New Short Strategy for the Synthesis of the Dibenz[b,f]oxepin Scaffold

Abstract
: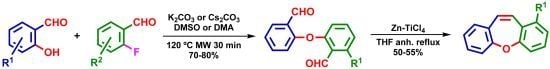
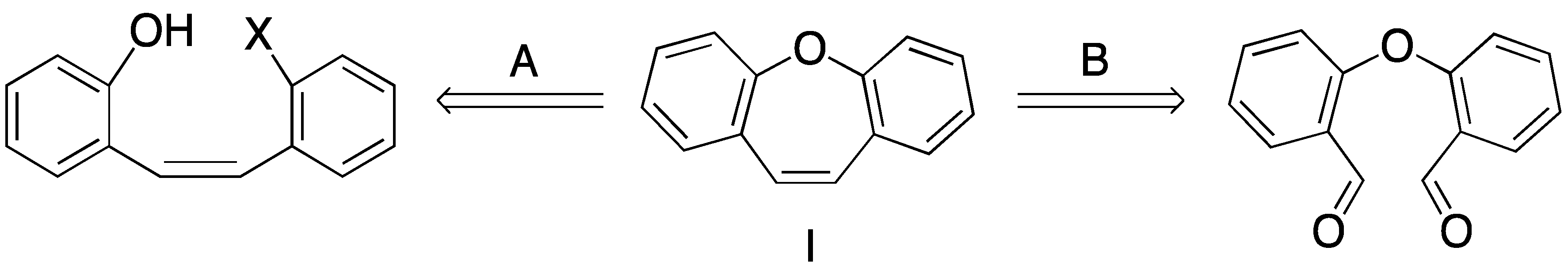
1. Introduction


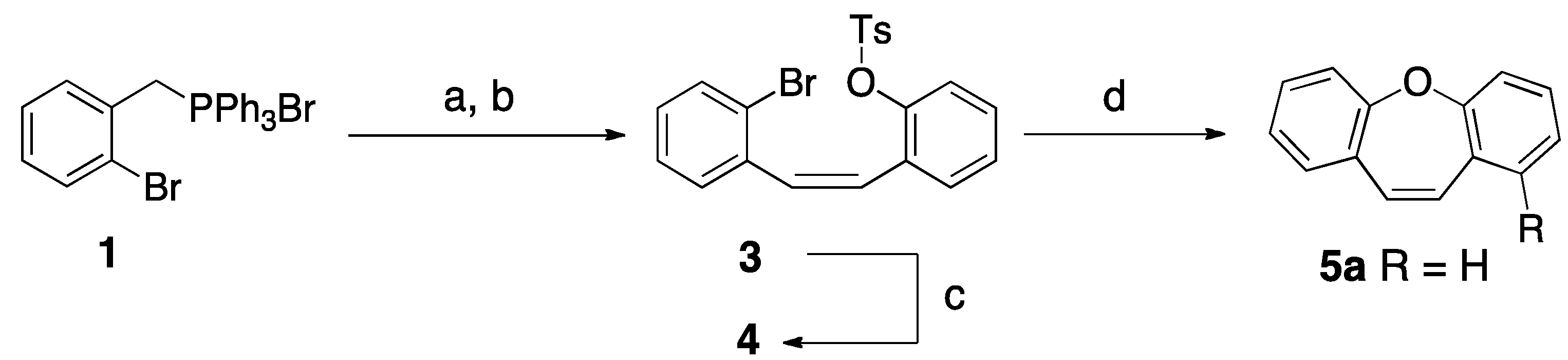
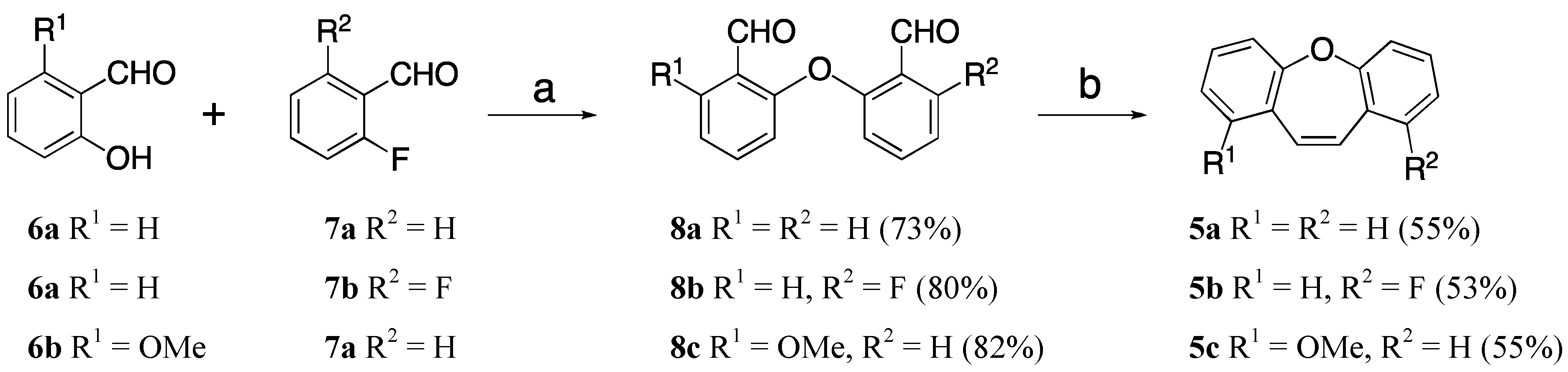
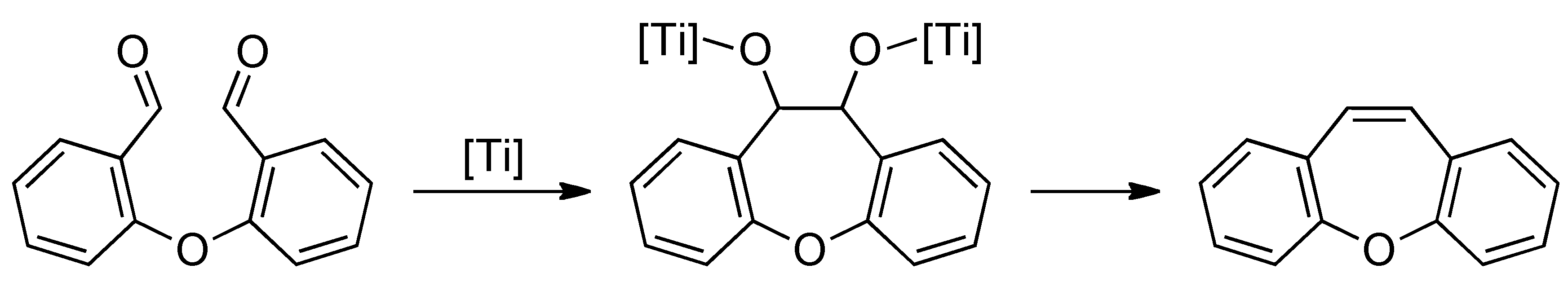
2. Results and Discussion


| Entry | Temp. (°C) | Base | Solvent | Time (min) | Yields (%) |
|---|---|---|---|---|---|
| 1 | 100 °C | K2CO3 | DMSO | 30 | 48% |
| 2 | 110 °C | K2CO3 | DMSO | 30 | 67% |
| 3 | 120 °C | K2CO3 | DMSO | 30 | 73% |
| 4 | 130 °C | K2CO3 | DMSO | 30 | 72% |
| 5 | 140 °C | K2CO3 | DMSO | 30 | 55% |
| 6 | 160 °C | K2CO3 | DMSO | 30 | 3% a |
| 7 | 100 °C | Cs2CO3 | DMSO | 30 | 50% |
| 8 | 110 °C | Cs2CO3 | DMSO | 30 | 65% |
| 9 | 120 °C | Cs2CO3 | DMSO | 30 | 73% |
| 10 | 130 °C | Cs2CO3 | DMSO | 30 | 70% |
| 11 | 140 °C | Cs2CO3 | DMSO | 30 | 45% |
| 12 | 160 °C | K2CO3 | DMA | 30 | 0% a |
| 13 | 120 °C | K2CO3 | DMA | 30 | 72% |
| 14 | 120 °C | Cs2CO3 | DMA | 30 | 73% |
| 15 | 120 °C | K2CO3 | DMA | 24 h | 71% b |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


3. Experimental
3.1. General
 max) were recorded on a Bruker Model Vector 22 spectrophotometer (Bruker Optik GmbH, Bremen, Germany). 1H- (400 MHz) and 13C-NMR (100 MHz) spectra were obtained on a Bruker AM-400 instrument (Bruker BioSpin GmbH, Rheinstetten, Germany), using tetramethylsilane as internal reference. Column chromatography was performed on silica gel Merck 60 (70–230 mesh) (Merck, Darmstadt, Germany). High-resolution mass spectrum was obtained using a Thermo Finnigan mass spectrometer Model MAT 95XP (Thermo Finnigan, San Jose, CA, USA). Microwave-assisted reactions were carried out in an Anton Paar Monowave 300 Microwave Synthesis Reactor (Anton Paar GmbH, Graz, Austria) in 30 mL sealed vials. THF was freshly distilled over sodium. DMSO and DMA were dried over 4 Å molecular sieves prior to use. Cs2CO3 and K2CO3 were dried overnight at 200 °C prior to use. All other reagents were used without further purification.max 3061, 1597, 1444, 1366, 1262, 1025, 810, 722, 671 cm−1. 