OxymaPure/DIC: An Efficient Reagent for the Synthesis of a Novel Series of 4-[2-(2-Acetylaminophenyl)-2-oxo-acetylamino] Benzoyl Amino Acid Ester Derivatives

 ,
,
Abstract
:1. Introduction

2. Results and Discussion



{kind=link}
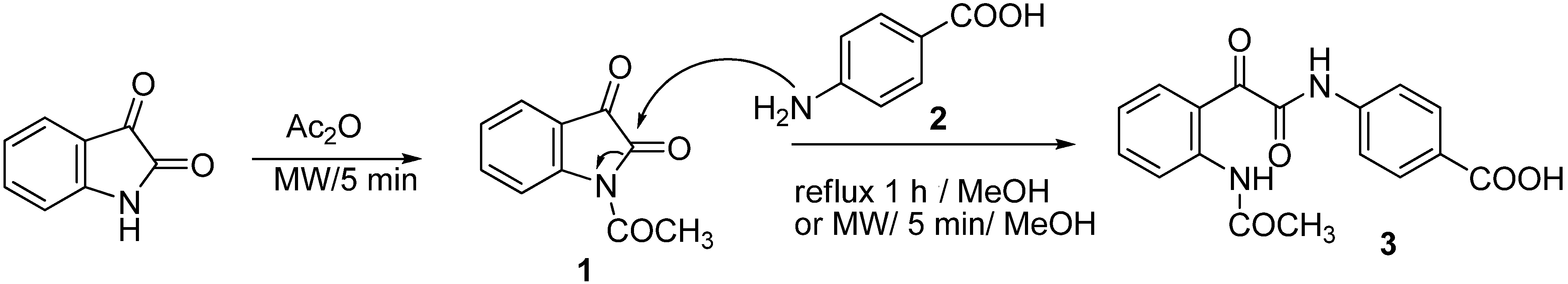{kind=link}
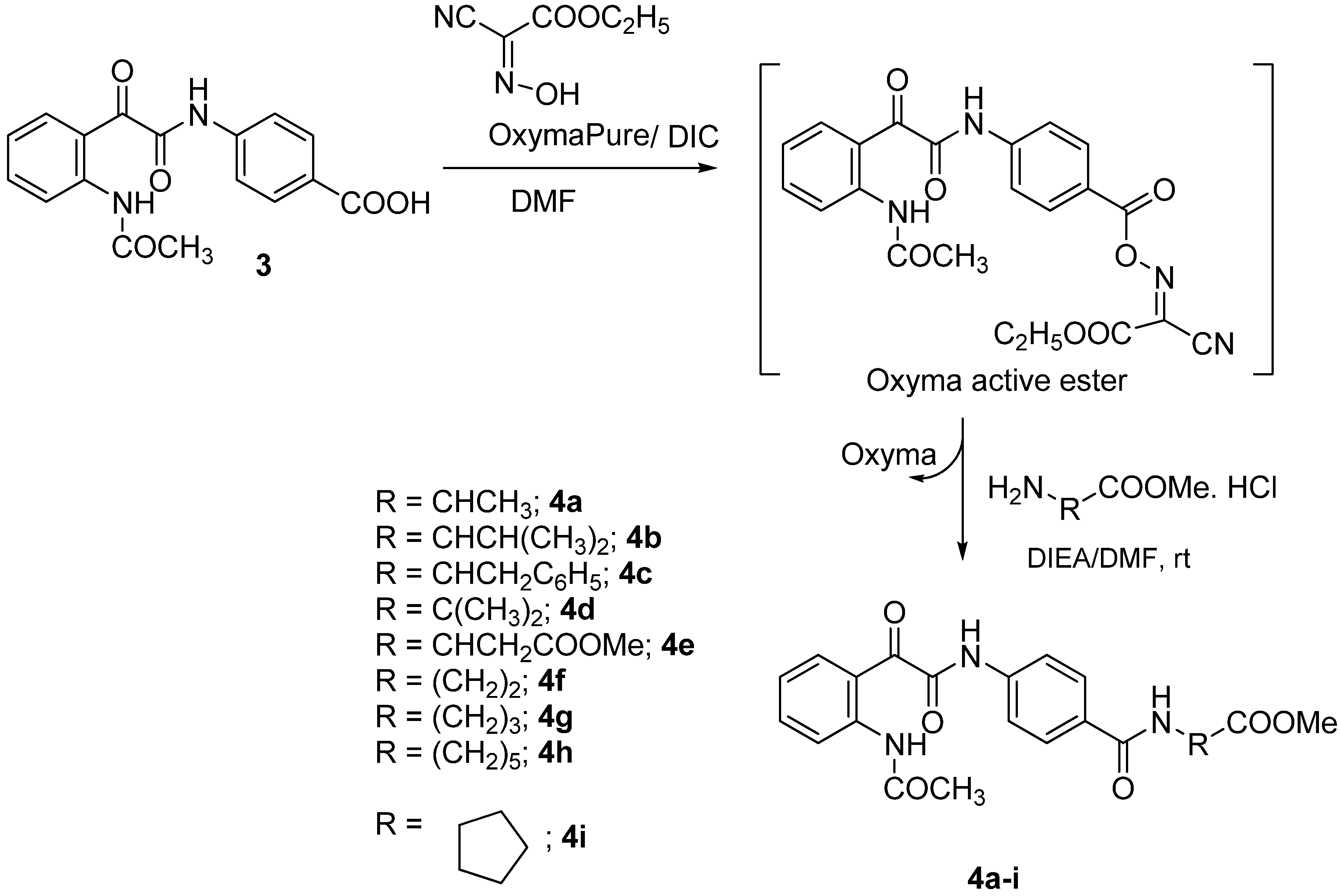{kind=link}
| Coupling Condition | Yield (%) | Mp (°C) |
|---|---|---|
| DIC/Oxyma | 88 | 174–176 |
| DCC/Oxyma * | 82 | 168–172 |
| DIC/HOBt | 72 | 172–175 |
| DCC/HOBt * | 70 | 170–174 |
| DIC | 60 | 170–173 |
| DCC * | 60 | 168–173 |
| Compd. No | Yield (%) | Mp (°C) | Elemental Analysis Calcd. (Found) | ||
|---|---|---|---|---|---|
| C | H | N | |||
| 4a | 88 | 174–176 | 61.31 (61.60) | 5.14 (5.34) | 10.21 (10.00) |
| 4b | 81 | 178–180 | 62.86 (63.02) | 5.73 (5.96) | 9.56 (9.581) |
| 4c | 83 | 168–170 | 66.52 (66.67) | 5.17 (5.24) | 8.62 (8.88) |
| 4d | 76 | 216–218 | 62.11 (62.38) | 5.45 (5.66) | 9.88 (10.07) |
| 4e | 82 | 154–156 | 66.52 (66.33) | 5.17 (5.26) | 8.62( 8.90) |
| 4f | 88 | 154–156 | 61.31 (61.09) | 5.14 (5.23) | 10.21 (10.48) |
| 4g | 86 | 118–120 | 62.11 (62.37) | 5.45 (5.67) | 9.88 (10.04) |
| 4h | 83 | 180–182 | 63.56 (63.38) | 6.00 (6.13) | 9.27 (9.53) |
| 4i | 78 | 238–240 | 63.85 (64.06) | 5.58 (5.65) | 9.31 (9.04) |
3. Experimental
3.1. General
3.2. Synthesis of 1-Acetyl-1H-indole-2,3-dione (N-acetylisatin) (1)
3.3. Synthesis of 4-(2-(2-Acetamidophenyl)-2-oxoacetamido)benzoic Acid (3)
3.4. General Method for the Synthesis of Amino Acid Esters
3.5. General Procedure for the Synthesis of 4a–i
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Li, Z.; Ortega-Vilain, A.C.; Patil, G.S.; Chu, D.L.; Foreman, J.E.; Eveleth, D.D.; Powers, J.C. Novel peptidyl α-Keto amide inhibitors of calpains and other cysteine proteases. J. Med. Chem. 1996, 39, 4089–4098. [Google Scholar] [CrossRef]
- James, D.A.; Koya, K.; Li, H.; Liang, G.; Xia, Z.; Ying, W.; Wu, Y.; Sun, L. Indole- and indolizine-glyoxylamides displaying cytotoxicity against multidrug resistant cancer cell lines. Bioorg. Med. Chem. Lett. 2008, 18, 1784–1787. [Google Scholar] [CrossRef]
- Montalban, A.G.; Boman, E.; Chang, C.D.; Ceide, S.C.; Dahl, R.; Dalesandro, D.; Delaet, N.G.J.; Erb, E.; Ernst, J.T.; Gibbs, A.; et al. The design and synthesis of novel α-ketoamide-based p38 MAP kinase inhibitors. J. Bioorg. Med. Chem. Lett. 2008, 18, 1772–1777. [Google Scholar] [CrossRef]
- Yu, P.-F.; Chen, H.; Wang, J.; He, C.-X.; Cao, B.; Li, M.; Yang, N.; Lei, Z.Y.; Cheng, M.-S. Design, synthesis and cytotoxicity of novel podophyllotoxin derivatives. Chem. Pharm. Bull. 2008, 56, 831–834. [Google Scholar] [CrossRef]
