Bio-Activity and Dereplication-Based Discovery of Ophiobolins and Other Fungal Secondary Metabolites Targeting Leukemia Cells

 ,
,
Abstract
:1. Introduction
2. Results and Discussion

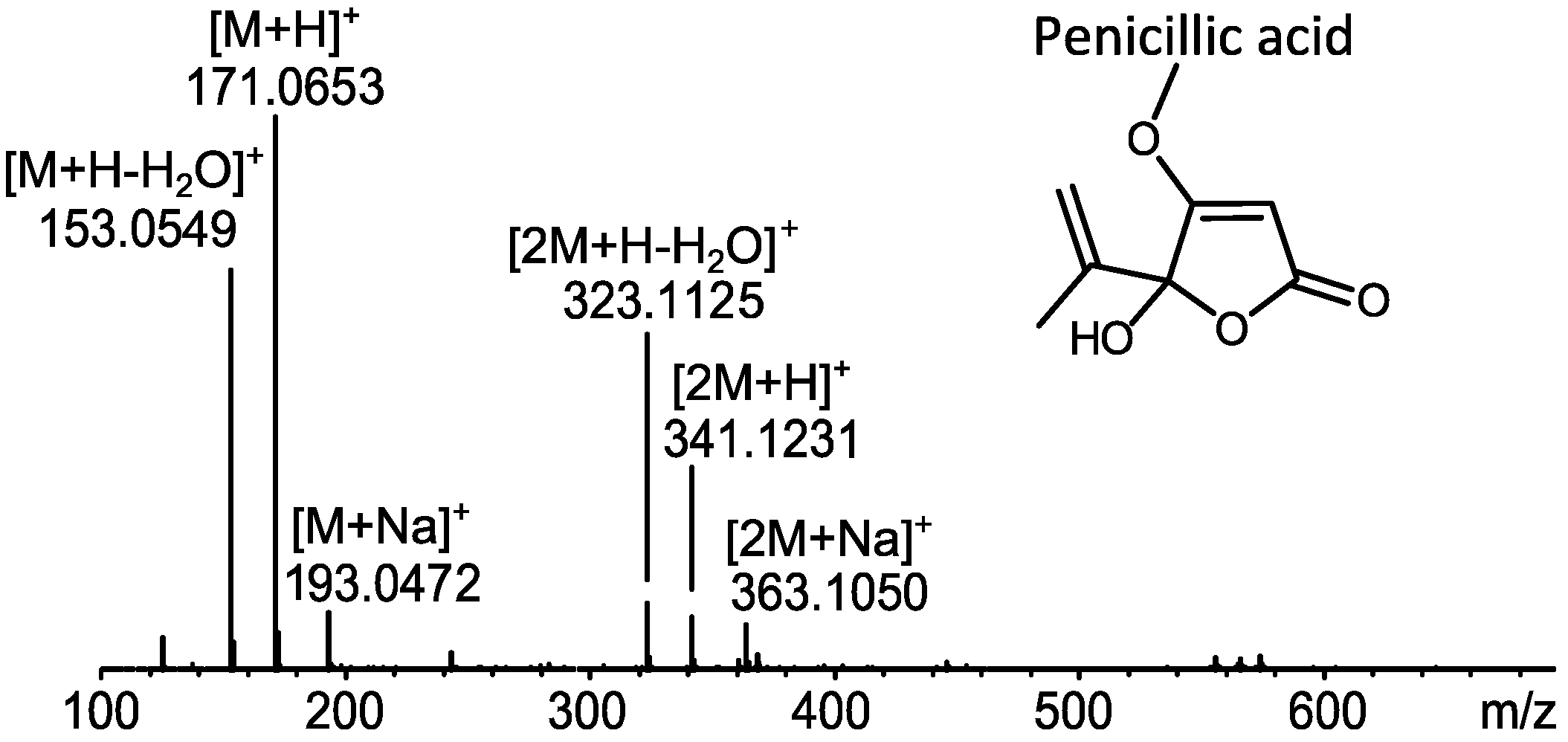
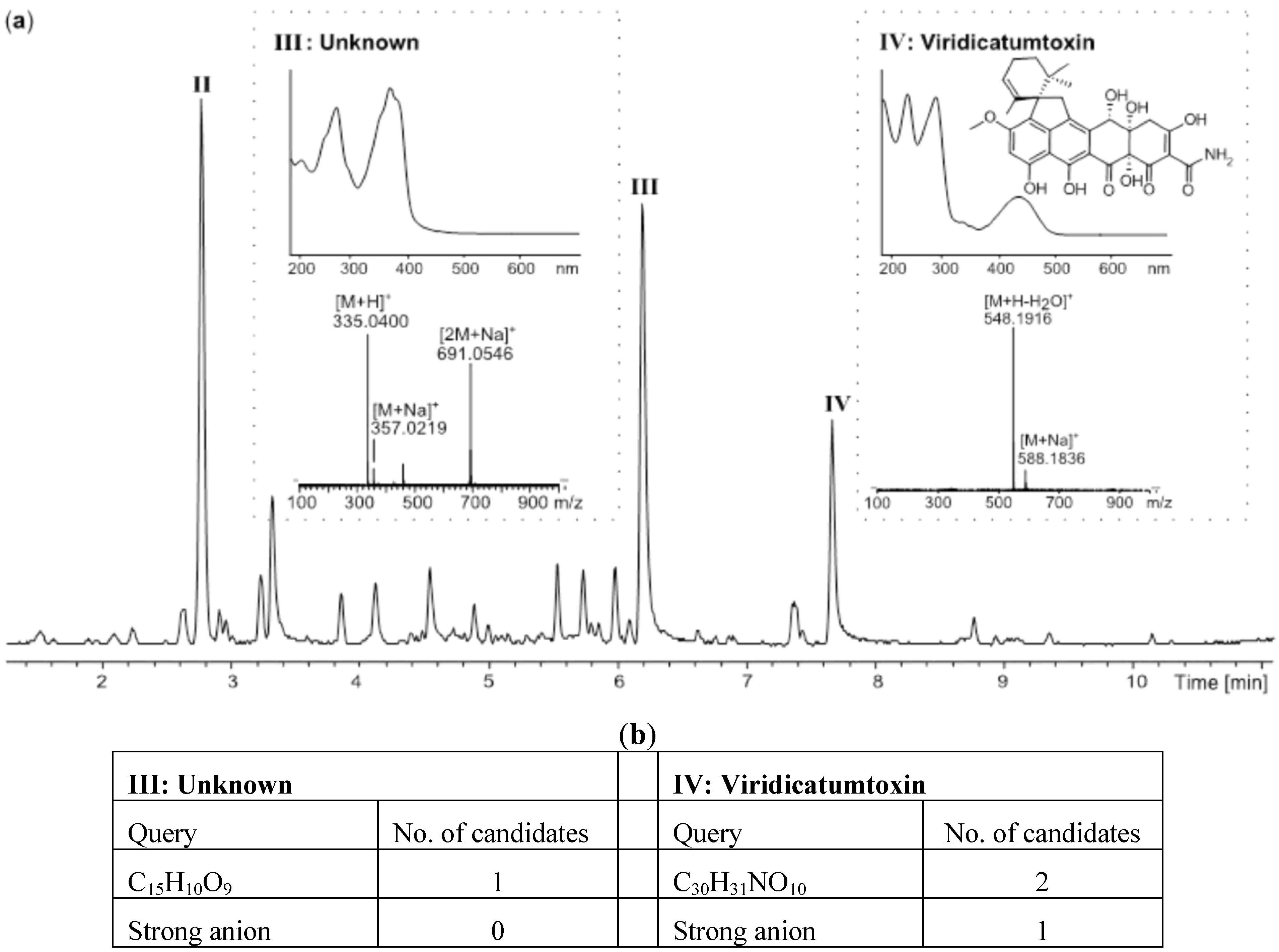
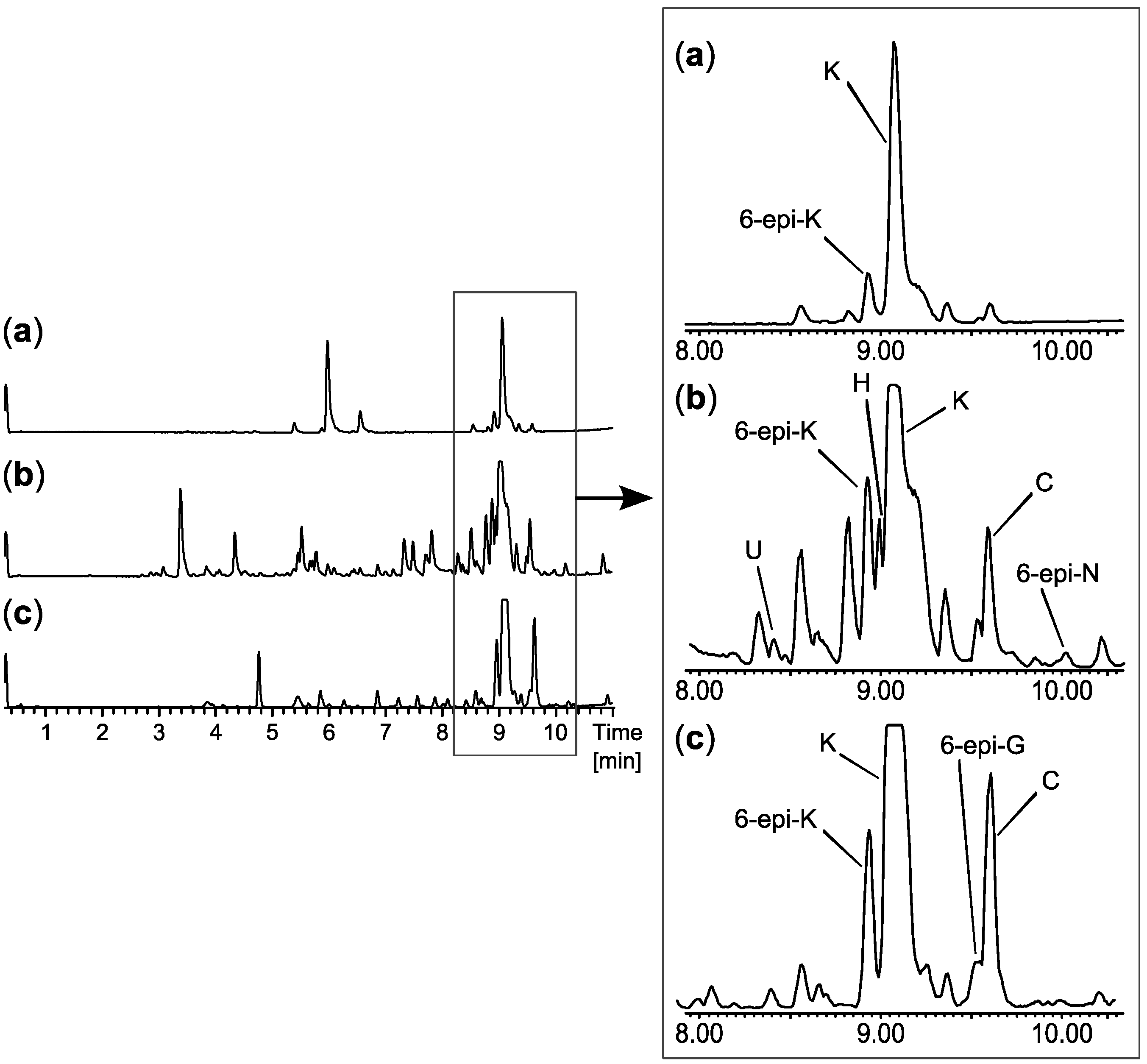
2.1. MS Based Dereplication of Penicillium pulvillorum Extract
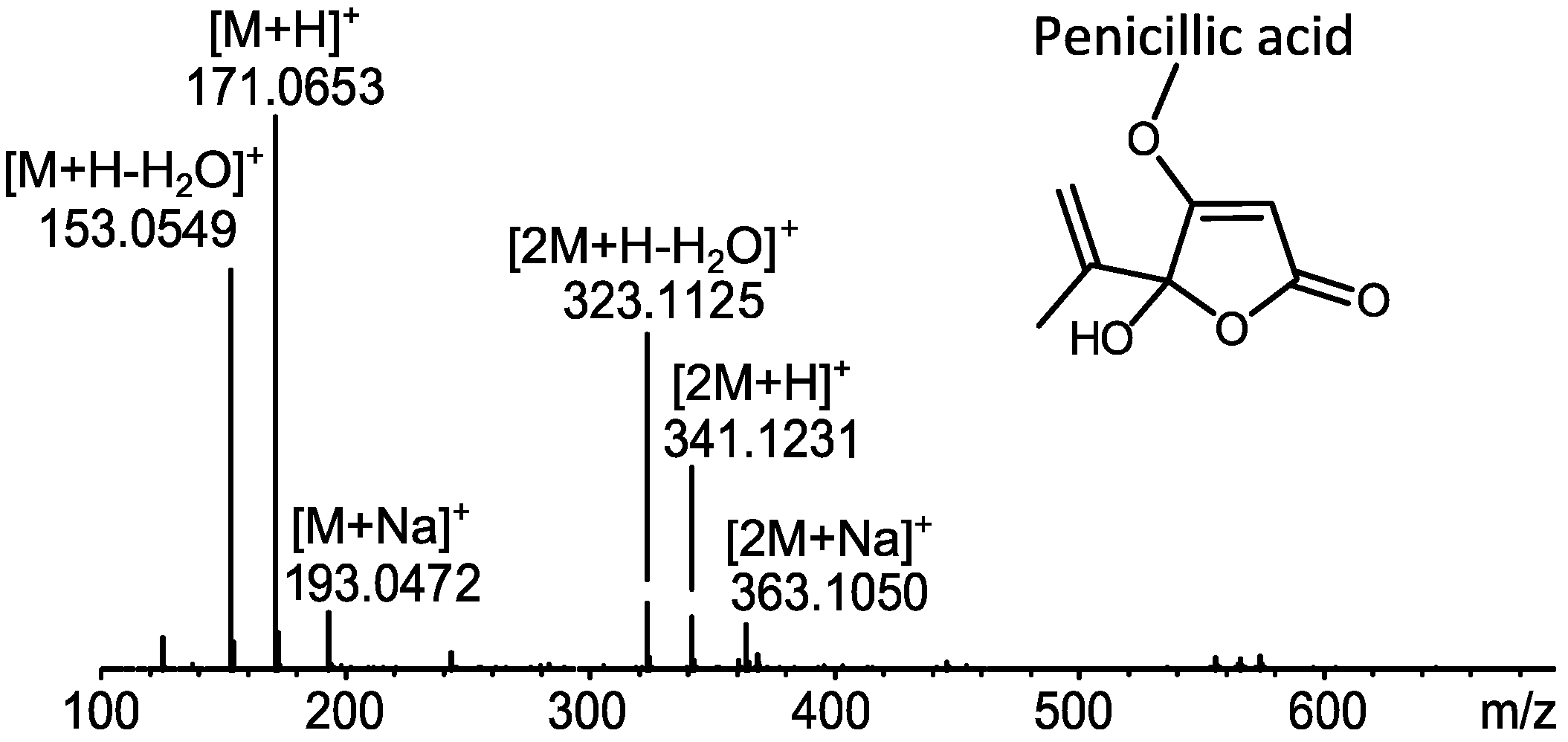
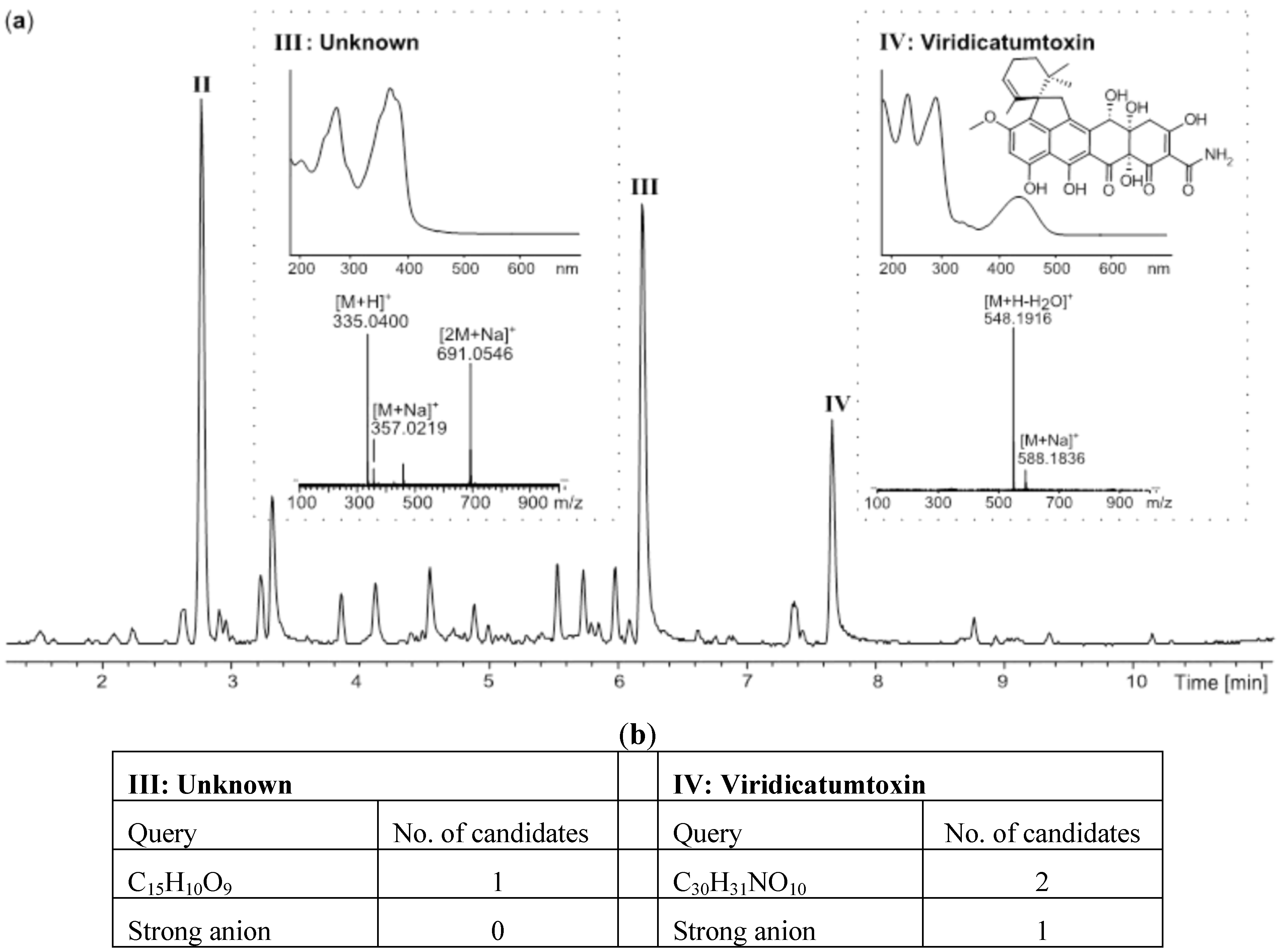
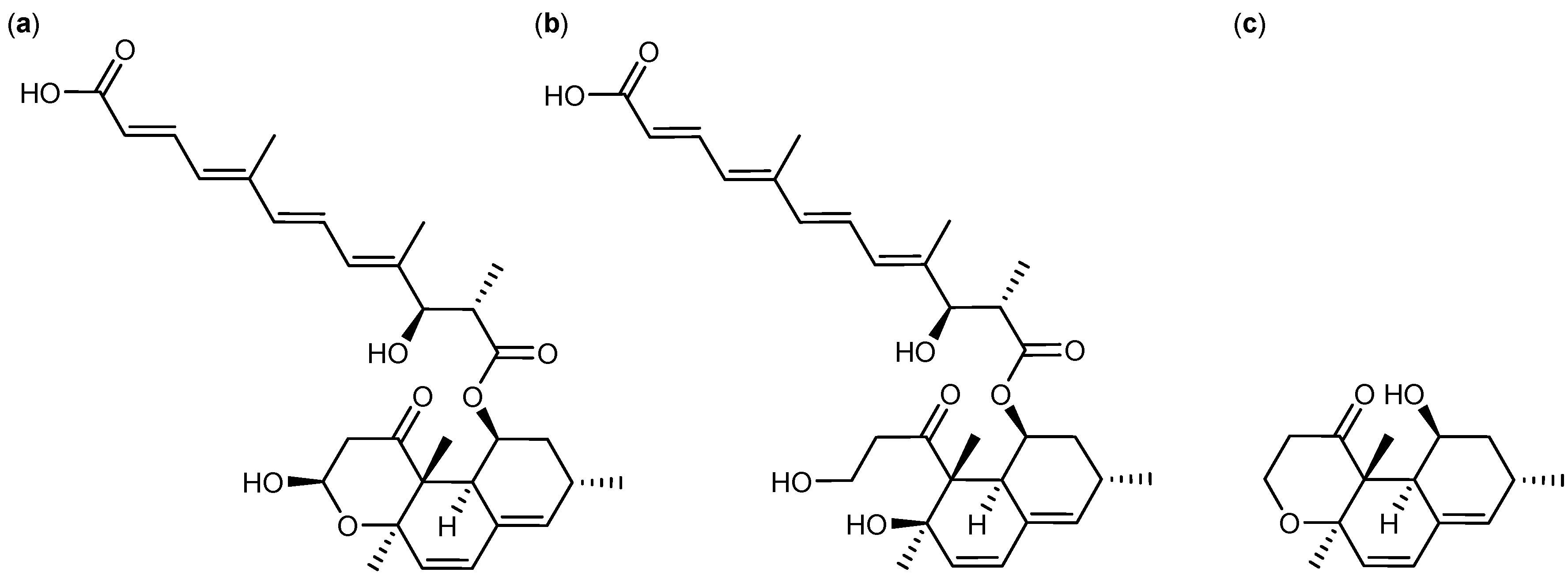
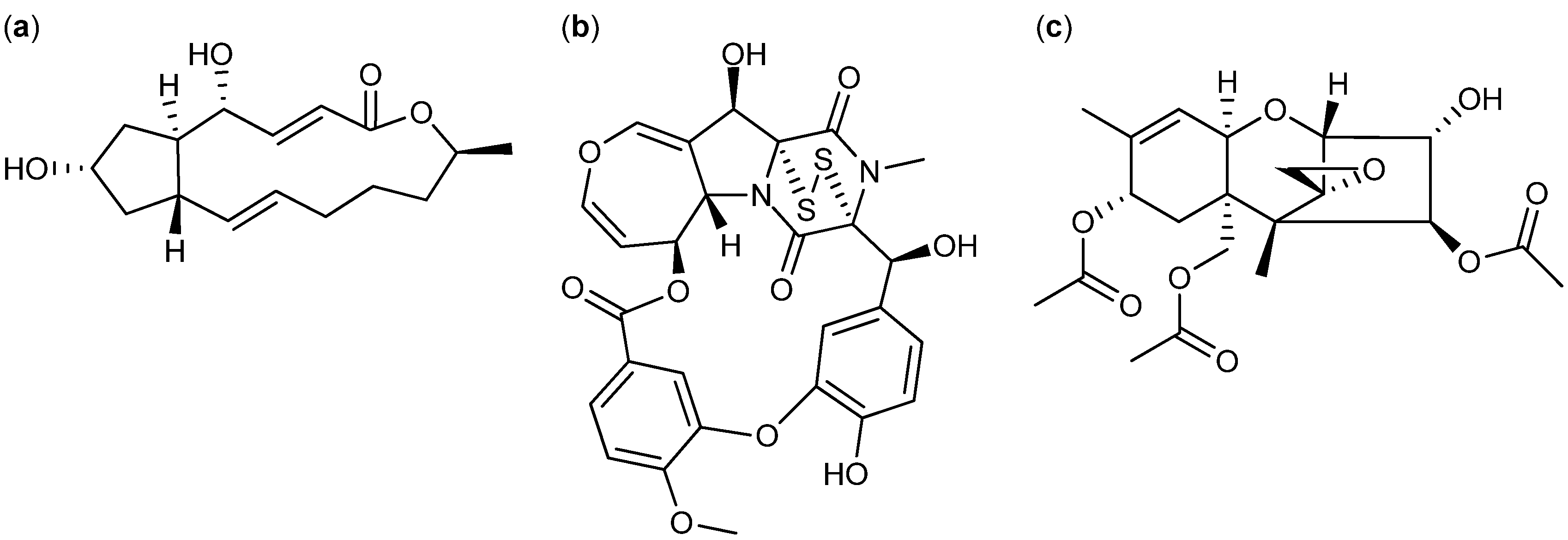
2.2. Comparative Dereplication Based on Explorative Solid Phase Extraction (E-SPE)

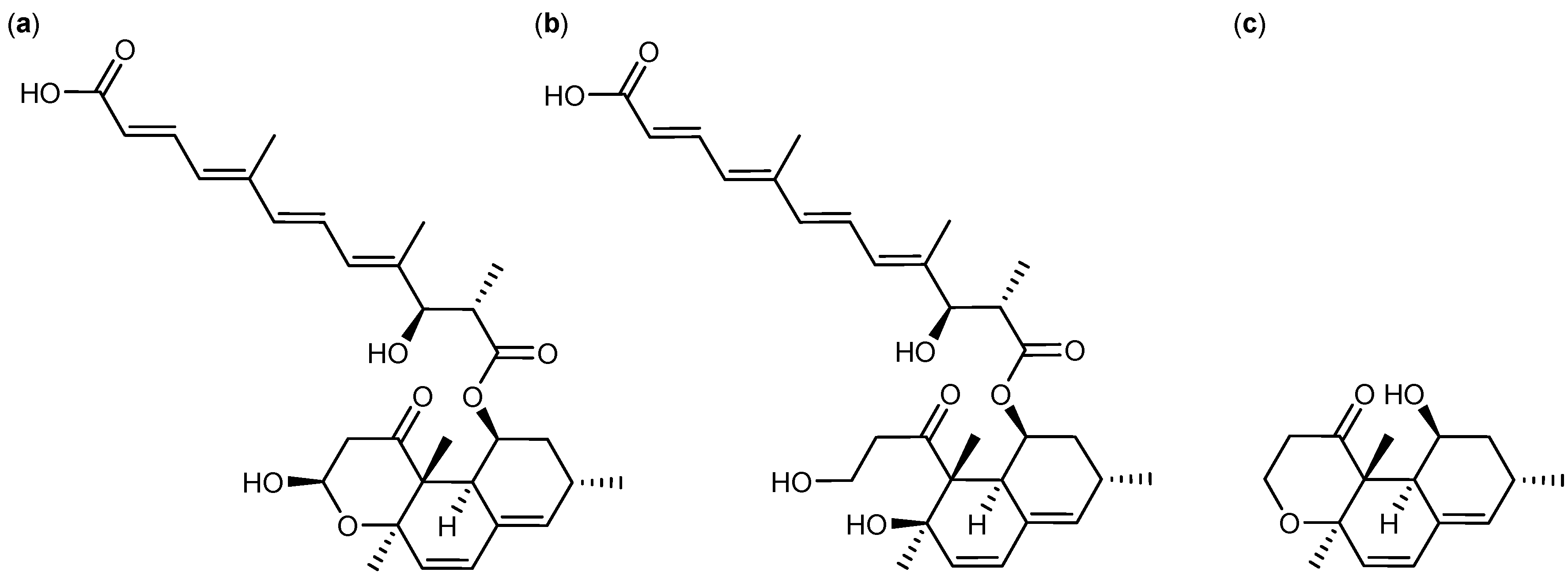

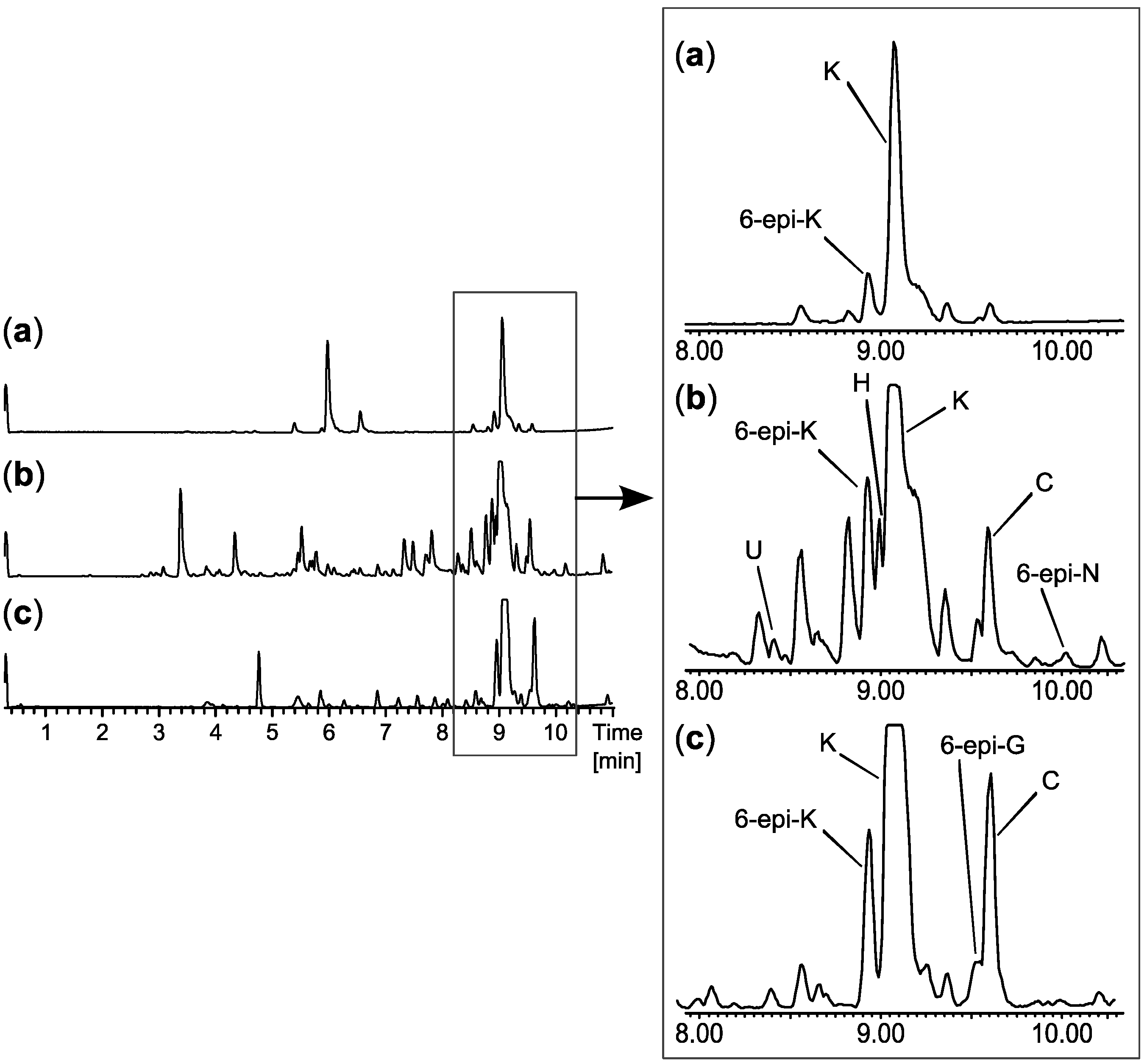
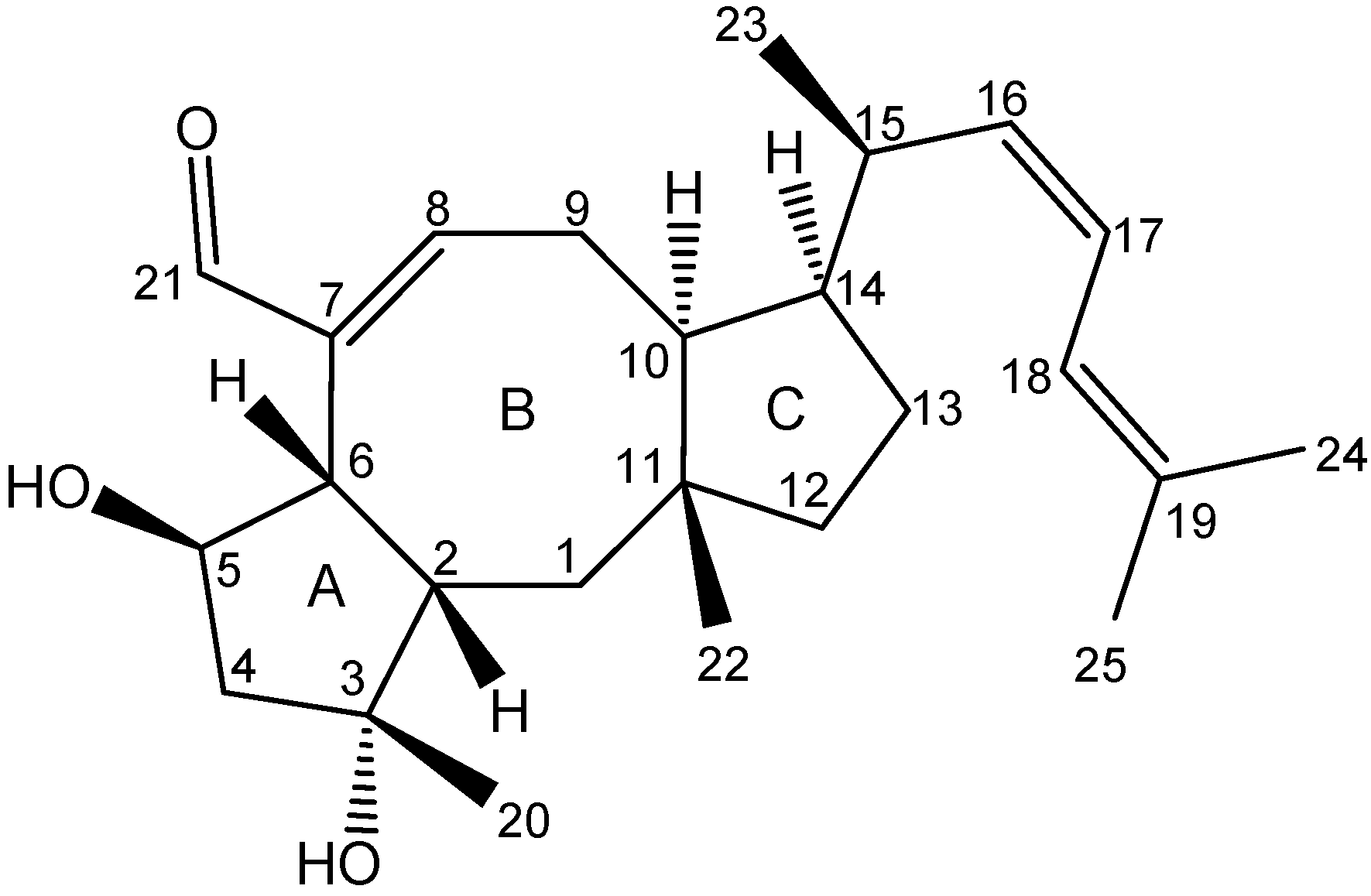
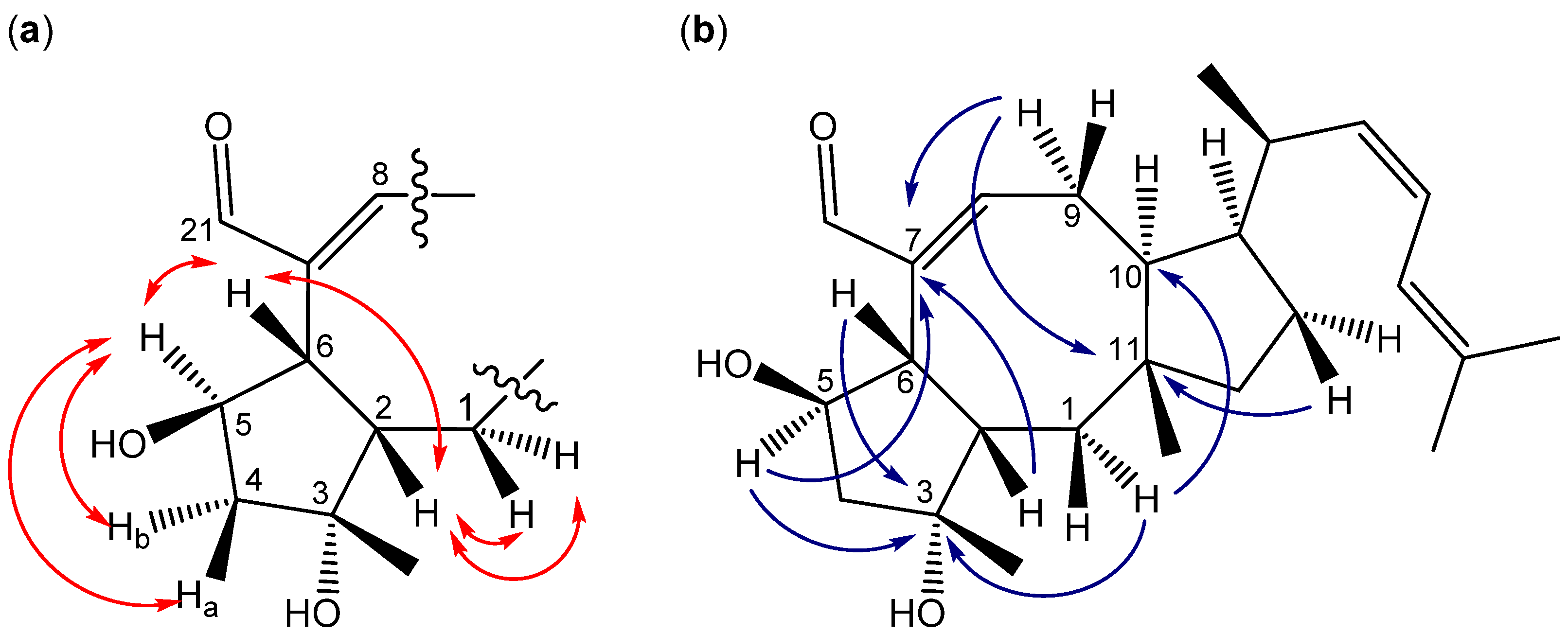
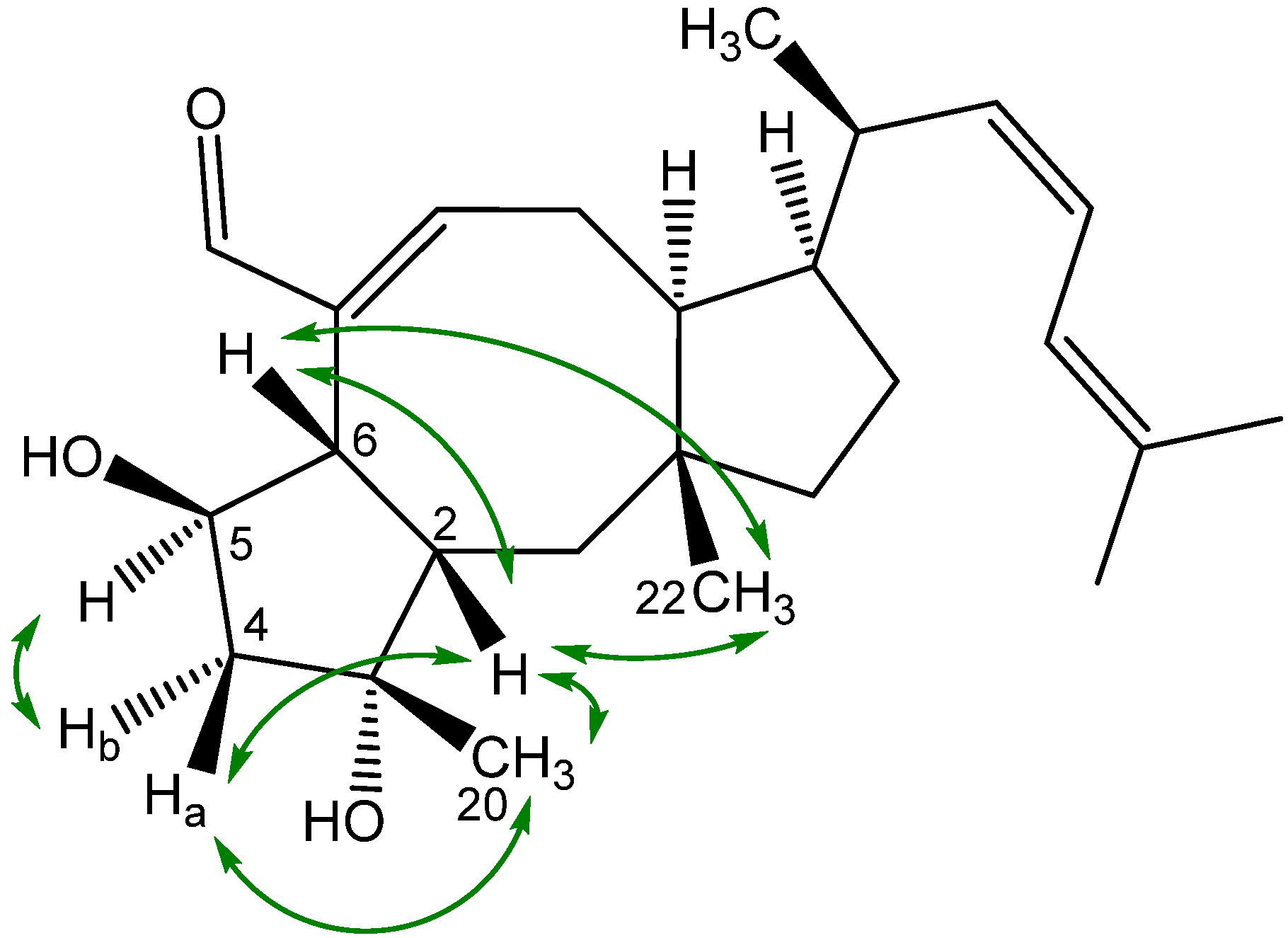
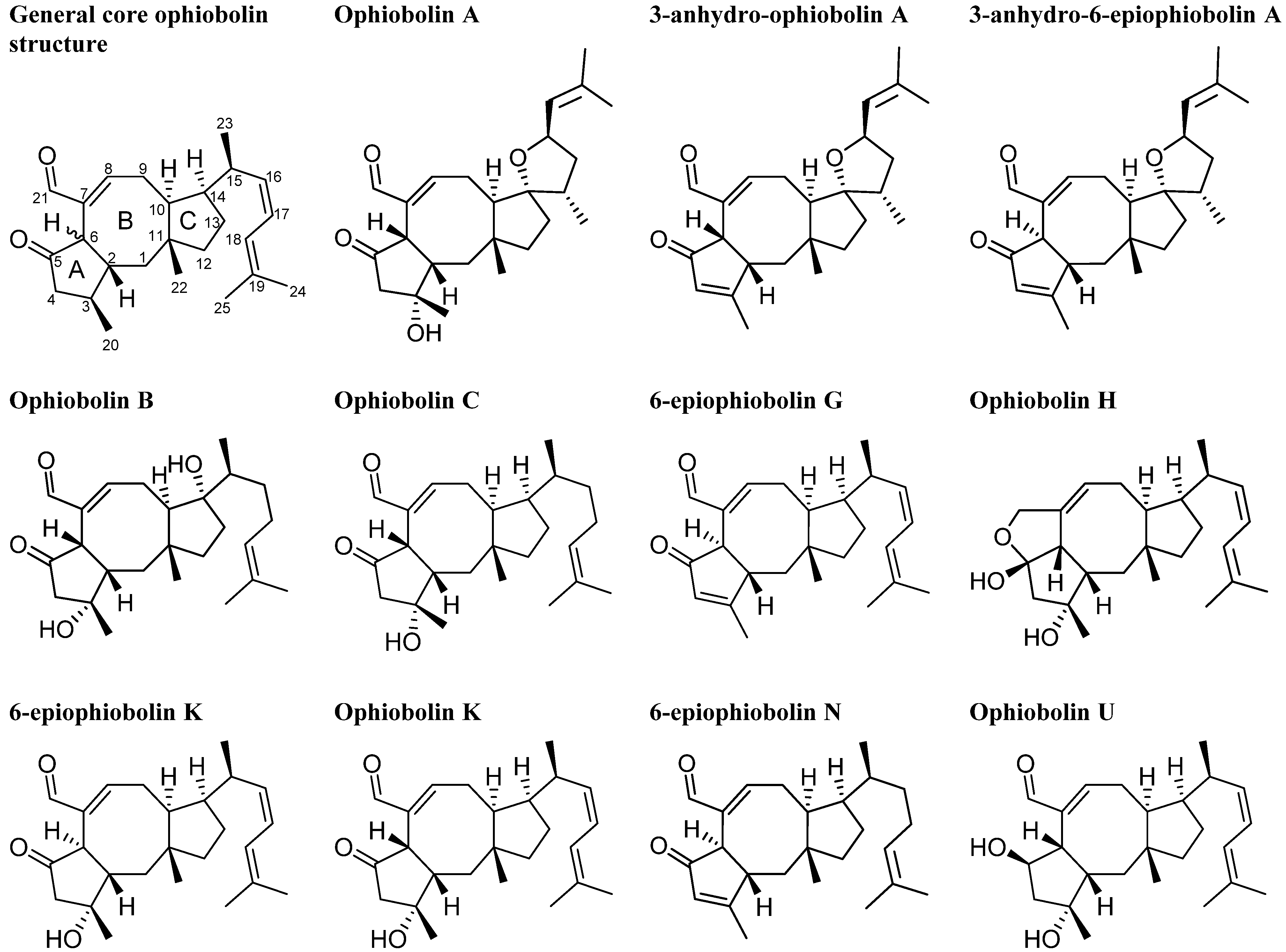
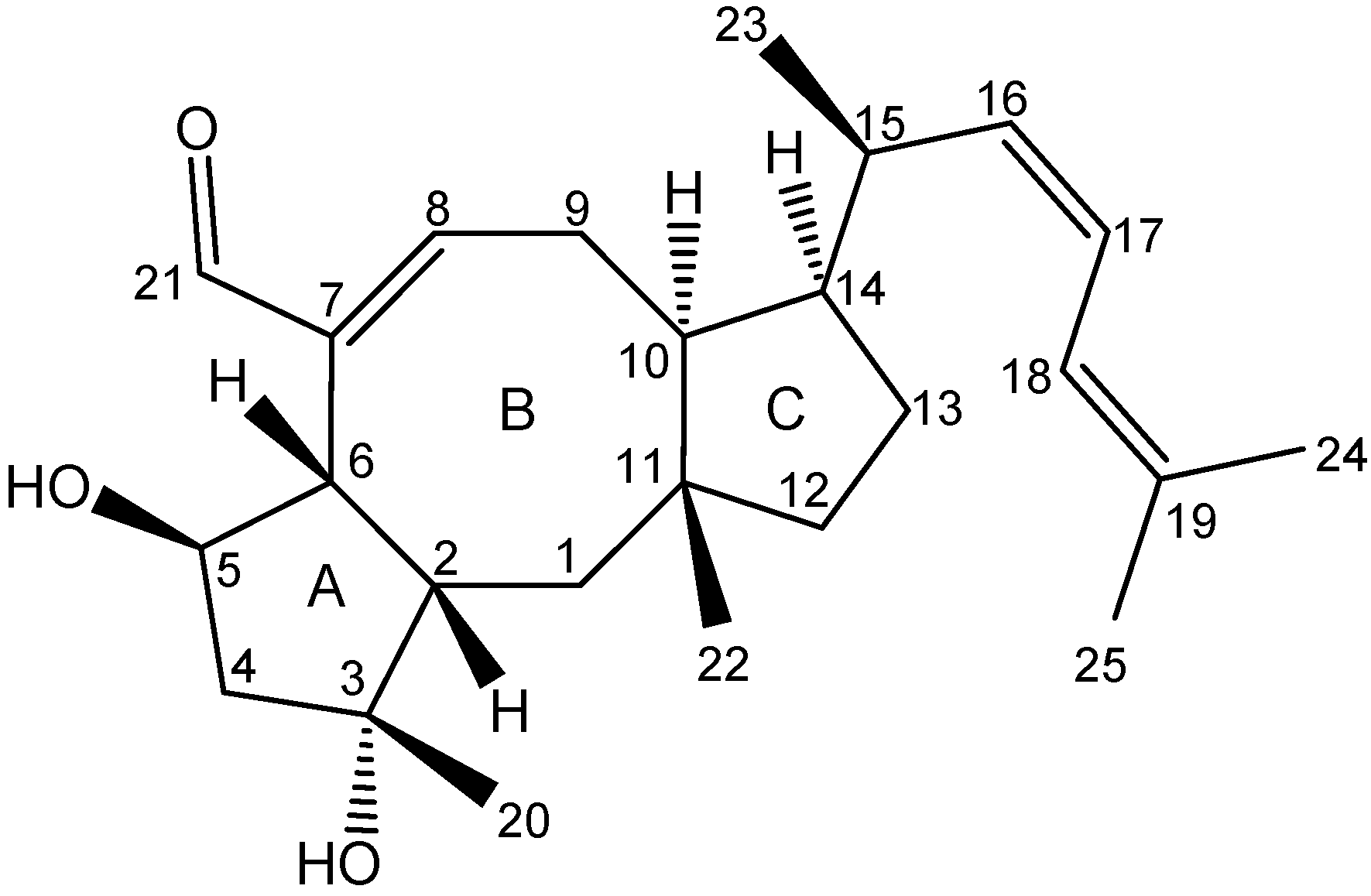
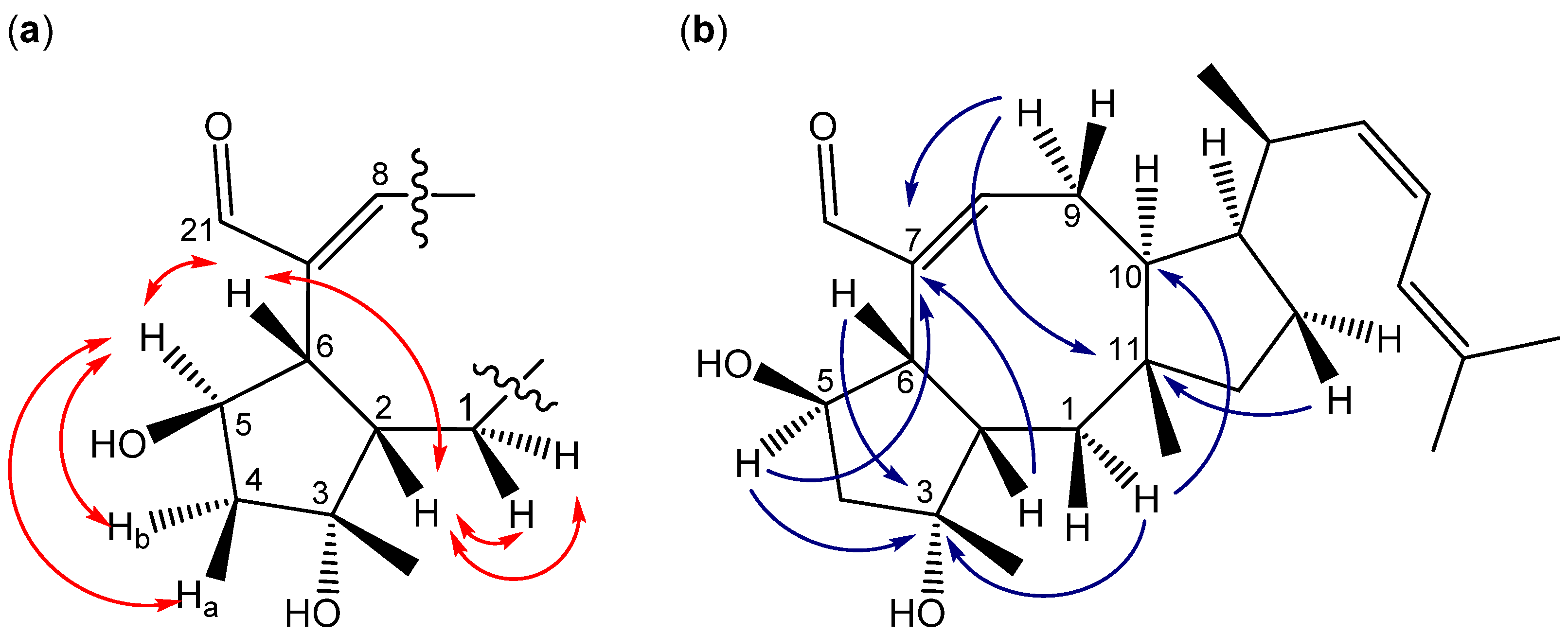