1H-NMR (CDCl3): δ 2.39 (s, 3H), 6.56 (d, J = 12 Hz, 1H), 6.61 (d, J = 12 Hz, 1H), 6.75 (d, J = 8 Hz, 1H), 6.90–7.10 (m, 6H), 7.29 (d, J = 8 Hz, 2H), 7.54 (d, J = 8 Hz, 1H), 7.80 (d, J = 8 Hz, 2H). 13C-NMR (acetone-d6): δ 22.1, 124.0, 124.8, 126.7, 128.0, 128.4, 130.0 (2C), 130.3, 130.6, 131.4 (2C), 131.7, 131.9, 132.1, 132.6, 133.9, 134.2, 138.3, 147.3, 149.0. HRMS (EI): m/z [M+] calcd for C21H17BrO3S: 428.0082; found: 428.0077. max 3069, 3044, 1483, 798 cm−1. 1H-NMR (acetone-d6) δ 6.82 (s, 2H), 7,19 (t, J = 7,8 Hz, 2H), 7.25 (d, J = 7.8 Hz, 2H), 7.30 (d, J = 7.8 Hz, 2H), 7.38 (t, J = 7.8 Hz, 2H). 13C-NMR (acetone-d6) δ 122.9, 126.6, 131.1, 131.6, 131.7, 132.3, 159.1.
max) were recorded on a Bruker Model Vector 22 spectrophotometer (Bruker Optik GmbH, Bremen, Germany). 1H- (400 MHz) and 13C-NMR (100 MHz) spectra were obtained on a Bruker AM-400 instrument (Bruker BioSpin GmbH, Rheinstetten, Germany), using tetramethylsilane as internal reference. Column chromatography was performed on silica gel Merck 60 (70–230 mesh) (Merck, Darmstadt, Germany). High-resolution mass spectrum was obtained using a Thermo Finnigan mass spectrometer Model MAT 95XP (Thermo Finnigan, San Jose, CA, USA). Microwave-assisted reactions were carried out in an Anton Paar Monowave 300 Microwave Synthesis Reactor (Anton Paar GmbH, Graz, Austria) in 30 mL sealed vials. THF was freshly distilled over sodium. DMSO and DMA were dried over 4 Å molecular sieves prior to use. Cs2CO3 and K2CO3 were dried overnight at 200 °C prior to use. All other reagents were used without further purification.max 3061, 1597, 1444, 1366, 1262, 1025, 810, 722, 671 cm−1. 1H-NMR (CDCl3): δ 2.39 (s, 3H), 6.56 (d, J = 12 Hz, 1H), 6.61 (d, J = 12 Hz, 1H), 6.75 (d, J = 8 Hz, 1H), 6.90–7.10 (m, 6H), 7.29 (d, J = 8 Hz, 2H), 7.54 (d, J = 8 Hz, 1H), 7.80 (d, J = 8 Hz, 2H). 