- Perni, R.B.; Farmer, L.J.; Cottrell, K.M.; Court, J.J.; Courtney, L.F.; Deininger, D.D.; Gates, C.A.; Harbeson, S.L.; Kim, J.L.; Lin, C.; et al. Inhibitors of hepatitis C virus NS3·4A protease. Part 3: P2 proline variants. Bioorg. Med. Chem. Lett. 2004, 14, 1939–1942. [Google Scholar] [CrossRef]
- Victor, F.; Lamar, J.; Snyder, N.; Yip, Y.; Guo, D.; Yumibe, N.; Johnson, R.B.; Wang, Q.M.; Glass, J.I.; Chen, S.H. P1 and P3 optimization of novel bicycloproline P2 bearing tetrapeptidyl α-ketoamide based HCV protease inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 257–261. [Google Scholar]
- Banfi, L.; Guanti, G.; Riva, R. Passerini multicomponent reaction of protected alpha-aminoaldehydes as a tool for combinatorial synthesis of enzyme inhibitors. J. Chem. Soc. Chem. Commun. 2000, 985–986. [Google Scholar] [CrossRef]
- Nakamura, M.; Inoue, J.; Yamada, T. A two-step, one-pot synthesis of diverse N-pyruvoyl amino acid derivatives using the Ugi reaction. Bioorg. Med. Chem. Lett. 2000, 10, 2807–2810. [Google Scholar] [CrossRef]
- Xu, P.; Lin, W.; Zhou, X. Synthesis of a peptidomimetic HCMV protease inhibitor library. Synthesis 2002, 8, 1017–1026. [Google Scholar]
- Chen, J.J.; Deshpande, V. Rapid synthesis of α-ketoamides using microwave irradiation simultaneous cooling method. Tetrahedron Lett. 2003, 44, 8873–8876. [Google Scholar] [CrossRef]
- Faggi, C.; Neo, A.G.; Marcaccini, S.; Menchi, G.; Revuelta, J. Ugi four-component condensation with two cleavable components: The easiest synthesis of 2,N-diarylglycines. Tetrahedron Lett. 2008, 49, 2099–2102. [Google Scholar] [CrossRef]
- Grassot, J.M.; Masson, G.; Zhu, J. Synthesis of α-ketoamides by a molecular-sieves-promoted formal oxidative coupling of aliphatic aldehydes with isocyanides. Angew. Chem. Int. Ed. 2008, 47, 947–950. [Google Scholar] [CrossRef]
- Chen, Y.H.; Zhang, Y.H.; Zhang, H.J.; Liu, D.Z.; Gu, M.; Li, J.Y.; Wu, F.; Zhu, X.Z.; Li, J.; Nan, F.J. Design, synthesis, and biological evaluation of isoquinoline-1,3,4-trione derivatives as potent Caspase-3 inhibitors. J. Med. Chem. 2006, 49, 1613–1623. [Google Scholar] [CrossRef]
- Song, B.; Wang, S.; Sun, C.; Deng, H.; Xu, B. Cesium carbonate promoted aerobic oxidation of arylacetamides: An efficient access to N-substituted α-keto amides. Tetrahedon Lett. 2007, 48, 8982–8986. [Google Scholar] [CrossRef]
- Chen, C.T.; Bettigeri, S.; Weng, S.S.; Pawar, V.D.; Lin, Y.H.; Liu, C.Y.; Lee, W.Z. Asymmetric aerobic oxidation of α-hydroxy acid derivatives by C4-Symmetric, Vanadate-Centered, Tetrakisvanadyl(V) clusters derived from N-Salicylidene-α-aminocarboxylates. J. Org. Chem. 2007, 72, 8175–8185. [Google Scholar] [CrossRef]
