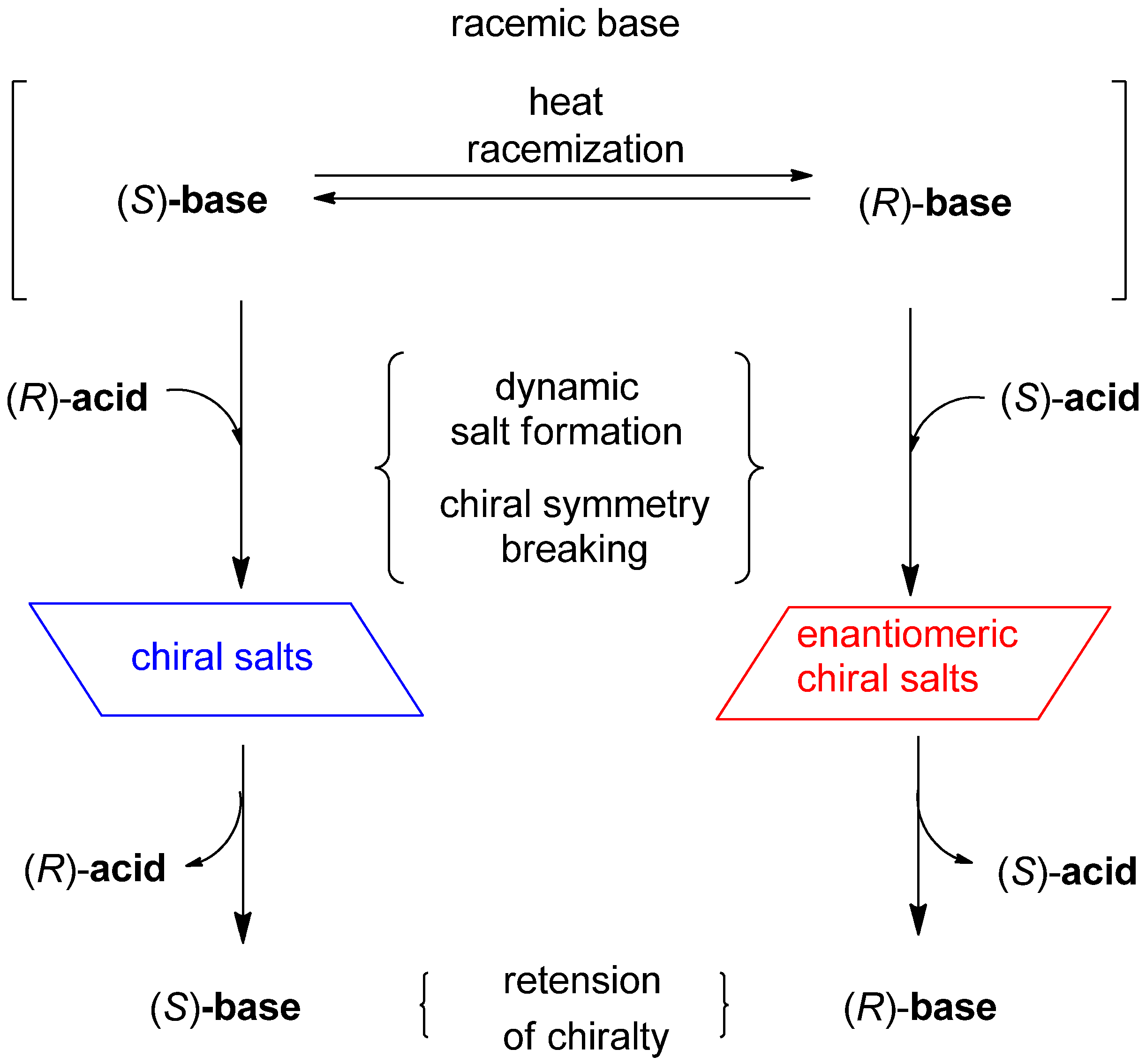
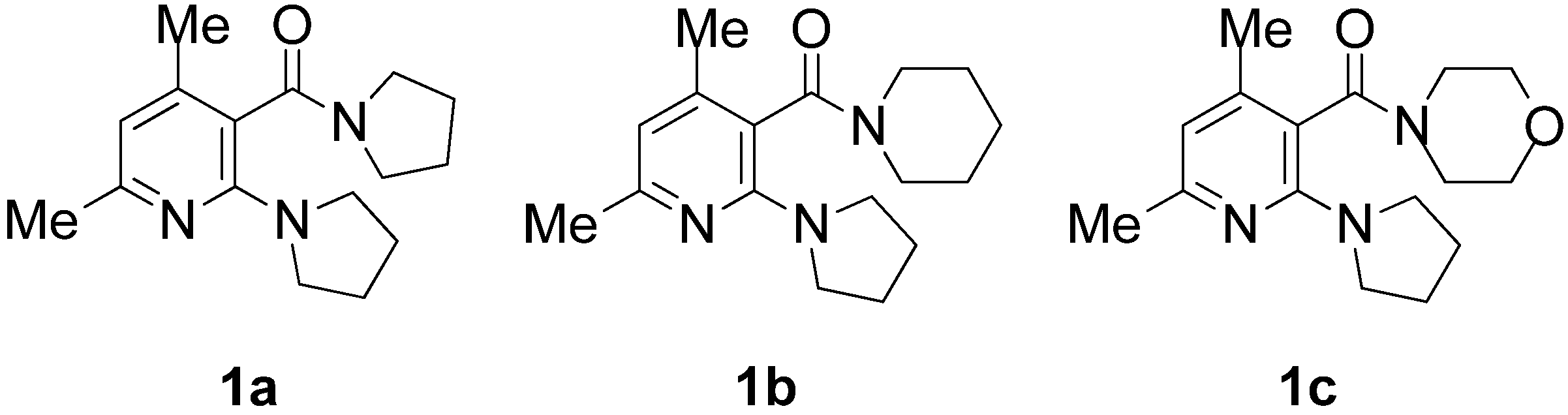
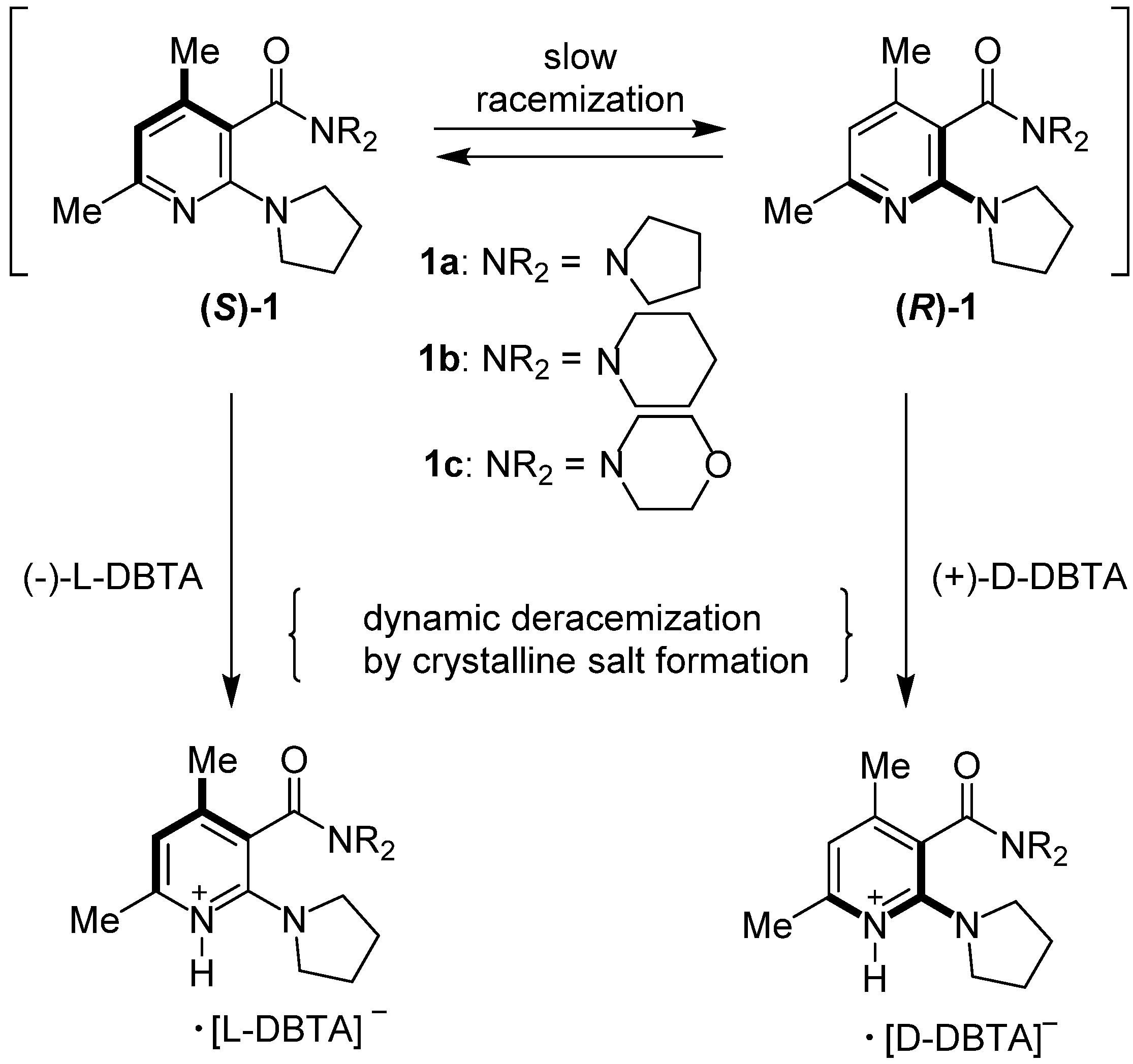

{kind=link}
{kind=link}
{kind=link}
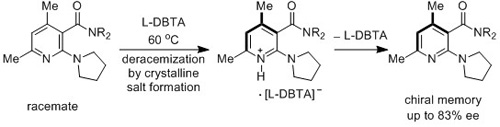
{kind=link}
{kind=link}
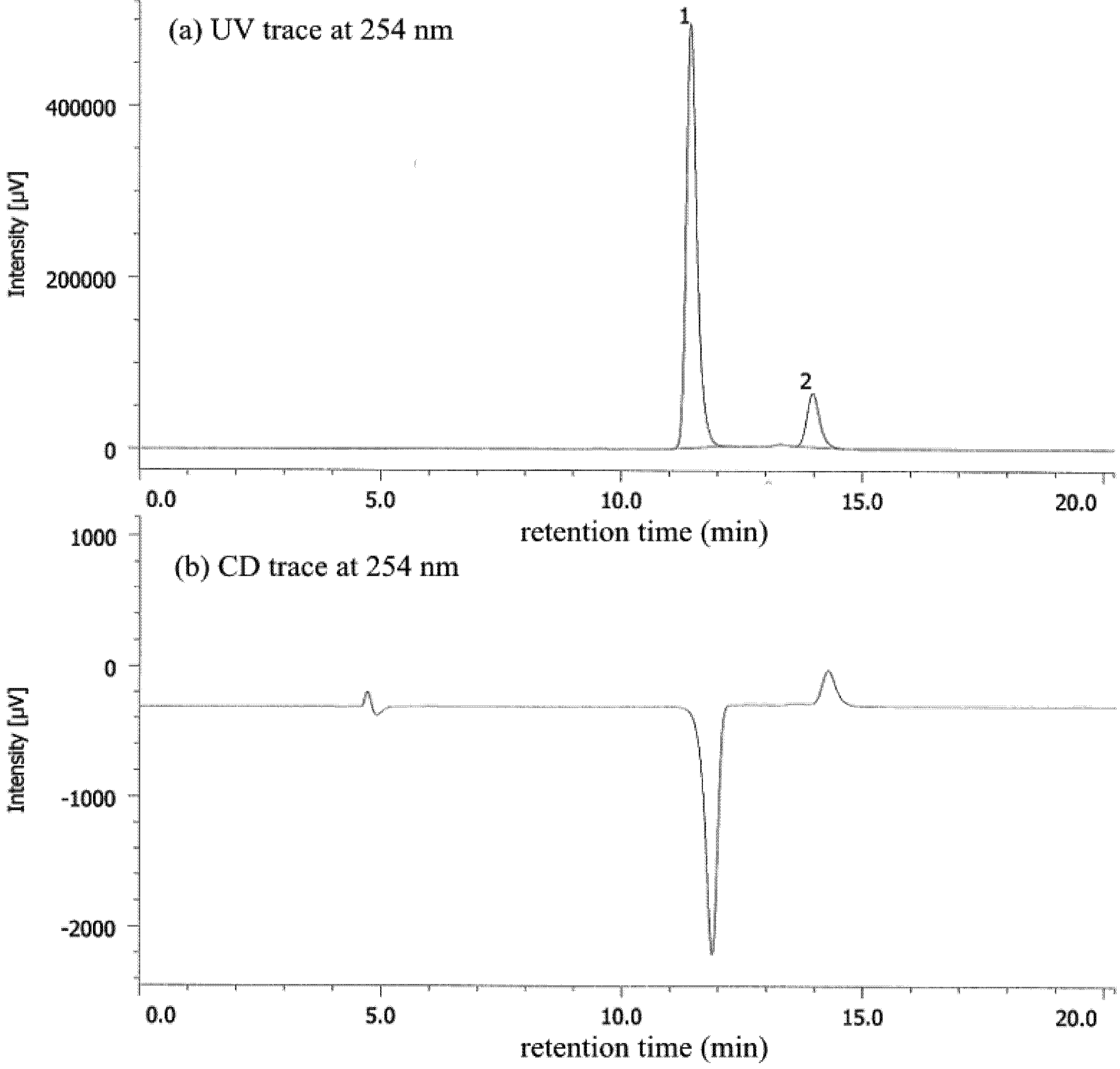
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
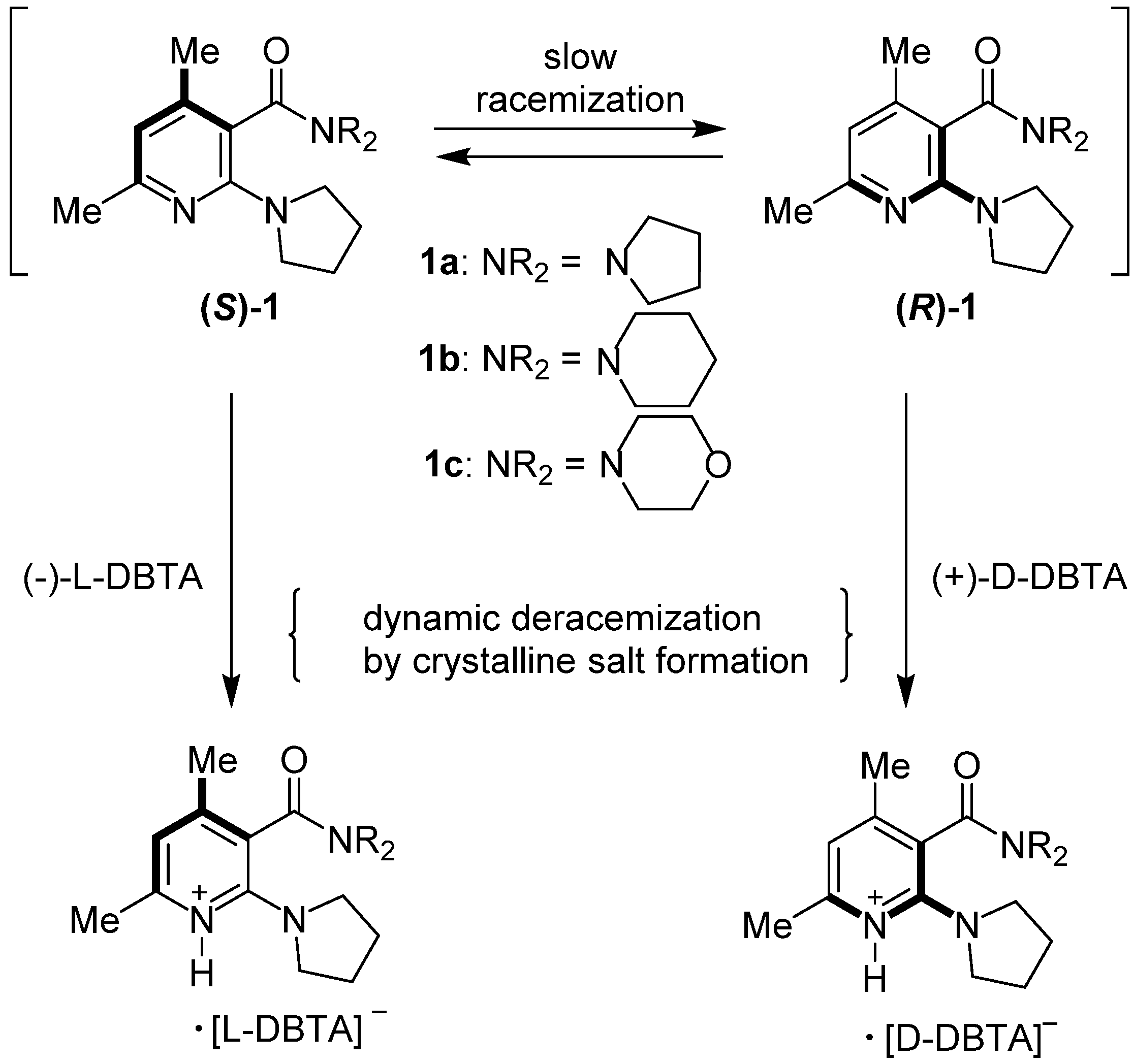{kind=link}
| Entry | Amide | ee (%) a | Abs. conf b |
|---|---|---|---|
| 1 c | 1a | 0–5 | - |
| 2 d | 1a | 60–67 | (−) f |
| 3 c | 1b | 0–6 | - |
| 4 d | 1b | 75–83 | (−)-( S) |
| 5 e | 1b | 76–82 | (+)-( R) |
| 6 c | 1c | 0–4 | - |
| 7 d | 1c | 51–60 | (−)-( S) |
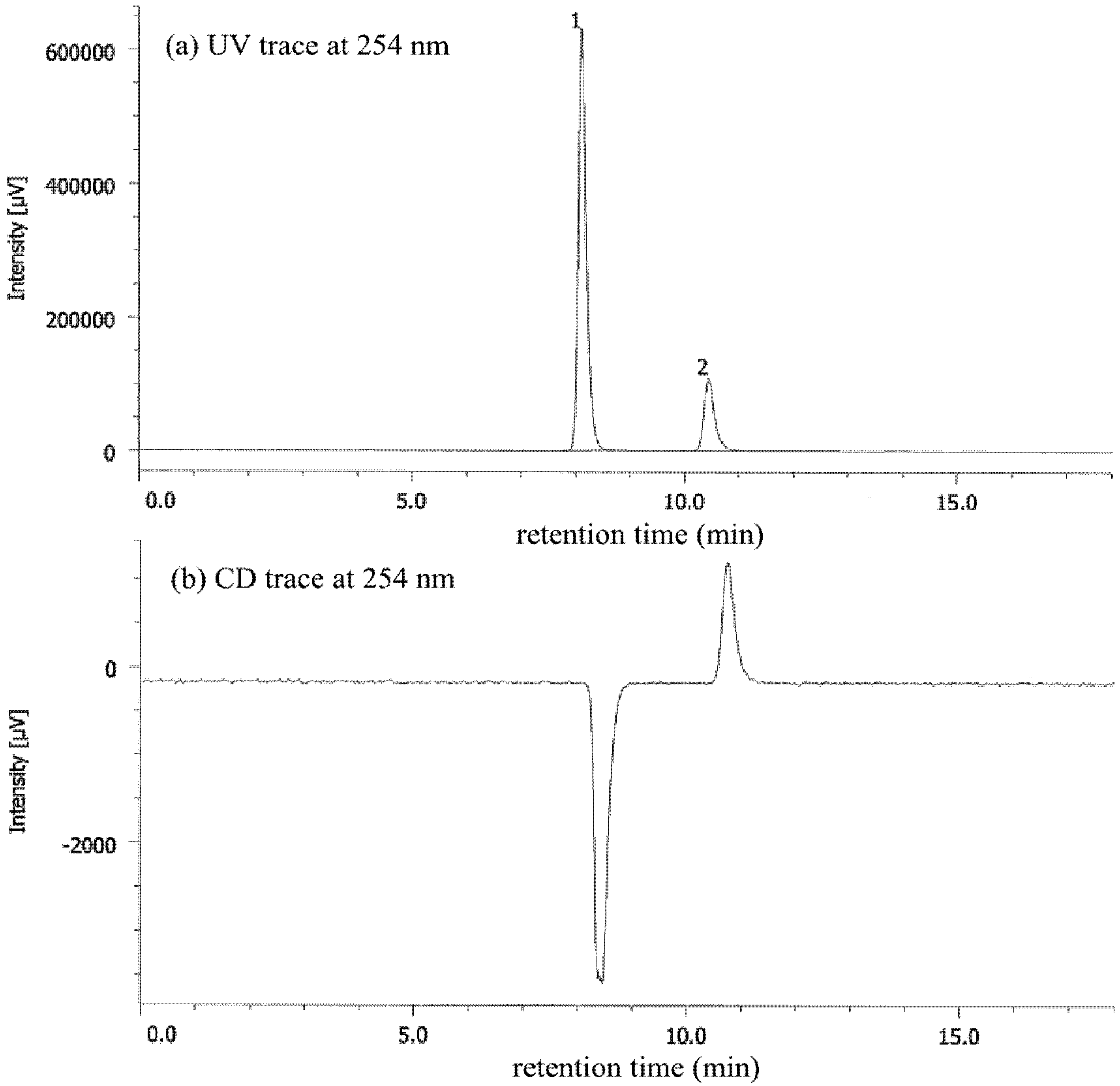
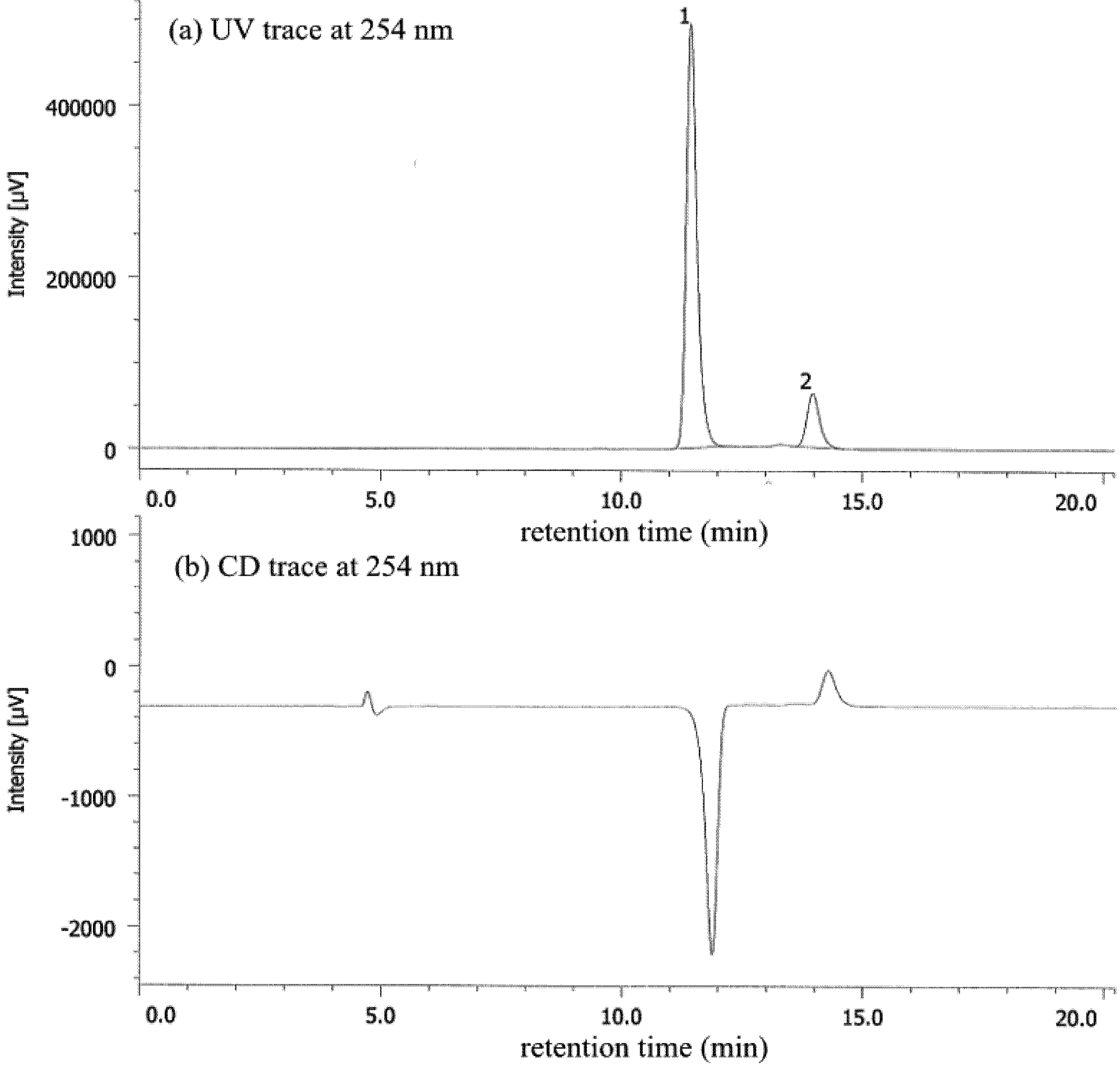
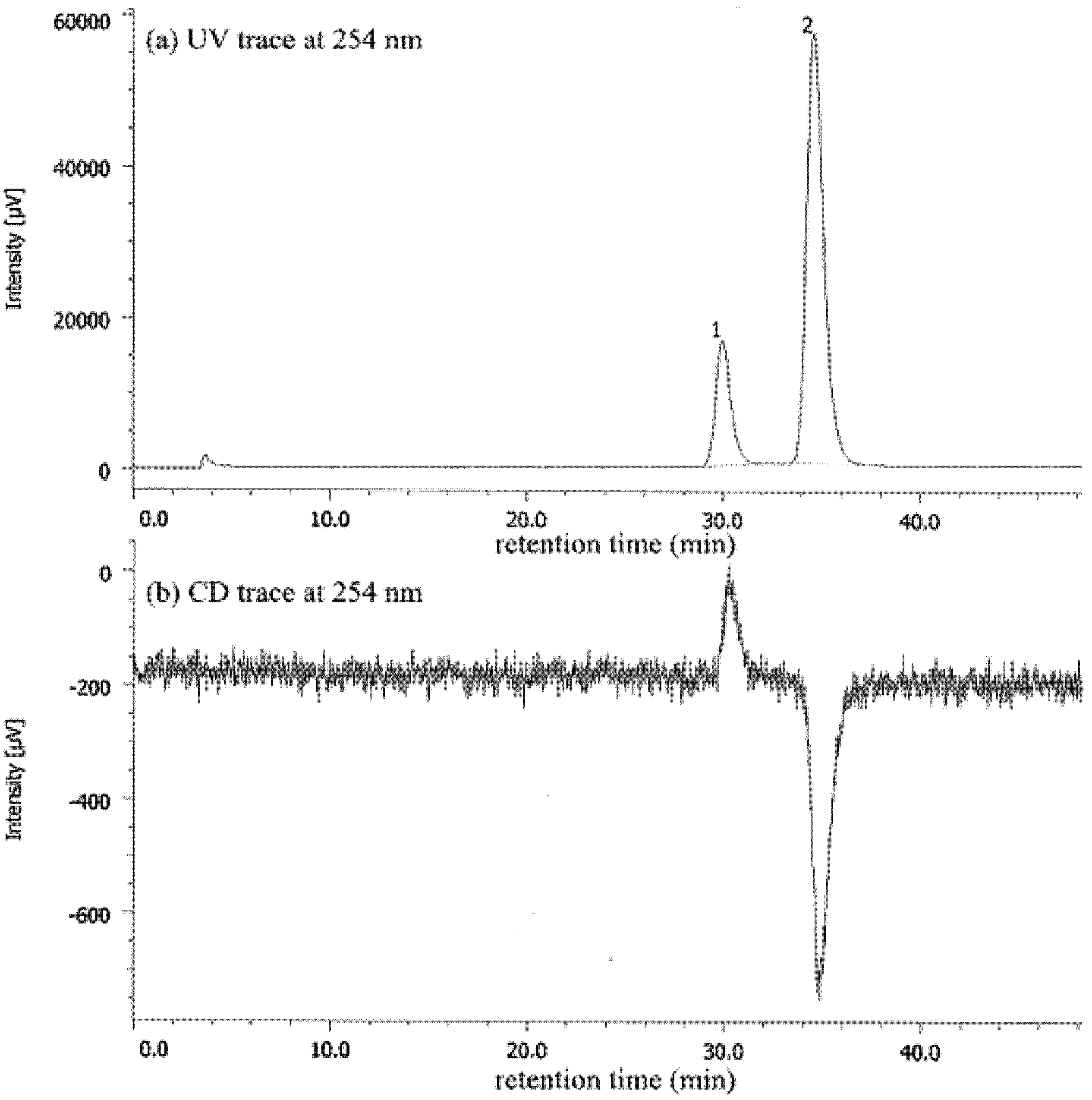


| Entry | Nicotinamide | Solvent | t1/2 b | krac c | ΔG‡ d |
|---|---|---|---|---|---|
| 1 | 1a | CHCl3 | 77.6 | 1.24 × 10−6 | 25.1 |
| 2 | 1a | MeCN | 56.9 | 1.69 × 10−6 | 24.9 |
| 3 | 1a e | MeOH | 27.7 | 3.47 × 10−6 | 25.3 |
| 4 | 1b | CHCl3 | 4.7 | 2.05 × 10−5 | 23.4 |
| 5 | 1b | MeCN | 6.2 | 1.56 × 10−5 | 23.6 |
| 6 | 1b | MeOH | 12.3 | 7.80 × 10−5 | 24.0 |
| 7 | 1b/DBTA f | MeOH | 20.2 | 4.76 × 10−6 | 24.2 |
| 8 | 1c | CHCl3 | 2.5 | 3.94 × 10−5 | 23.1 |
| 9 | 1c | MeCN | 2.6 | 3.71 × 10−5 | 23.1 |
| 10 | 1c | MeOH | 7.2 | 1.34 × 10−5 | 23.7 |
| 11 | 1c/DBTA f | MeOH | 9.4 | 3.06 × 10−5 | 23.2 |
3. Experimental
3.1. General
3.2. Preparation of Nicotinamides 1a–c
3.3. Deracemization of Nicotinamides 1a–c by Crystalline Salt Formation
3.4. Determination of the ee Value of 1 after Removing Chiral Acid
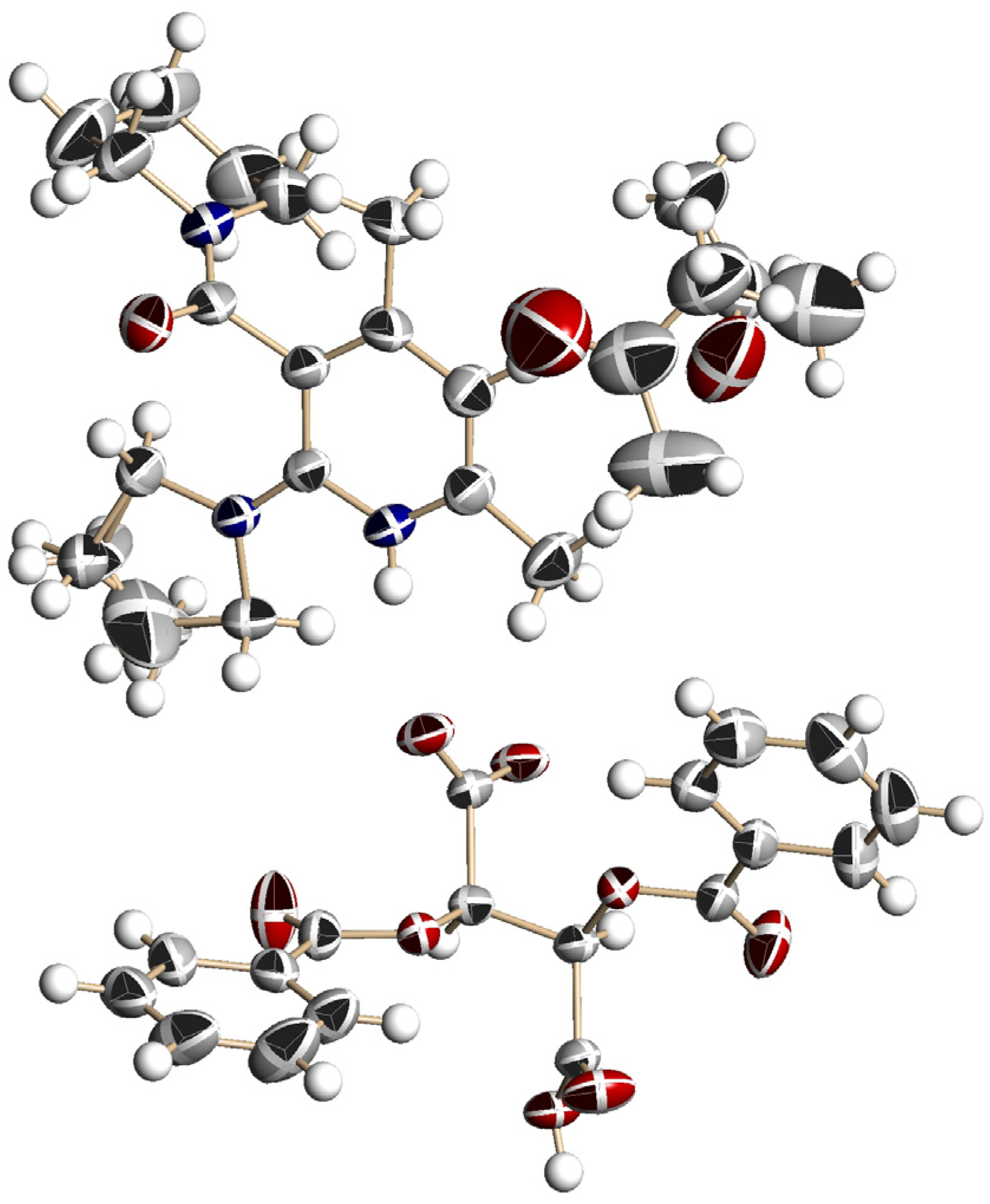
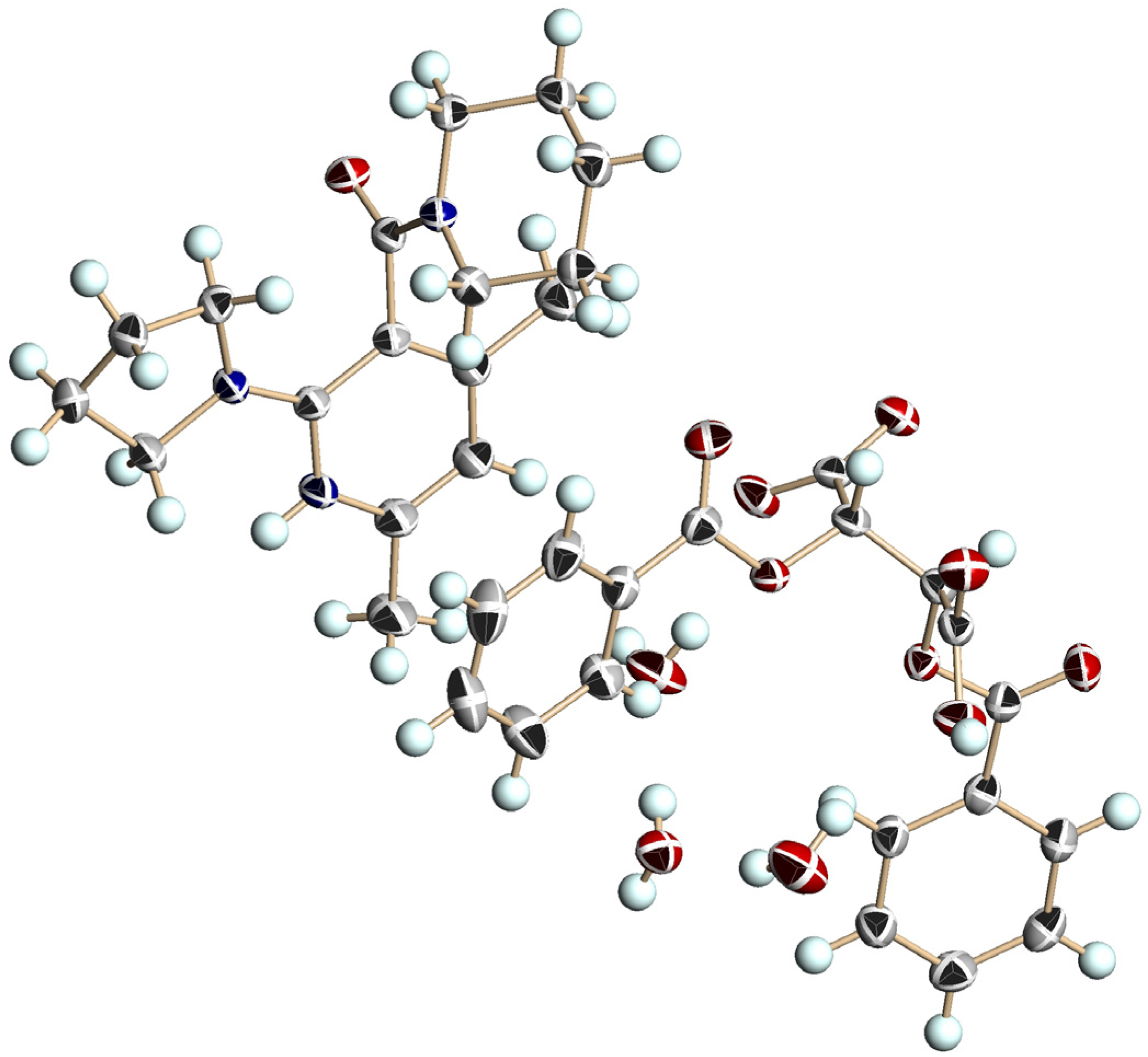
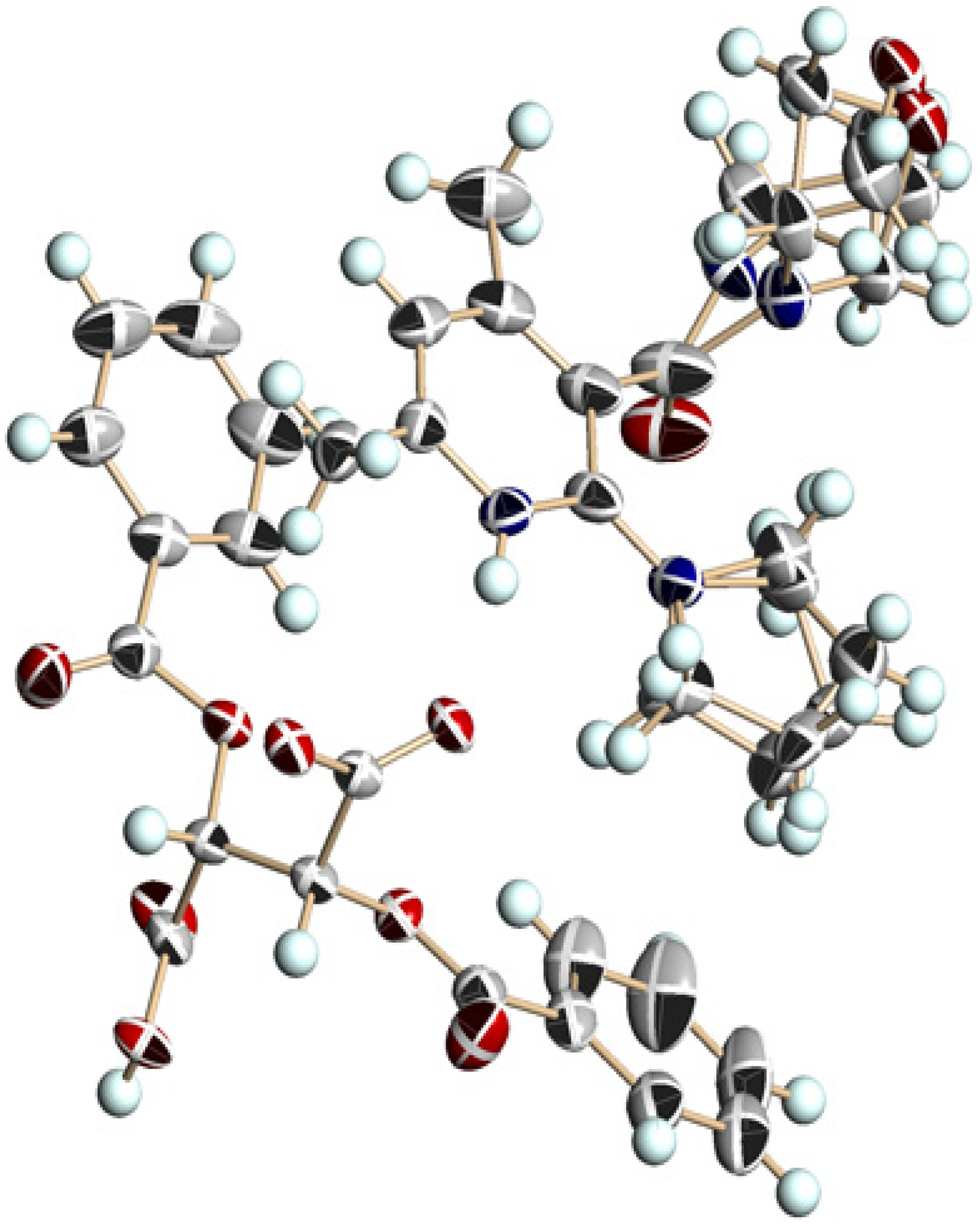
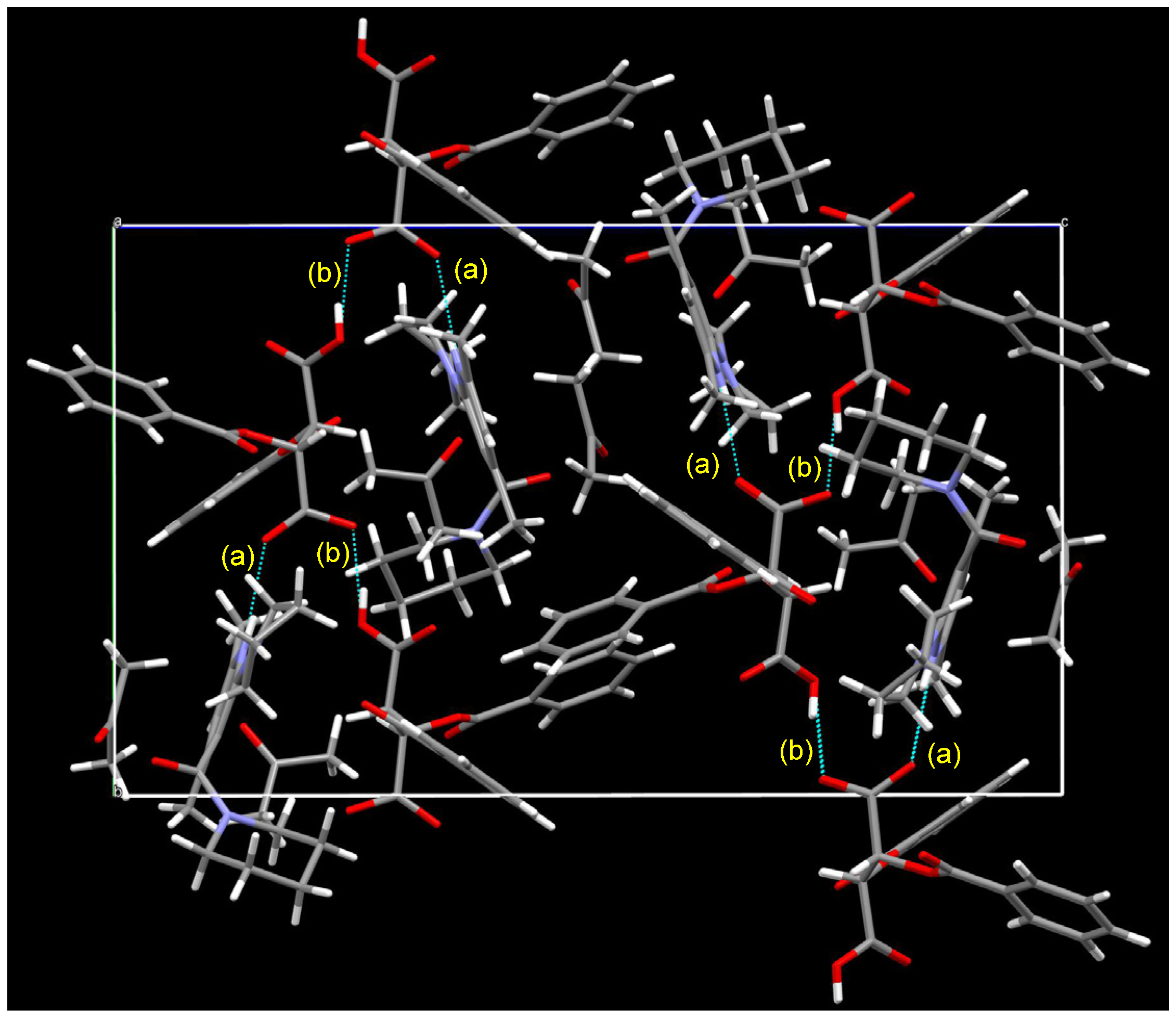
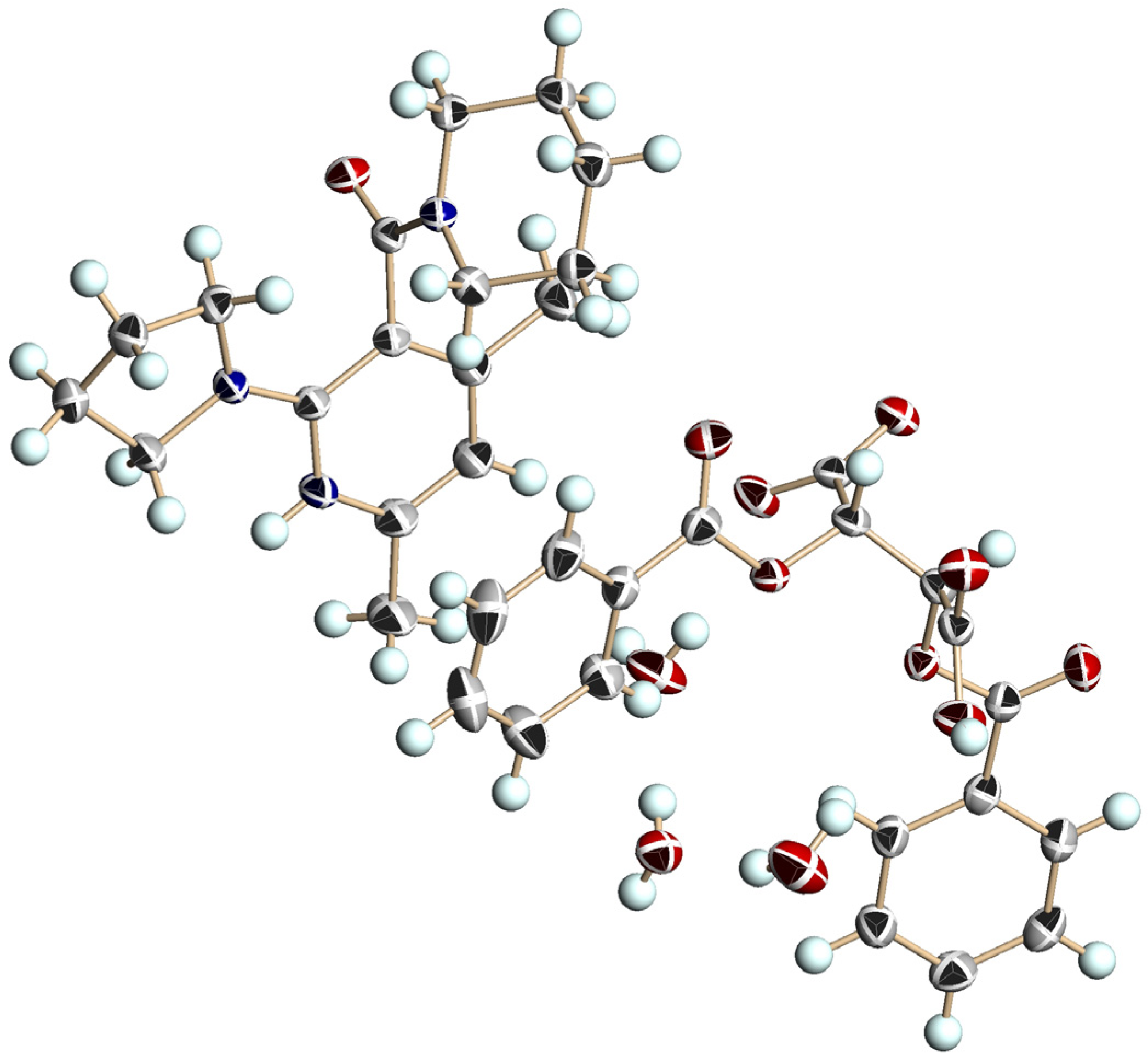
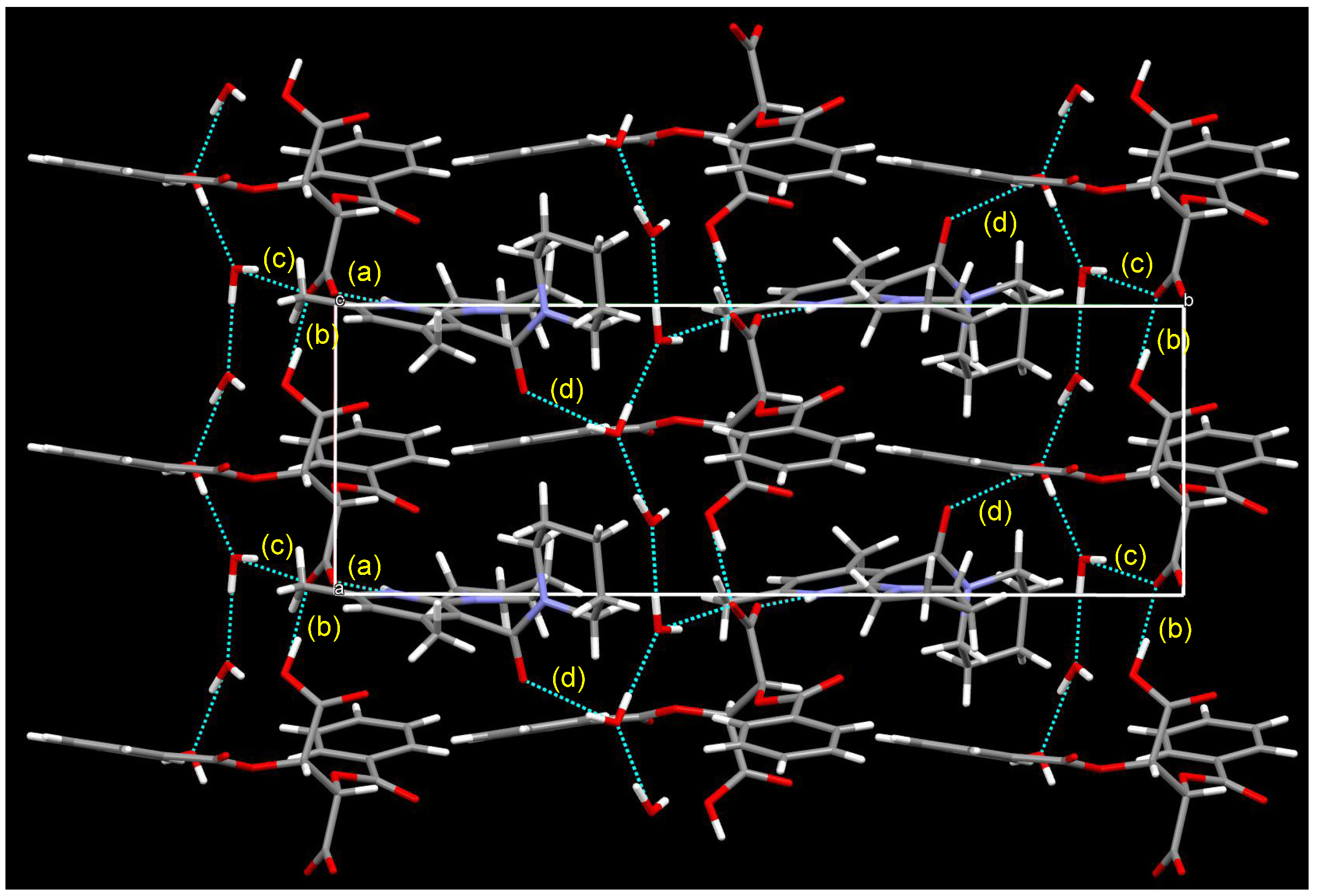
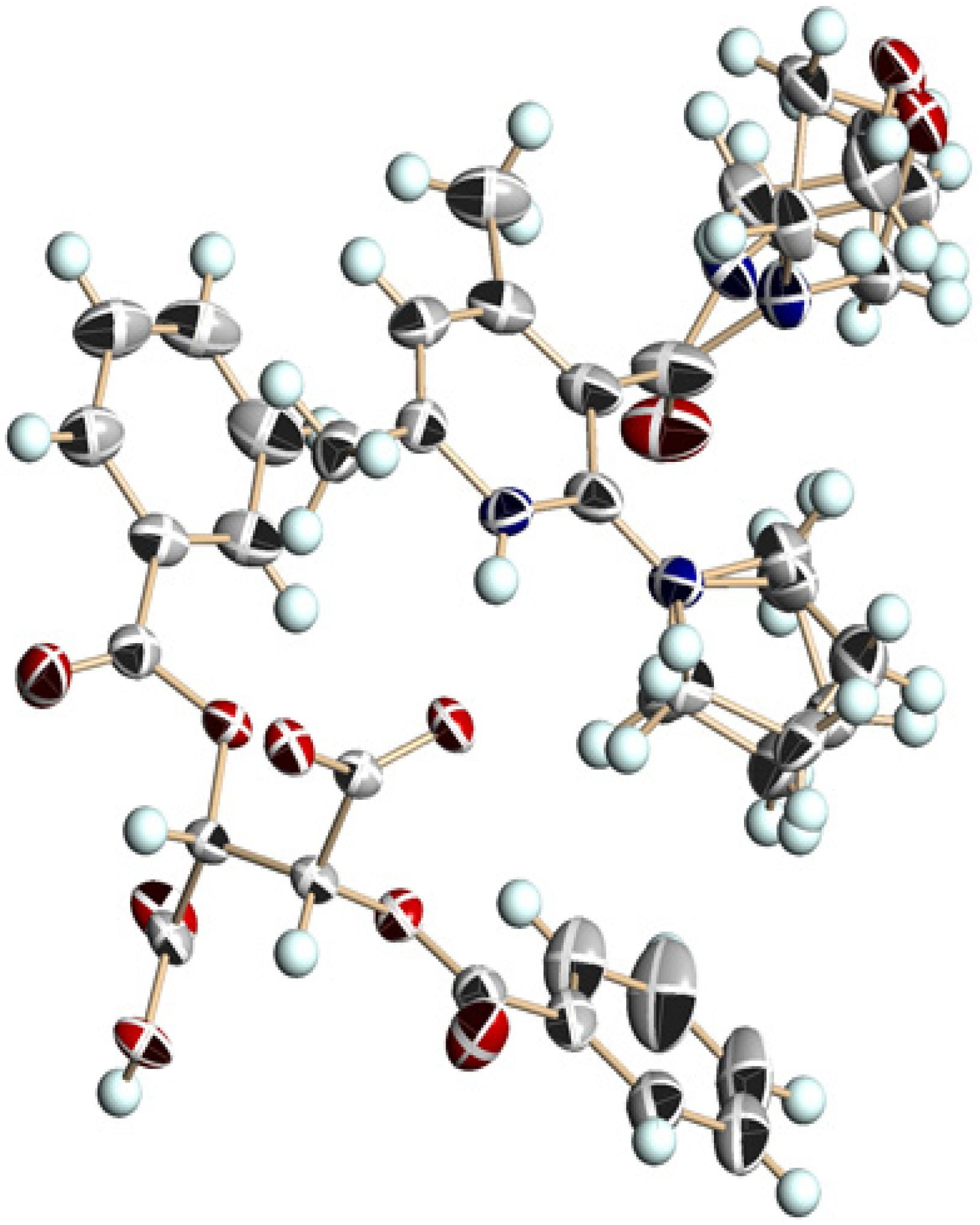
3.5. Crystal Structure of Chiral Salts
3.6. Kinetic Studies for Racemization of 1