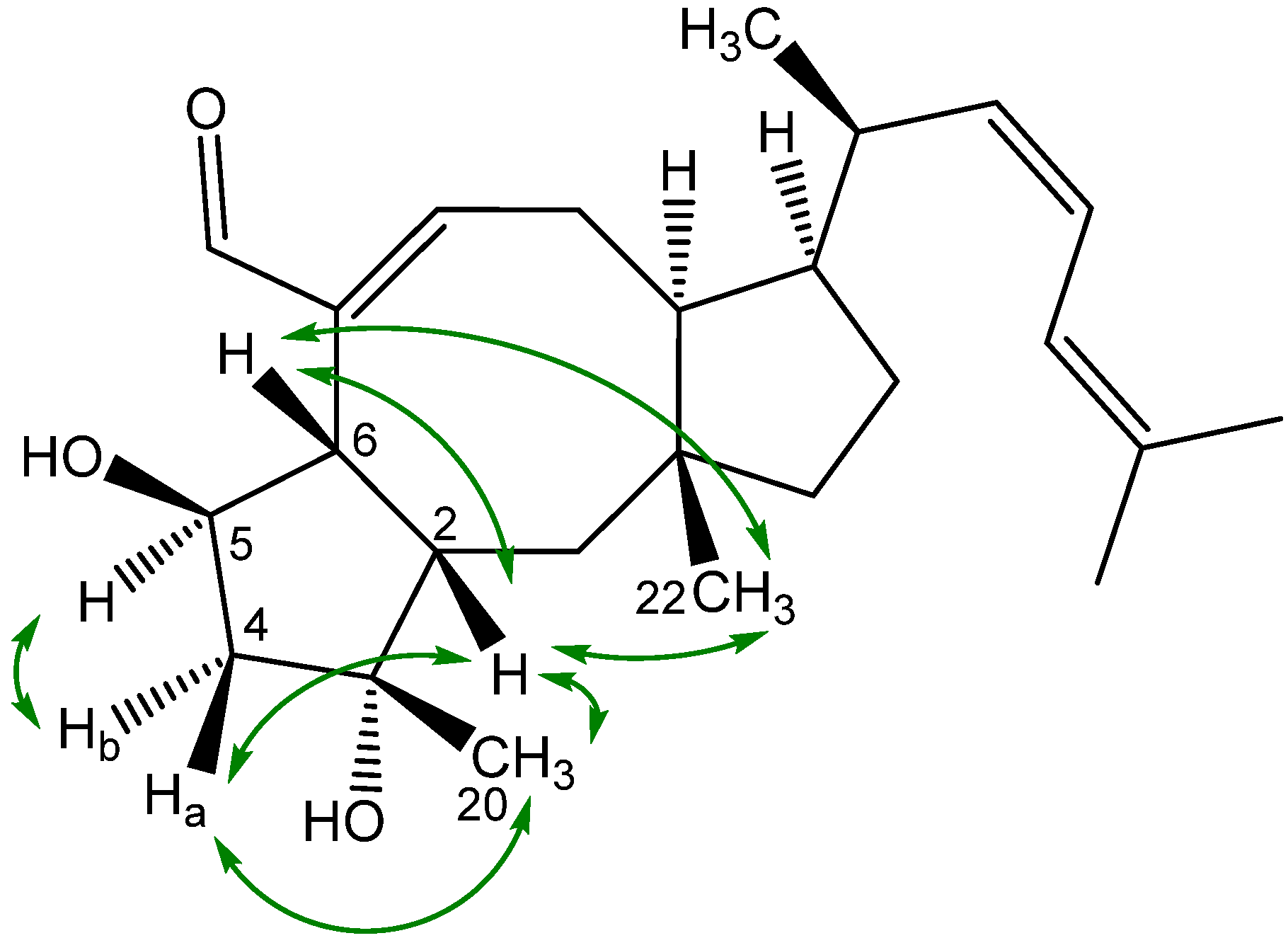
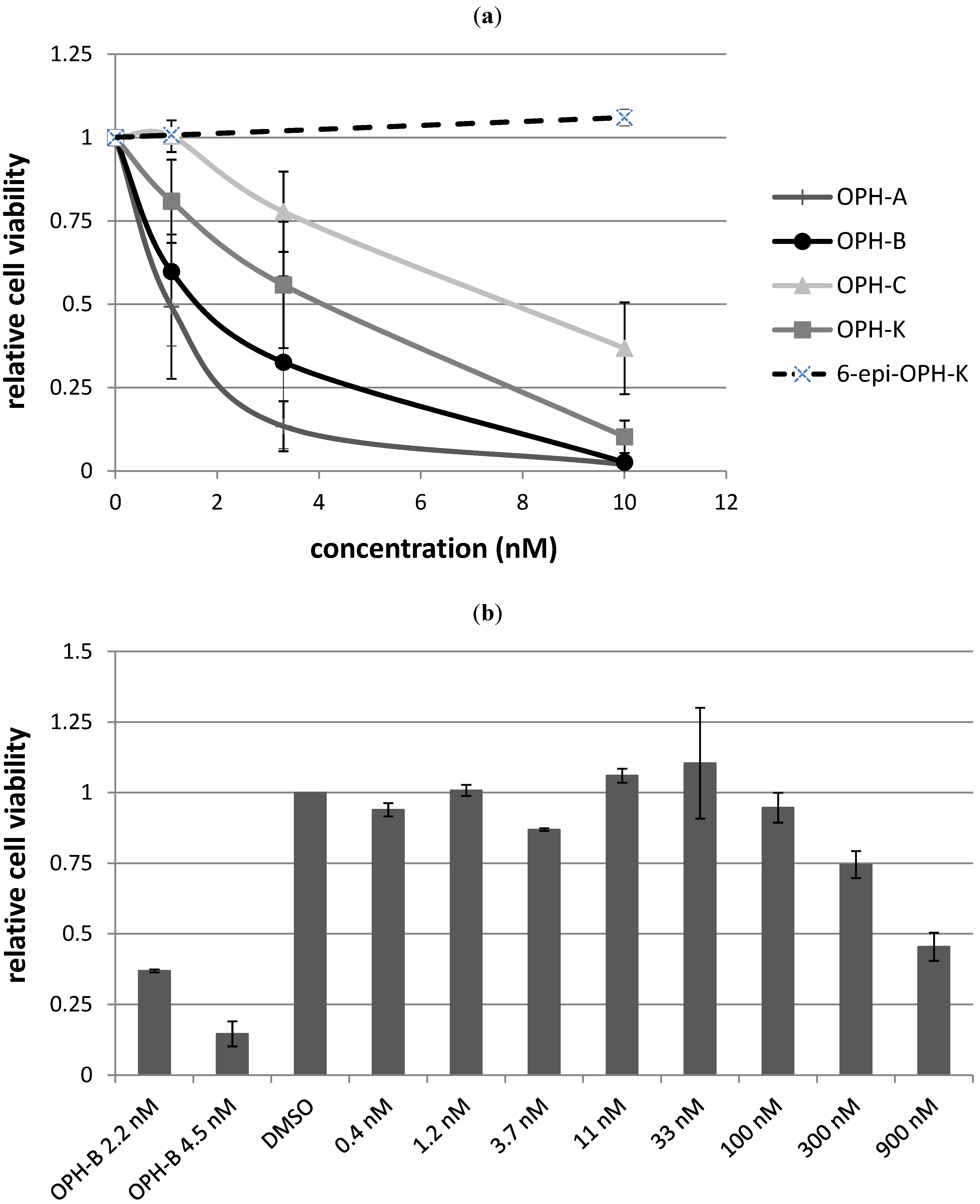
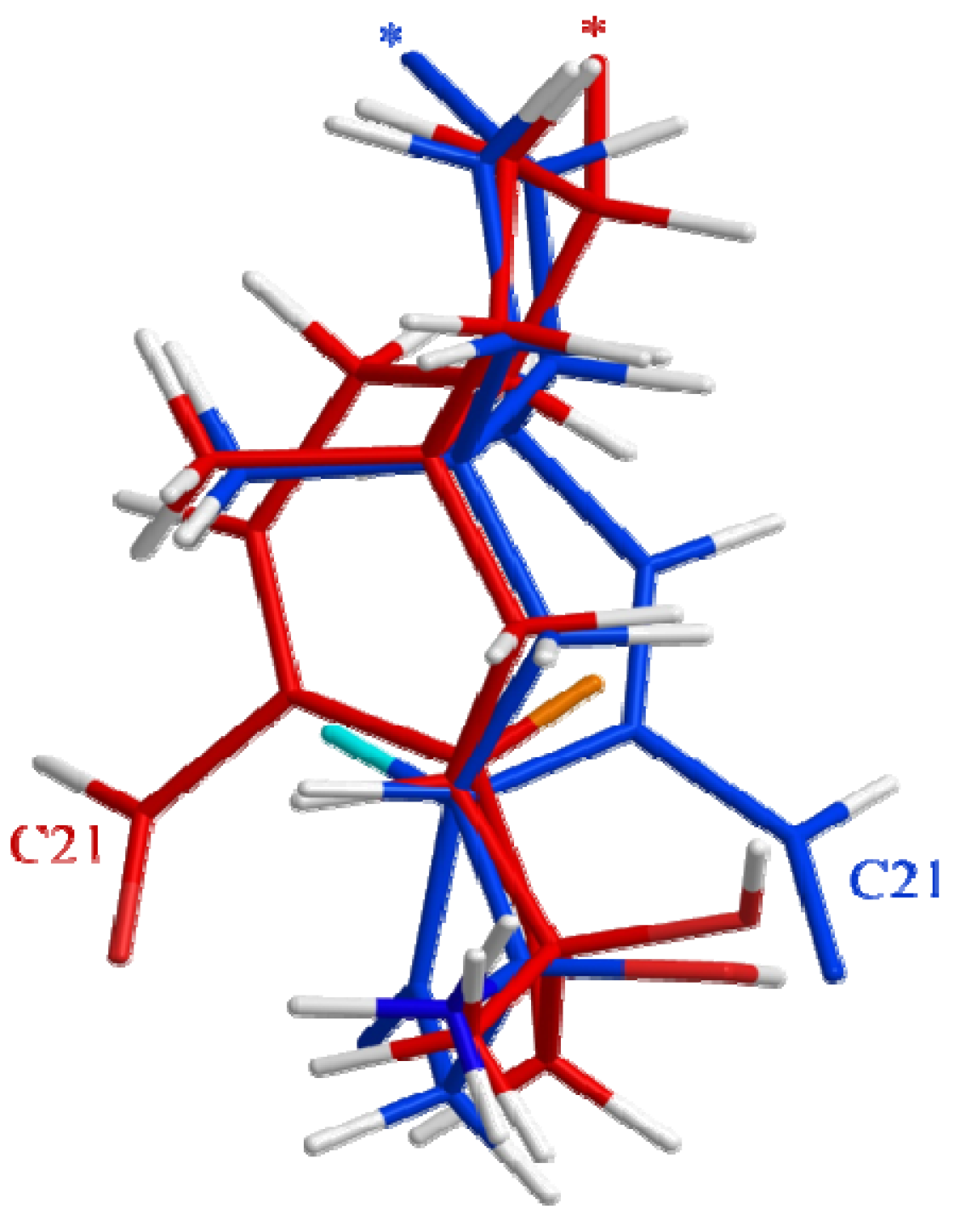
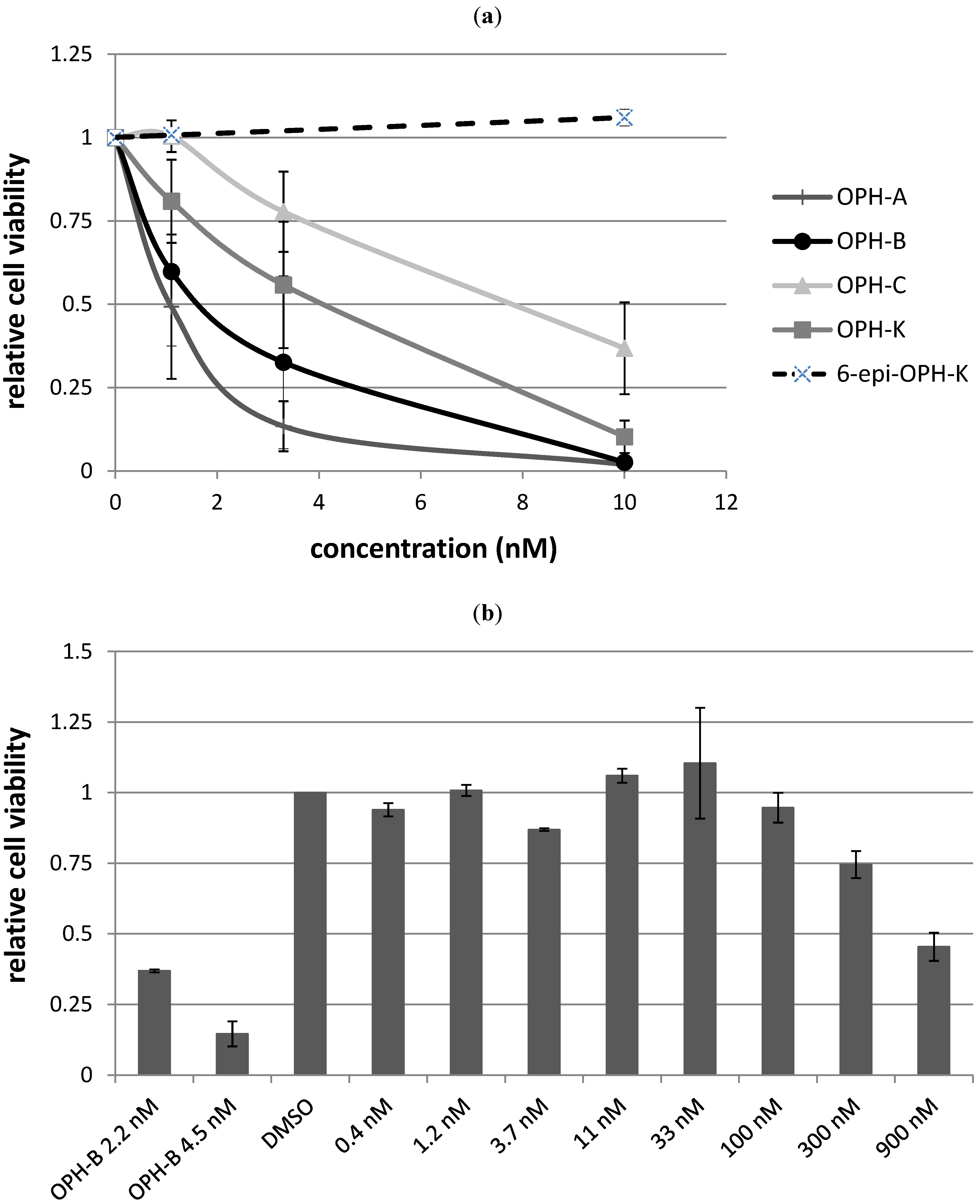
2.3. Biological Structure-Activity Relationship of Ophiobolins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | LC50 |
|---|---|
| Ophiobolin A | 1 nM |
| 3-anhydro-ophiobolin A | Inactive |
| 3-anhydro-6-epiophiobolin A | Inactive |
| Ophiobolin B | 2 nM |
| Ophiobolin C | 8 nM |
| 6-epiophiobolin G | Inactive |
| Ophiobolin H | Inactive |
| Ophiobolin K | 4 nM |
| 6-epiophiobolin K | Inactive |
| 6-epiophiobolin H | Inactive |

3. Experimental
3.1. General
3.2. Micro Extraction for Initial Screen
3.3. Cultivation and Extraction
3.4. Bioassay-Guided Fractionation
3.4.1. E-SPE
3.4.2. Ophiobolins
3.5. CLL Cells, Cell Viability and Apoptosis Assays