13C-NMR (acetone-d6): δ 22.1, 124.0, 124.8, 126.7, 128.0, 128.4, 130.0 (2C), 130.3, 130.6, 131.4 (2C), 131.7, 131.9, 132.1, 132.6, 133.9, 134.2, 138.3, 147.3, 149.0. HRMS (EI): m/z [M+] calcd for C21H17BrO3S: 428.0082; found: 428.0077. max 3069, 3044, 1483, 798 cm−1. 1H-NMR (acetone-d6) δ 6.82 (s, 2H), 7,19 (t, J = 7,8 Hz, 2H), 7.25 (d, J = 7.8 Hz, 2H), 7.30 (d, J = 7.8 Hz, 2H), 7.38 (t, J = 7.8 Hz, 2H). 13C-NMR (acetone-d6) δ 122.9, 126.6, 131.1, 131.6, 131.7, 132.3, 159.1.3.2. General Procedure for the Preparation of Diarylethers 8
max 1686, 1574, 1473, 1454, 1393, 1301, 1224, 760 cm−1. 1H-NMR (acetone-d6): δ 7.11 (d, J = 8.3 Hz, 2H), 7.39 (t, J = 7.7 Hz, 2H), 7.72 (m, 2H), 7.96 (dd, J = 7.7, 1.5 Hz, 2H), 10.53 (s, 2H), 13C-NMR (acetone-d6): δ 121.6, 126.7, 129.6, 131.0, 138.4, 161.2, 190.9. max 1685, 1611, 1598, 1577, 1396, 787 cm−1. 1H-NMR (CDCl3) δ 6.73 (d, J = 8.3 Hz, 1H), 6.99 (m, 2H), 7.33 (t, J = 7.5 Hz, 1H), 7.54 (m, 1H), 7.62 (ddd, J = 8.3, 7.5, 1.5 Hz, 1H), 7.99 (dd, J = 7.5, 1.5 Hz, 1H), 10.44 (s, 1H), 10.51 (s, 1H), 10.43 (s, 1H). 13C-NMR (CDCl3) δ 112.5, 112.7, 115.0, 115.1, 119.7, 125.3, 127.7, 129.8, 136.4, 136.5, 158.5, 159.0, 186.2, 188.9. HRMS (EI): m/z [M+] calcd for C14H9FO3: 244.0536; found: 244.0532. max 2914, 2862, 2762, 1687, 1600, 1279, 1236, 758, 737 cm−1. 1H-NMR (CDCl3) δ 3.97 (s, 3H), 6.54 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 8.4 Hz, 1H), 6.86 (dd, J = 8.4, 1.8 Hz, 1H), 7.22 (tt, J = 7.8, 1.8 Hz, 1H), 7.48 (t, J = 8.4 Hz, 1H), 7,52 (ddd, J = 8.4, 7.8, 1.8 Hz, 1H), 7.95 (dd, J = 7.8, 1.8 Hz, 1H), 10.48 (s, 1H), 10.52 (s, 1H). 13C-NMR (CDCl3) δ 56.7, 108.0, 112.5, 117.4, 118.8, 124.3, 127.4, 129.1, 136.1, 136.2, 158.3, 159.7, 163.1, 188.4, 189.5. HRMS (EI): m/z [M+] calcd for C15H12O4: 256.0736; found: 256.0733.3.3. General Procedure for the McMurry Reaction