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Clayden, N.A. New classes of chiral reagents, Auxiliaries, and ligands? Angew. Chem. Int. Ed. Engl. 1997, 36, 949–951. [Google Scholar] [CrossRef]
- Curran, P.; Qi, H.; Geib, S.J.; DeMello, N.C. Atroposelective thermal reactions of axially twisted Amides and imides. J. Am. Chem. Soc. 1994, 116, 3131–3132. [Google Scholar] [CrossRef]
- Guthrie, B.; Geib, S.J.; Curran, D.P. Synthesis of highly enantioenriched 3,4-dihydroquinolin-2-ones by 6-exo-trig radical cyclizations of axially chiral R-halo-ortho-alkenylanilides. J. Am. Chem. Soc. 2009, 131, 15492–15500. [Google Scholar] [CrossRef]
- Hughes, D.; Price, D.A; Shishkin, O.; Simpkins, N.S. Diastereoselective enolate chemistry using atropisomeric amides. Tetrahedron Lett. 1996, 37, 7607–7610. [Google Scholar] [CrossRef]
- Kitagawa, H.I.; Taguchi, T.; Shiro, M. An Efficient Synthesis of Optically active axially chiral anilide and its application to iodine-mediated asymmetric Diels-Alder reaction. Tetrahedron Lett. 1997, 38, 4447–4450. [Google Scholar] [CrossRef]
- Clayden, D.; Mitjans, L.H.Y. Lithium-Sulfoxide-Lithium exchange for the asymmetric synthesis of atropisomers under thermodynamic control. J. Am. Chem. Soc. 2002, 124, 5266–5267. [Google Scholar] [CrossRef]
- Curran, D.P.; Geib, S.; DeMello, N. Rotational features of carbon-nitrogen bonds in N-Aryl maleimides. Atroposelective reactions of o-tert-Butylphenylmaleimides. Tetrahedron 1999, 55, 5681–5704. [Google Scholar] [CrossRef]
- Kondo, K.; Fujita, H.; Suzuki, T.; Murakami, Y. A new chiral axis due to N(open-chain imide) -- Ar bond: Unexpected racemization effect of an Acyl group. Tetrahedron Lett. 1999, 40, 5577–5580. [Google Scholar] [CrossRef]
- Shimizu, K.D.; Freyer, H.O.; Adams, R.D. Synthesis, resolution and structure of axially chiral atropisomeric N-arylimides. Tetrahedron Lett. 2000, 41, 543–5434. [Google Scholar] [CrossRef]