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Zenz, T.; Mertens, D.; Küppers, R.; Döhner, H.; Stilgenbauer, S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2010, 10, 37–50. [Google Scholar]
- Burger, J.A.; Montserrat, E. Coming full circle: 70 years of chronic lymphocytic leukemia cell redistribution, from glucocorticoids to inhibitors of B-cell receptor signaling. Blood 2013, 121, 1501–1509. [Google Scholar] [CrossRef]
- Nielsen, K.F.; Månsson, M.; Rank, C.; Frisvad, J.C.; Larsen, T.O. Dereplication of microbial natural products by LC-DAD-TOFMS. J. Nat. Prod. 2011, 74, 2338–2348. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Smedsgaard, J.; Larsen, T.O.; Samson, R.A. Mycotoxins, drugs and other extrolites produced by species in Penicillium subgenus Penicillium. Stud. Mycol. 2004, 49, 201–241. [Google Scholar]
- Bladt, T.T.; Frisvad, J.C.; Knudsen, P.B.; Larsen, T.O. Anticancer and antifungal compounds from Aspergillus, Penicillium and other filamentous fungi. Molecules 2013, 18, 11338–11376. [Google Scholar] [CrossRef]
- Delgado, J.; Baumann, T.; Ghita, G.; Montserrat, E. Chronic lymphocytic leukemia therapy: Beyound chemoimmunotherapy. Curr. Pharm. Des. 2012, 18, 3356–3362. [Google Scholar] [CrossRef]
- Tsimberidou, A.-M.; Keating, M.J. Treatment of patients with fludarabine-refractory chronic lymphocytic leukemia: need for new treatment options. Leuk. Lymphoma 2010, 51, 1188–1199. [Google Scholar]
- Isfort, S.; Cramer, P.; Hallek, M. Novel and emerging drugs for chronic lymphocytic leukemia. Curr. Cancer Drug Tar. 2012, 12, 471–483. [Google Scholar]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Aquila, M.D.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar]
- Munk Pedersen, I.; Reed, J. Microenvironmental interactions and survival of CLL B-cells. Leuk. Lymphoma 2004, 45, 2365–2372. [Google Scholar] [CrossRef]
- Lagneaux, L.; Delforge, A.; Bron, D.; de Bruyn, C.; Stryckmans, P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood 1998, 91, 2387–2396. [Google Scholar]