- Lamberth, C.; Jeanguenat, A.; Cederbaum, F.; de Mesmaecker, A.; Zeller, M.; Kempf, H.J.; Zeun, R. Multicomponent reactions in fungicide research: The discovery of mandipropamid. Bioorg. Med. Chem. 2008, 16, 1531–1545. [Google Scholar] [CrossRef]
- Sanz, R.; Castroviejo, M.P.; Guilarte, V.; Pérez, A.; Fañanás, F.J. Regioselective synthesis of 4- and 7-Alkoxyindoles from 2,3-Dihalophenols: Application to the preparation of indole inhibitors of phospholipase A2. J. Org. Chem. 2007, 72, 5113–5118. [Google Scholar] [CrossRef]
- Maresh, J.J.; Giddings, L.-A.; Friedrich, A.; Loris, E.A.; Panjikar, S.; Trout, B.L.; Stöckigt, J.; Peters, B.; O’Connor, S.E. Strictosidine synthase: Mechanism of a pictet−spengler catalyzing enzyme. J. Am. Chem. Soc. 2008, 130, 710–723. [Google Scholar] [CrossRef]
- Hua, R.; Takeda, H.A.; Abe, Y.; Tanaka, M. Reactions of a carbamoylstannane with acid chlorides: Highly efficient synthesis of α-Oxo amides. J. Org. Chem. 2004, 69, 974–976. [Google Scholar] [CrossRef]
- Arasappan, A.; Venkatraman, S.; Padilla, A.I.; Wu, W.; Meng, T.; Jin, Y.; Wong, J.; Prongay, A.; Girijavallabhan, V.; Njoroge, F.G. Practical and efficient method for amino acid derivatives containing β-quaternary center: Application toward synthesis of hepatitis C virus NS3 serine protease inhibitors. Tetrahedron Lett. 2007, 48, 6343–6347. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, F.; Li, Y.; Sun, X.; Liu, X.; Huang, Y.; Zhang, L.-H.; Ye, X.-S.; Xiao, J. Rapid synthesis of iminosugar derivatives for cell-based in situ screening: Discovery of “Hit” compounds with anticancer activity. ChemMedChem 2007, 2, 1594–1597. [Google Scholar] [CrossRef]
- González, J.F.; de la Cuesta, E.; Avendaño, C. Atom-efficient synthesis of 2,6-diazacyclophane compounds through alcoholysis/reduction of 3-nitroarylmethylene-2,5-piperazinediones. Tetrahedron 2008, 64, 2762–2771. [Google Scholar] [CrossRef]
- Popp, F.D.; Piccirilli, M. The reaction of N-acetylisatin with amines. J. Heterocycl. Chem. 1971, 8, 473–475. [Google Scholar] [CrossRef]
- Obafemi, C.A.; Taiwo, F.O.; Iwalewai, E.O.; Akinpelu, D.A. Synthesis, antibacterial and anti-inflammatory activities of some 2-phenylglyoxylic acid derivatives. Int. J. Life Sci. 2013, 2, 22–36. [Google Scholar]
- Andreani, A.; Burnelli, S.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Cremonini, M.A.; Placucci, G.; et al. New isatin derivatives with antioxidant activity. Eur. J. Med. Chem. 2010, 45, 1374–1378. [Google Scholar] [CrossRef]