- Sakamoto, M.; Iwamoto, T.; Nono, N.; Ando, M.; Arai, W.; Mino, T.; Fujita, T. Memory of chirality generated by spontaneous crystallization and asymmetric synthesis using the frozen chirality. J. Org. Chem. 2003, 68, 942–946. [Google Scholar] [CrossRef]
- Koide, H.; Uemura, M. Synthesis of axially chiral N,N-diethyl 2,6-disubstituted benzamides utilizing planar chiral (Arene) chromium complexes. Chem. Commun. 1998, 2483–2484. [Google Scholar] [CrossRef]
- Koide, H.; Hata, T.; Uemura, M. Asymmetric synthesis of axially chiral benzamides and anilidesbyenantiotopic lithiation of prochiral arene chromium complexes. J. Org. Chem. 2002, 67, 1929–1935. [Google Scholar] [CrossRef]
- Clayden, J.; Johnson, P.; Pink, J.H.; Helliwell, M. Atropisomeric amides as chiral ligands: Using (−)-sparteine-directed enantioselective silylation to control the conformation of a stereogenic axis. J. Org. Chem. 2000, 65, 7033–7040. [Google Scholar] [CrossRef]
- Mai, T.T.; Branca, M.; Gori, D.; Guillot, R.; Kouklovsky, C.; Alezra, V. Absolute asymmetric synthesis of tertiary α-amino acids. Angew. Chem., Int. Ed.Engl. 2012, 51, 4981–4984. [Google Scholar] [CrossRef]
- Rios, R.; Jimeno, C.; Carroll, P.J.; Walsh, P.J. Kinetic resolution of atropisomeric amides. J. Am. Chem. Soc. 2002, 124, 10272–10273. [Google Scholar] [CrossRef]
- Thayumanavan, S.; Beak, P.; Curran, D.P. Asymmetric deprotonation of N,N-dihexyl-l-naphthamides to provide atropisomers of N,N-dihexyl-2-alkyl-l-naphthamides. Tetrahedron Lett. 1996, 37, 2899–2902. [Google Scholar] [CrossRef]
- Clayden, J.; Lai, L.W. Enantioselective synthesis of atropisomeric amides by dynamic resolution: Thermodynamic control with a proline-derived diamine resolving agent. Angew. Chem. Int. Ed. Engl. 1999, 38, 2556–2558. [Google Scholar] [CrossRef]