- Seiffert, M.; Stilgenbauer, S.; Döhner, H.; Lichter, P. Efficient nucleofection of primary human B cells and B-CLL cells induces apoptosis, which depends on the microenvironment and on the structure of transfected nucleic acids. Leukemia 2007, 21, 1977–1983. [Google Scholar] [CrossRef]
- Panayiotidis, P.; Jones, D.; Ganeshaguru, K.; Foroni, L.; Hoffbrand, A.V. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br. J. Haematol. 1996, 92, 97–103. [Google Scholar] [CrossRef]
- Schulz, A.; Toedt, G.; Zenz, T.; Stilgenbauer, S.; Lichter, P.; Seiffert, M. Inflammatory cytokines and signaling pathways are associated with survival of primary chronic lymphocytic leukemia cells in vitro: A dominant role of CCL2. Haematologica 2011, 96, 408–416. [Google Scholar] [CrossRef]
- Knudsen, P.B.; Hanna, B.; Ohl, S.; Sellner, L.; Zenz, T.; Stilgenbauer, S.; Larsen, T.O.; Lichter, P.; Seiffert, M. Chaetoglobosin A preferentially induces apoptosis in chronic lymphocytic leukemia cells by targeting the cytoskeleton. Leukemia 2013, in press. [Google Scholar]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. ChemBioChem 2002, 3, 619–627. [Google Scholar] [CrossRef]
- Rebacz, B.; Larsen, T.O.; Clausen, M.H.; Rønnest, M.H.; Löffler, H.; Ho, A.D.; Krämer, A. Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype-based screen. Cancer Res. 2007, 67, 6342–6350. [Google Scholar] [CrossRef]
- Liao, W.-Y.; Shen, C.-N.; Lin, L.-H.; Yang, Y.-L.; Han, H.-Y.; Chen, J.-W.; Kuo, S.-C.; Wu, S.-H.; Liaw, C.-C. Asperjinone, a nor-neolignan, and terrein, a suppressor of ABCG2-expressing breast cancer cells, from thermophilic Aspergillus terrus. J. Nat. Prod. 2012, 75, 630–635. [Google Scholar] [CrossRef]
- Larsen, T.O.; Smedsgaard, J.; Nielsen, K.F.; Hansen, M.E.; Frisvad, J.C. Phenotypic taxonomy and metabolite profiling in microbial drug discovery. Nat. Prod. Rep. 2005, 22, 672–695. [Google Scholar] [CrossRef]
- Smedsgaard, J.; Nielsen, J. Metabolite profiling of fungi and yeast: From phenotype to metabolome by MS and informatics. J. Exp. Bot. 2005, 56, 273–286. [Google Scholar] [CrossRef]
- Månsson, M.; Phipps, R.K.; Gram, L.; Munro, M.H.G.; Larsen, T.O.; Nielsen, K.F. Explorative solid-phase extraction (E-SPE) for accelerated microbial natural product discovery, dereplication, and purification. J. Nat. Prod. 2010, 73, 1126–1132. [Google Scholar] [CrossRef]
- Smedsgaard, J. Micro-scale extraction procedure for standardized screening of fungal metabolite production in cultures. J. Chromatogr. A 1997, 760, 264–270. [Google Scholar] [CrossRef]
- Laatsch, H. (Ed.) AntiBase 2012; Wiley-VCH: Weinheim, Germany, 2012. Available online: http://eu.wiley.com/WileyCDA/WileyTitle/productCd-3527334068.html (accessed on 5 September 2013).