max 3051, 1613, 1571, 1442, 1259, 1007, 774 cm−1. 1H-NMR (acetone-d6) δ 6.93 (d, J = 11.5 Hz, 1H), 6.97 (d, J = 11.5 Hz, 1H), 7.03 (ddd, J = 9.6, 8.4, 1.2 Hz, 1H), 7.13 (dt, J = 8.4, 1.2 Hz, 1H), 7.23 (dd, J = 7.6, 1.2 Hz, 1H), 7.35 (dd, J = 7.6, 1.7 Hz, 1H). 13C-NMR (acetone-d6) δ 112.3, 117.4, 120.0, 121.8, 122.6, 126.6, 129.9, 130.4, 130.5, 130.9, 131.4, 157.7, 159.5, 161.5. HRMS (EI): m/z [M+] calcd for C14H9FO: 212.0637; found: 212.0640.max 1599, 1570, 1464, 1075, 778 cm−1. 1H-NMR (acetone-d6) δ 3.90 (s, 3H), 6.84 (d, J = 11.6 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 7.06 (d, J = 11.6 Hz, 1H), 7.20 (td, J = 7.4, 1.6 Hz, 1H), 7.24 (m, 1H), 7.30 (dd, J = 7.6, 1.6 Hz, 2H), 7.35 (t, J = 8.4 Hz, 1H), 7.38 (ddd, J = 8.4, 7.4, 1.6 Hz, 1H). 13C-NMR (CDCl3) δ 56.2, 107.3, 114.1, 120.1, 121.6, 125.1, 125.2, 129.4, 129.5, 129.9, 130.3, 131.5, 157.5, 157.8, 159.53. HRMS (EI): m/z [M+] calcd for C15H12O2: 224.0837; found: 224.0831.4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Ong, H.H.; Profitt, J.A.; Anderson, V.B.; Spaulding, T.C.; Wilker, J.C.; Geyer, H.M., 3rd; Kruse, H. Tricyclics with analgesic and antidepressant activity. 1. [[(Alkylamino)ethyl]thio]dibenz[b,f]oxepins and 10,11-dihydro derivatives. J. Med. Chem. 1980, 23, 494–501. [Google Scholar] [CrossRef]
- Trabanco, A.A.; Alonso, J.M.; Andres, J.I.; Cid, J.M.; Fernandez, J.; Iturrino, L.; Megens, A. Synthesis of 2-N,N-dimethylaminomethyl-2,3,3a,12b-tetrahydrodibenzo-[b,f]furo[2,3-d]oxepin derivatives as potential anxiolytic agents. Chem. Pharm. Bull. 2004, 52, 262–265. [Google Scholar] [CrossRef]
- Harris, T.W.; Smith, H.E.; Mobley, P.L.; Manier, D.H.; Sulser, F. Affinity of 10-(4-methylpiperazino)dibenz[b,f]oxepins for clozapine and spiroperidol binding sites in rat brain. J. Med. Chem. 1982, 25, 855–858. [Google Scholar] [CrossRef]
- Phillips, S.T.; de Paulis, T.; Neergaard, J.R.; Baron, B.M.; Siegel, B.W.; Seeman, P.; van Tol, H.H.; Guan, H.C.; Smith, H.E. Binding of 5H-dibenzo[a,d]cycloheptene and dibenz[b,f]oxepin analogues of clozapine to dopamine and serotonin receptors. J. Med. Chem. 1995, 38, 708–714. [Google Scholar] [CrossRef]