- Dai, W.M.; Zhang, Y. Synthesis of atropisomeric 2,8-dioxygenated N,N-diisopropyl-1-naphthamides via kinetic resolution under sharpless asymmetric dihydroxylation conditions. Tetrahedron Asymmetry 2004, 15, 525–535. [Google Scholar] [CrossRef]
- Sakamoto, M.; Unosawa, A.; Kobaru, S.; Saito, A.; Mino, T.; Fujita, T. Asymmetric photocycloaddition in solution of a chiral crystallized naphthamide. Angew. Chem. Int. Ed. Engl. 2005, 44, 5523–5526. [Google Scholar] [CrossRef]
- Sakamoto, M.; Kato, M.; Aida, Y.; Fujita, K.; Mino, T.; Fujita, T. Photosensitized 2 + 2 cycloaddition reaction using homochirality generated by spontaneous crystallization. J. Am. Chem. Soc. 2008, 130, 1132–1133. [Google Scholar] [CrossRef]
- Yagishita, F.; Mino, T.; Fujita, T.; Sakamoto, M. Two-Step asymmetric reaction using the frozen chirality generated by spontaneous crystallization. Org. Lett. 2012, 14, 2638–2641. [Google Scholar] [CrossRef]
- Shaw, S.A; Aleman, P.; Christy, J.; Kampf, J.W.; Va, P.; Vedejs, E. Enantioselective TADMAP-catalyzed carboxyl migration reactions for the synthesis of stereogenic quaternary carbon. J. Am. Chem. Soc. 2006, 128, 925–934. [Google Scholar] [CrossRef]
- Kawabata, T.; Stragies, R.; Fukaya, T.; Fuji, K. Remote chirality transfer in nucleophilic catalysis with N-(4-pyridinyl)-L-proline derivatives. Chirality 2003, 15, 71–76. [Google Scholar] [CrossRef]
- Spivey, A.C.; Zhu, F.; Mitchell, M.B.; Davey, S.G.; Jarvest, R.L. Concise synthesis, preparative resolution, absolute configuration determination, and applications of anatropisomeric biaryl catalyst for asymmetric acylation. J. Org. Chem. 2003, 68, 7379–7385. [Google Scholar] [CrossRef]
- Spivey, A.C.; Leese, D.P.; Zhu, F.; Davey, S.G.; Jarvest, R.L. New atropisomeric biaryl derivatives of 4-aminopyridine-identification of an improved nucleophilic catalyst for asymmetric acylation of sec-alcohols. Tetrahedron 2004, 60, 4513–4525. [Google Scholar] [CrossRef]