- Pohland, A.E.; Schuller, P.L.; Steyn, P.S. Physicochemical data for some selected mycotoxins. Pure Appl. Chem. 1982, 54, 2219–2284. [Google Scholar] [CrossRef]
- Raju, M.S.; Wu, G.-S.; Gard, A.; Rosazza, J.P. Microbial transformations of natural antitumor agents. 20. Glucosylation of viridicatumtoxin. J. Nat. Prod. 1982, 45, 321–327. [Google Scholar] [CrossRef]
- Shao, R.G.; Shimizu, T.; Pommier, Y. Brefeldin A is a potent inducer of apoptosis in human cancer cells independently of p53. Exp. Cell Res. 1996, 227, 190–196. [Google Scholar] [CrossRef]
- Seya, H.; Nakajima, S.; Kawai, K.-I.; Udagawa, S.-I. Structure and absolute configuration of emestrin, a new macrocyclic epidithiodioxopiperazine from Emericella striata. J. Chem. Soc. Chem. Comm. 1985, 739, 657–658. [Google Scholar]
- Seya, H.; Nozawa, K.; Nakajima, S.; Kawai, K.-I.; Udagawa, S.-I. Studies on fungal products. Part 8. Isolation and structure of emestrin, a novel antifungal macrocyclic epidithiodioxopiperazine from Emericeella striata. X-Ray molecular structure of emestrin. J. Chem. Soc. Perk. T. 1 1986, 67, 109–116. [Google Scholar]
- Ueno, Y.; Umemori, K.; Nilmi, E.; Tanuma, S.; Nagata, S.; Sugamata, M.; Ihara, T.; Sekljlma, M.; Kawai, K.-I.; Ueno, I.; et al. Induction of apoptosis by T-2 toxin and other natural toxins in HL-60 human promyelotic leukemia cells. Nat. Toxins 1995, 3, 129–137. [Google Scholar] [CrossRef]
- Terao, K.; Ito, E.; Kawai, K.; Nozawa, K.; Udagawa, S. Experimental acute poisoning in mice inducedby emestrin, a new mycotoxin isolated from Emericella species. Mycopathologia 1990, 112, 71–79. [Google Scholar] [CrossRef]
- Lansden, J.A.; Cole, R.J.; Dorner, J.W.; Cox, R.H.; Cutler, H.G.; Clark, J.D. A new trichothecene mycotoxin isolated from Fusarium tricinctum. J. Agric. Food Chem. 1978, 26, 242–244. [Google Scholar] [CrossRef]
- Singh, S.B.; Smith, J.L.; Sabnis, G.S.; Dombrowski, A.W.; Schaeffer, J.M.; Goetz, M.A.; Bills, G.F. Structure and conformation of ophiobolin K and 6- epiophiobolin K from Aspergillus ustus as a nematocidal agent. Tetrahedron 1991, 47, 6931–6938. [Google Scholar] [CrossRef]
- Krizsán, K.; Bencsik, O.; Nyilasi, I.; Galgóczy, L.; Vágvölgyi, C.; Papp, T. Effect of the sesterterpene-type metabolites, ophiobolins A and B, on zygomycetes fungi. FEMS Microbiol. Lett. 2010, 313, 135–140. [Google Scholar]
- Zhang, D.; Fukuzawa, S.; Satake, M.; Li, X.; Kuranaga, T.; Niitsu, A.; Yoshizawa, K.; Tachibana, K. Ophiobolin O and 6-epi-ophiobolin O, two new cytotoxic sesterterpenes from the marine derived fungus Aspergillus sp. Nat. Prod. Commun. 2012, 7, 1411–1414. [Google Scholar]
- Yang, T.; Lu, Z.; Meng, L.; Wei, S.; Hong, K.; Zhu, W.; Huang, C. The novel agent ophiobolin O induces apoptosis and cell cycle arrest of MCF-7 cells through activation of MAPK signaling pathways. Bioorg. Med. Chem. Lett. 2012, 22, 579–585. [Google Scholar] [CrossRef]
- Wang, Q.-X.; Yang, J.-L.; Qi, Q.-Y.; Bao, L.; Yang, X.-L.; Liu, M.-M.; Huang, P.; Zhang, L.-X.; Chen, J.-L.; Cai, L.; et al. 3-Anhydro-6-hydroxy-ophiobolin A, a new sesterterpene inhibiting the growth of methicillin-resistant Staphylococcus aureus and inducing the cell death by apoptosis on K562, from the phytopathogenic fungus Bipolaris oryzae. Bioorgan. Med. Chem. Lett. 2013, 23, 3547–3550. [Google Scholar] [CrossRef]
- Wang, Q.-X.; Bao, L.; Yang, X.-L.; Liu, D.-L.; Guo, H.; Dai, H.-Q.; Song, F.-H.; Zhang, L.-X.; Guo, L.-D.; Li, S.-J.; et al. Ophiobolins P-T, five new cytotoxic and antibacterial sesterterpenes from the endolichenic fungus Ulocladium sp. Fitoterapia 2013, 90, 220–227. [Google Scholar] [CrossRef]
- Au, T.K.; Chick, W.S.; Leung, P.C. The biology of ophiobolins. Life Sci. 2000, 67, 733–742. [Google Scholar] [CrossRef]
- Nozoe, S.; Morisaki, M.; Tsuda, K.; Takahashi, N.; Tamura, S.; Ishibashi, K.; Schirasaka, M. The structure of ophiobolin, a C25 terpenoid having a novel skeleton. J. Am. Chem. Soc. 1965, 87, 4968–4970. [Google Scholar]
- Cutler, H.G.; Crumley, F.G.; Cox, R.H.; Springer, J.P.; Arrendale, R.F.; Cole, R.J.; Cole, P.D. Ophiobolins G and H: New fungal metabolites from a novel source, Aspergillus ustus. J. Agric. Food Chem. 1984, 32, 778–782. [Google Scholar]
- Chiba, R.; Minami, A.; Gomi, K.; Oikawa, H. Identification of Ophiobolin F synthase by a genome mining approach: A sesterterpene synthase from Aspergillus clavatus. Org. Lett. 2013, 15, 594–597. [Google Scholar] [CrossRef]
- Shen, X.; Krasnoff, S.B.; Lu, S.W.; Dunbar, C.D.; O’Neal, J.; Turgeon, B.G.; Yoder, O.C.; Gibson, D.M.; Hamann, M.T. Characterization of 6-epi-3-anhydroophiobolin B from Cochliobolus heterostrophus. J. Nat. Prod. 1999, 62, 895–897. [Google Scholar] [CrossRef]
- Wei, H.; Itoh, T.; Kinoshita, M.; Nakai, Y.; Kurotaki, M.; Kobayashi, M. Cytotoxic sesterterpenes, 6-epi-ophiobolin G and 6-epi-ophiobolin N, from marine derived fungus Emericella variecolor GF10. Tetrahedron 2004, 60, 6015–6019. [Google Scholar] [CrossRef]
- Fujiwara, H.; Matsunaga, K.; Kumagai, H.; Ishizuka, M.; Ohizumi, Y. Ophiobolin A, a novel apoptosis-inducing agent from fungus strain f-7438. Pharm. Pharmacol. Commun. 2000, 6, 427–431. [Google Scholar] [CrossRef]
- Bury, M.; Novo-Uzal, E.; Andolfi, A.; Cimini, S.; Wauthoz, N.; Heffeter, P.; Lallemand, B.; Avolio, F.; Delporte, C.; Cimmino, A.; et al. Ophiobolin A, a sesterterpenoid fungal phytotoxin, displays higher in vitro growth-inhibitory effects in mammalian than in plant cells and displays in vivo antitumor activity. Int. J. Oncol. 2013, 43, 575–585. [Google Scholar]
- Samson, R.A.; Varga, J.; Meijer, M.; Frisvad, J.C. New taxa in Aspergillus section Usti. Stud. Mycol. 2011, 69, 81–97. [Google Scholar] [CrossRef]
- Nozoe, S.; Hirai, K.; Tusda, K. The structure of zizanin-A and -B, C25-terpenoids isolated from Helminthosporium zizaniae. Tetrahedron Lett. 1966, 20, 2211–2216. [Google Scholar] [CrossRef]
- Li, E.; Clark, A.M.; Rotella, D.P.; Hufford, C.D. Microbial metabolites of ophiobolin A and antimicrobial evaluation of ophiobolins. J. Nat. Prod. 1995, 58, 74–81. [Google Scholar] [CrossRef]
- Tsipouras, A.; Adefarati, A.A.; Tkacz, J.S.; Frazier, E.G.; Rohrer, S.P.; Birzin, E.; Rosegay, A.; Zink, D.L.; Goetz, M.A.; Singh, S.B.; et al. Ophiobolin M and analogues, noncompetitive inhibitors of ivermectin binding with nematocidal activity. Bioorg. Med. Chem. 1996, 4, 531–536. [Google Scholar] [CrossRef]
- Canonica, L.; Fiecchi, A.; Galli Kienle, M.; Ranzi, B.M.; Scala, A. The biosynthesis of ophiobolins. Tetrahedron Lett. 1967, 35, 3371–3376. [Google Scholar]
- De Vries-van Leeuwen, I.J.; Kortekaas-Thijssen, C.; Mandouckou, J.A.N.; Kas, S.; Evidente, A.; de Boer, A.H. Fusicoccin-A selectively induces apoptosis in tumor cells after interferon-alpha priming. Cancer Lett. 2010, 293, 198–206. [Google Scholar] [CrossRef]
- Evidente, A.; Andolfi, A.; Cimmino, A.; Vurro, M.; Fracchiolla, M.; Charudattan, R. Herbicidal potential of ophiobolins produced by Drechslera gigantea. J. Agric. Food Chem. 2006, 54, 1779–1783. [Google Scholar] [CrossRef]
- Seiffert, M.; Schulz, A.; Ohl, S.; Döhner, H.; Stilgenbauer, S.; Lichter, P. Soluble CD14 is a novel monocyte-derived survival factor for chronic lymphocytic leukemia cells, which is induced by CLL cells in vitro and present at abnormally high levels in vivo. Blood 2010, 116, 4223–4230. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bladt, T.T.; Dürr, C.; Knudsen, P.B.; Kildgaard, S.; Frisvad, J.C.; Gotfredsen, C.H.; Seiffert, M.; Larsen, T.O. Bio-Activity and Dereplication-Based Discovery of Ophiobolins and Other Fungal Secondary Metabolites Targeting Leukemia Cells. Molecules 2013, 18, 14629-14650. https://doi.org/10.3390/molecules181214629
Bladt TT, Dürr C, Knudsen PB, Kildgaard S, Frisvad JC, Gotfredsen CH, Seiffert M, Larsen TO. Bio-Activity and Dereplication-Based Discovery of Ophiobolins and Other Fungal Secondary Metabolites Targeting Leukemia Cells. Molecules. 2013; 18(12):14629-14650. https://doi.org/10.3390/molecules181214629
Chicago/Turabian StyleBladt, Tanja Thorskov, Claudia Dürr, Peter Boldsen Knudsen, Sara Kildgaard, Jens Christian Frisvad, Charlotte Held Gotfredsen, Martina Seiffert, and Thomas Ostenfeld Larsen. 2013. "Bio-Activity and Dereplication-Based Discovery of Ophiobolins and Other Fungal Secondary Metabolites Targeting Leukemia Cells" Molecules 18, no. 12: 14629-14650. https://doi.org/10.3390/molecules181214629