- Kiyama, R.; Honma, T.; Hayashi, K.; Ogawa, M.; Hara, M.; Fujimoto, M.; Fujishita, T. Novel angiotensin II receptor antagonists. Design, synthesis, and in vitro evaluation of dibenzo[a,d]cycloheptene and dibenzo[b,f]oxepin derivatives. Searching for bioisosteres of biphenylyltetrazole using a three-dimensional search technique. J. Med. Chem. 1995, 38, 2728–2741. [Google Scholar] [CrossRef]
- Nagai, Y.; Irie, A.; Nakamura, H.; Hino, K.; Uno, H.; Nishimura, H. Nonsteroidal antiinflammatory agents. 1. 10,11-Dihydro-11-oxodibenz[b,f]oxepinacetic acids and related compounds. J. Med. Chem. 1982, 25, 1065–1070. [Google Scholar] [CrossRef]
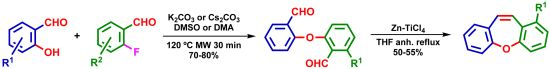
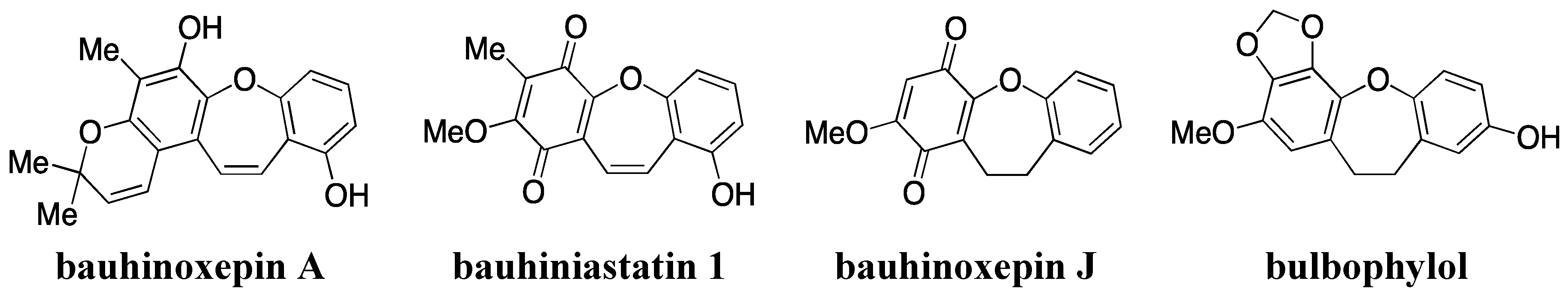
- Filho, V.C. Chemical composition and biological potential of plants from the genus Bauhinia. Phytother. Res. 2009, 23, 1347–1354. [Google Scholar] [CrossRef]
- Kittakoop, P.; Nopichai, S.; Thongon, N.; Charoenchai, P.; Thebtaranonth, Y. Bauhinoxepins A and B: New antimycobacterial dibenzo[b,f]oxepins from Bauhinia saccocalyx. Helv. Chim. Acta 2004, 87, 175–179. [Google Scholar] [CrossRef]
- Pettit, G.R.; Numata, A.; Iwamoto, C.; Usami, Y.; Yamada, T.; Ohishi, H.; Cragg, G.M. Antineoplastic agents. 551. Isolation and structures of bauhiniastatins 1–4 from Bauhinia purpurea. J. Nat. Prod. 2006, 69, 323–327. [Google Scholar] [CrossRef]
- Boonphong, S.; Puangsombat, P.; Baramee, A.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Bioactive compounds from Bauhinia purpurea possessing antimalarial, antimycobacterial, antifungal, anti-inflammatory, and cytotoxic activities. J. Nat. Prod. 2007, 70, 795–801. [Google Scholar] [CrossRef]
- Wu, B.; He, S.; Pan, Y.J. New dihydrodibenzoxepins from Bulbophyllum kwangtungense. Planta Med. 2006, 72, 1244–1247. [Google Scholar] [CrossRef]