- Yamada, S.; Yamashita, K. Dynamic kinetic resolution of hemiaminals using a novel DMAP catalyst. Tetrahedron Lett. 2008, 49, 32–35. [Google Scholar] [CrossRef]
- De Kok, P.M.T.; Buck, H.M. Regio- and Stereo-selective hydride uptake in model systems related to 3-carbamoyl pyridinium compounds. J. Chem. Soc. Chem. Commun. 1985, 1009–1010. [Google Scholar] [CrossRef]
- De Kok, P.M.T.; Donkersloot, M.C.A.; van Lier, P.M.; Meulendijks, G.H.W.M.; Bastiaansen, L.A.M.; van Hooff, H.J.G.; Kanters, J.A.; Buck, H.M. Stereoselective hydride uptake in modelsystem related to the redox-couple NAD+/NADH. Tetrahedron 1986, 42, 941–959. [Google Scholar] [CrossRef]
- Ohno, A.; Oda, S.; Ishikawa, Y; Yamazaki, N. NAD(P)+-NAD(P)H Models. 90. Stereoselection controlled by electronic effect of a carbonyl group in oxidation of NAD(P)H analog. J. Org. Chem. 2000, 65, 6381–6387. [Google Scholar] [CrossRef]
- Mikata, Y.; Mizukami, K.; Hayashi, K.; Matsumoto, S.; Yano, S.; Yamazaki, N.; Ohno, A. NAD/NADH models with axial/central chiralities: Superiority of the quinoline ring system. J. Org. Chem. 2001, 66, 1590–1599. [Google Scholar] [CrossRef]
- Ishichi, Y.; Ikeura, Y.; Natsugari, H. Amide-based atropisomers in Tachykinin NK1-receptor antagonists: Synthesis and antagonistic activity of axially chiral N-benzylcarboxamide derivatives of 2,3,4,5-tetrahydro-6Hpyrido[2,3-b][1,5]oxazocin-6-one. Tetrahedron 2004, 60, 4481–4490. [Google Scholar] [CrossRef]
- Planar chiral nicotinamides, see: Kanomata, N.; Nakata, T. A compact chemical miniature of a holoenzyme, coenzyme NADH linked dehydrogenase. Design and synthesis of bridged NADH models and their highly enantioselective reduction. J. Am. Chem. Soc. 2000, 122, 4563–4568. [Google Scholar] [CrossRef]
- Kanomata, N.; Mishima, G.; Onozato, J. Synchronized stereocontrol of planar chirality by crystallization-induced asymmetric transformation. Tetrahedron Lett. 2009, 50, 409–412. [Google Scholar] [CrossRef]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates, and Resolutions; John Wiley & Sons: New York, NY, USA, 1981; (reprint edition 1991, reissued with corrections; Krieger Publishing Company: Malabar, FL, USA, 1994). [Google Scholar]