- Boechat, N.; Kover, W.B.; Bastos, M.M.; Pinto, A.C.; Maciel, L.C.; Mayer, L.M.U.; da Silva, F.S.Q.; Sá, P.M.; Mendonça, J.S.; Wardella, S.M.S.V.; et al. N-Acyl-3,3-difluoro-2-oxoindoles as versatile intermediates for the preparation of different 2,2-difluorophenylacetic derivatives. J. Braz. Chem. Soc. 2008, 19, 445–457. [Google Scholar] [CrossRef]
- Ghazzali, M.; El-Faham, A.; Abdel-Megeed, A.; Al-Farhan, K. Microwave-assisted synthesis, structural elucidation and biological assessment of 2-(2-acetamidophenyl)-2-oxo-N phenyl acetamide and N-(2-(2-oxo-2(phenylamino)acetyl)phenyl)propionamide derivatives. J. Mol. Struct. 2012, 1013, 163–167. [Google Scholar]
- Cheah, W.C.; Black, D.S.; Goh, W.K.; Kumar, N. Synthesis of anti-bacterial peptidomimetics derived from N-acylisatins. Tetrahedron Lett. 2008, 49, 2965–2968. [Google Scholar] [CrossRef]
- Cheah, W.C.; Wood, K.; Black, D.S.; Kumar, N. Facile ring-opening of N-acylisatins for the development of novel peptidomimetics. Tetrahedron 2011, 67, 7603–7610. [Google Scholar] [CrossRef]
- Hossain, M.M.; Islam, R.M.; Saha, S.K.; Islam, M.K. An efficient microwave-assisted synthesis of dihydropyrazinones and bis-benzoylketones. Tetrahedron Lett. 2010, 51, 1155–1157. [Google Scholar] [CrossRef]
- Somogy, L. Transformation of Isatin 3-Acylhydrazones under acetylating conditions: Synthesis and structure elucidation of 1,5'-Disubstituted 3'-Acetylspiro[oxindole-3,2'-[1,3,4]oxadiazolines]. Bull. Chem. Soc. Jpn. 2001, 74, 873–881. [Google Scholar] [CrossRef]
- Pandey, S.K. Synthesis and evaluation of anti-inflammatory activity of 3-substituted indole derivatives. J. Pharm. Res. 2010, 3, 2738–2741. [Google Scholar]
- Verma, M.; Pandeya, S.N.; Singh, K.N.; Stables, J. Anticonvulsant activity of Schiff bases of isatin derivatives. Acta Pharm. 2004, 54, 49–56. [Google Scholar]
- Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An efficient additive for peptide synthesis to replace benzotriazole-based HOBt and HOAt with a lower risk of explosion. Chem. Eur. J. 2009, 15, 9394–9403. [Google Scholar] [CrossRef]
- Subirós-Funosas, R.; Khattab, S.N.; Nieto-Rodriguez, L.; El-Faham, A.; Albericio, F. Oxyma as cyanooxime building block for the construction of versatile reagents assisting acylation reactions in peptide and organic chemistry. Aldrichimica Acta 2013, 40, 21–40. [Google Scholar]
- Jad, Y.E.; Khattab, S.N.; El-Faham, A.; Albericio, F. Oxime-based carbonates as useful reagents for both N-protection and peptide coupling. Molecules 2012, 17, 14361–14376. [Google Scholar] [CrossRef]