- Chiral symmetry breaking of Binaphthyl, see: Pincock, R.E.; Perkins, R.R.; Ma, A.S.; Wilson, K.R. Probability distribution of enantiomorphous forms in spontaneous generation of optically active substances. Science 1971, 174, 1018–1020. [Google Scholar]
- Chiral symmetry breaking of binaphthyl, see: Kondepudi, D.K.; Laudadio, J.; Asakura, K. Chiral symmetry breaking in stirred crystallization of 1,1′-binaphthyl melt. J. Am. Chem. Soc. 1999, 121, 1448–1451. [Google Scholar] [CrossRef]
- Chiral symmetry breaking of pyrimidine-2(1H)-ones and thiones, see: Sakamoto, M.; Utsumi, N.; Ando, M.; Saeki, M.; Mino, T.; Fujita, T.; Katoh, A.; Nishio, T.; Kashima, C. Breaking the Symmetry of Axially Chiral N-Aryl-2(1H)-pyrimidinones by Spontaneous Crystallization. Angew. Chem. Int. Ed.Engl. 2003, 42, 4360–4363. [Google Scholar] [CrossRef]
- Sakamoto, M.; Yaghishita, F.; Ando, M.; Sasahara, Y.; Kamataki, N.; Ohta, M.; Mino, T.; Kasashima, Y.; Fujta, T. Generation and amplification of optical activity of axially chiral n-(1-naphthyl)-2(1h)-pyrimidinethione by crystallization. Org. Biomol. Chem. 2010, 8, 5418–5422. [Google Scholar] [CrossRef]
- Sakamoto, M.; Mino, T. Asymmetric Reaction Using Molecular Chirality Controlled by Spontaneous Crystallization. In Advances in Crystallization Processes; Mastai, Y., Ed.; InTech: Rijeka, Croatia, 2012; pp. 59–80. [Google Scholar]
- Yagishita, F.; Okamoto, K.; Kamataki, N.; Kanno, S.; Mino, T.; Kasashima, Y.; Sakamoto, M. Chiral symmetry breaking of axially chiral nicotinamide by crystallization from the melt. Chem. Lett. 2013, in press. [Google Scholar]
- Sakamoto, M. Spontaneous chiral crystallization of achiral materials and absolute asymmetric photochemical transformation using the chiral crystalline environment. J. Photochem. Photobiol. C 2007, 7, 183–196. [Google Scholar] [CrossRef]
- Koshima, H.; Matsuura, T. Chiral crystallization of achiral organic compounds: Generation of chirality without chiral environment. J. Syn. Org. Chem. 1998, 56, 268–279. [Google Scholar] [CrossRef]
- Kozma, D. (Ed.) CRC Handbook of Optical Resolution via Diastereomeric Salt Formation; CRC Press: Boca Raton, FL, USA, 2002.
- Nohira, H.; Sakai, K. Optical Resolution by Means of Crystallization. In Enantiomer Separation; Toda, F., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2005; pp. 165–191. [Google Scholar]
- Coquerel, G. Preferential crystallization. Top. Curr. Chem. 2007, 269, 1–51. [Google Scholar] [CrossRef]
- Yoshioka, R. Racemization, optical resolution and crystallization-induced asymmetric transformation of amino acids and pharmaceutical intermediates. Top. Curr. Chem. 2007, 269, 83–132. [Google Scholar] [CrossRef]
- Collet, A. Separation and purification of enantiomers by crystallization methods. Enantiomer 1999, 4, 157–172. [Google Scholar]
- Brands, K.M.J.; Davies, A.J. Crystallization-Induced diastereomer transformations. Chem. Rev. 2006, 106, 2711–2733. [Google Scholar] [CrossRef]
- Yashima, E.; Maeda, K.; Okamoto, Y. Memory of macromolecular helicity assisted by interaction with achiral small molecules. Nature 1999, 399, 449–451. [Google Scholar] [CrossRef]
- Miyagawa, T.; Furuko, A.; Maeda, K.; Katagiri, H.; Furusho, Y.; Yashima, E. Dual memory of enantiomeric helices in a polyacetylene induced by a single enantiomer. J. Am. Chem. Soc. 2005, 127, 5018–5019. [Google Scholar] [CrossRef]
- Ousaka, N.; Inai, Y.; Kuroda, R. Chain-Terminus triggered chiral memory in an optically inactive 310-helical peptide. J. Am. Chem. Soc. 2008, 130, 12266–12267. [Google Scholar] [CrossRef]