- Khattab, S.N.; Subirós-Funosas, R.; El-Faham, A.; Albericio, F. Screening of N-alkyl-cyanoacetamido oximes as substitutes for N-Hydroxysuccinimide. ChemistryOpen 2012, 1, 147–152. [Google Scholar] [CrossRef]
- Subiros-Funosas, R.; El-Faham, A.; Albericio, F. Use of Oxyma as pH modulatory agent to be used in the prevention of base-driven side reactions and its effect on 2-chlorotrityl chloride resin. Biopolymer 2012, 98, 89–97. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef]
- Subiros-Funosas, R.; El-Faham, A.; Albericio, F. Aspartimide formation in peptide chemistry: Occurrence, prevention strategies and the role of N-hydroxylamines. Tetrahedron 2011, 67, 8595–8606. [Google Scholar] [CrossRef]
- El-Faham, A.; Subiros-Funosas, R.; Albericio, F. A novel family of onium salts based upon isonitroso Meldrum’s acid proves useful as peptide coupling reagents. Eur. J. Org. Chem. 2010, 2010, 3641–3649. [Google Scholar] [CrossRef]
- Subiros-Funosas, R.; Acosta, G.A.; El-Faham, A.; Albericio, F. Microwave irradiation and COMU: A superior tool for solid phase peptide synthesis. Tetrahedron Lett. 2009, 50, 6200–6202. [Google Scholar] [CrossRef]
- Kudelko, A.; Zieliński, W. An efficient synthesis of new 2-aminomethyl-1,3,4-oxadiazoles from enantiomeric phenylglycine hydrazides. Tetrahedron 2009, 65, 1200–1206. [Google Scholar] [CrossRef]
- Li, J.; Sha, Y. A convenient synthesis of amino acid methyl esters. Molecules 2008, 13, 1111–1119. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compounds are available from authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
El-Faham, A.; Al Marhoon, Z.; Abdel-Megeed, A.; Albericio, F. OxymaPure/DIC: An Efficient Reagent for the Synthesis of a Novel Series of 4-[2-(2-Acetylaminophenyl)-2-oxo-acetylamino] Benzoyl Amino Acid Ester Derivatives. Molecules 2013, 18, 14747-14759. https://doi.org/10.3390/molecules181214747
El-Faham A, Al Marhoon Z, Abdel-Megeed A, Albericio F. OxymaPure/DIC: An Efficient Reagent for the Synthesis of a Novel Series of 4-[2-(2-Acetylaminophenyl)-2-oxo-acetylamino] Benzoyl Amino Acid Ester Derivatives. Molecules. 2013; 18(12):14747-14759. https://doi.org/10.3390/molecules181214747
Chicago/Turabian StyleEl-Faham, Ayman, Zainab Al Marhoon, Ahmed Abdel-Megeed, and Fernando Albericio. 2013. "OxymaPure/DIC: An Efficient Reagent for the Synthesis of a Novel Series of 4-[2-(2-Acetylaminophenyl)-2-oxo-acetylamino] Benzoyl Amino Acid Ester Derivatives" Molecules 18, no. 12: 14747-14759. https://doi.org/10.3390/molecules181214747