- Pijper, D.; Jongejan, M.G.M.; Meetsma, A.; Feringa, B.L. Light-Controlled supramolecular helicity of a liquid crystalline phase using a helical polymer functionalized with a single chiroptical molecular switch. J. Am. Chem. Soc. 2008, 108, 4541–4552. [Google Scholar]
- Lautrette, G.; Kauffmann, B.; Ferrand, Y.; Aube, C.; Chandramouli, N.; Dubreuil, D.; Huc, I. Structure Elucidation of Host-Guest Complexes of Tartaric and Malic Acids by Quasi-Racemic Crystallography. Angew. Chem. Int. Ed. 2013. [Google Scholar] [CrossRef]
- Ferrand, Y.; Kendhale, A.M.; Kauffmann, B.; Grelard, A.; Marie, C.; Blot, V.; Pipelier, M.; Dubreuil, D.; Huc, I. Diastereoselective encapsulation of tartaric acid by a helical aromatic oligoamide. J. Am. Chem. Soc. 2010, 132, 7858–7859. [Google Scholar] [CrossRef]
- Dyadyuchenko, L.V.; Strelkov, V.D.; Mikhailichenko, S.N.; Zaplishny, V.N. Synthesis of some halogen-and nitro-substituted nicotinic acids and their fragmentation under electron impact. Chem. Heterocycl. Comp. 2004, 40, 308–314. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yagishita, F.; Kamataki, N.; Okamoto, K.; Kanno, S.; Mino, T.; Masu, H.; Sakamoto, M. Deracemization of Axially Chiral Nicotinamides by Dynamic Salt Formation with Enantiopure Dibenzoyltartaric Acid (DBTA). Molecules 2013, 18, 14430-14447. https://doi.org/10.3390/molecules181114430
Yagishita F, Kamataki N, Okamoto K, Kanno S, Mino T, Masu H, Sakamoto M. Deracemization of Axially Chiral Nicotinamides by Dynamic Salt Formation with Enantiopure Dibenzoyltartaric Acid (DBTA). Molecules. 2013; 18(11):14430-14447. https://doi.org/10.3390/molecules181114430
Chicago/Turabian StyleYagishita, Fumitoshi, Norifumi Kamataki, Kazuma Okamoto, Shota Kanno, Takashi Mino, Hyuma Masu, and Masami Sakamoto. 2013. "Deracemization of Axially Chiral Nicotinamides by Dynamic Salt Formation with Enantiopure Dibenzoyltartaric Acid (DBTA)" Molecules 18, no. 11: 14430-14447. https://doi.org/10.3390/molecules181114430