- Kraus, G.A.; Thite, A.; Liu, F. Intramolecular radical cyclizations onto quinones. A direct synthesis of Bauhinoxepin J. Tetrahedron Lett. 2009, 50, 5303–5304. [Google Scholar] [CrossRef]
- Narita, K.; Nakamura, K.; Abe, Y.; Katoh, T. Total synthesis of bauhinoxepin J: A biologically active dibenzo[b,f]oxepin isolated from Bauhinia purpurea. Eur. J. Org. Chem. 2011, 2011, 4985–4988. [Google Scholar]
- Lin, J.S.; Zhang, W.; Jiang, N.; Niu, Z.; Bao, K.; Zhang, L.; Liu, D.L.; Pan, C.; Yao, X.S. Total synthesis of Bulbophylol-B. J. Nat. Prod. 2008, 71, 1938–1941. [Google Scholar] [CrossRef]
- Olivera, R.; SanMartin, R.; Churruca, F.; Dominguez, E. Dibenzo[b,f]oxepines: Syntheses and applications. A review. Org. Prep. Proced. Int. 2004, 36, 297–330. [Google Scholar] [CrossRef]
- Choi, Y.L.; Lim, H.S.; Lim, H.J.; Hee, J.N. One-pot transition-metal-free synthesis of dibenzo[b,f]oxepins from 2-halobenzaldehydes. Org. Lett. 2012, 14, 5102–5105. [Google Scholar] [CrossRef]
- Arnold, L.A.; Luo, W.C.; Guy, R.K. Synthesis of medium ring heterocycles using an intramolecular Heck reaction. Org. Lett. 2004, 6, 3005–3007. [Google Scholar] [CrossRef]
- Salas, C.; Tapia, R.A.; Ciudad, K.; Armstrong, V.; Orellana, M.; Kemmerling, U.; Ferreira, J.; Maya, J.D.; Morello, A. Trypanosoma cruzi: Activities of lapachol and alpha- and beta-lapachone derivatives against epimastigote and trypomastigote forms. Bioorg. Med. Chem. 2008, 16, 668–674. [Google Scholar] [CrossRef]
- Salas, C.O.; Faundez, M.; Morello, A.; Maya, J.D.; Tapia, R.A. Natural and synthetic naphthoquinones active against Trypanosoma cruzi: An initial step towards new drugs for Chagas disease. Curr. Med. Chem. 2011, 18, 144–161. [Google Scholar] [CrossRef]
- Pitsinos, E.N.; Vidali, V.P.; Couladouros, E.A. Diaryl ether formation in the synthesis of natural products. Eur. J. Org. Chem. 2011, 2011, 1207–1222. [Google Scholar]
- Mcmurry, J.E. Carbonyl-coupling reactions using low-valent titanium. Chem. Rev. 1989, 89, 1513–1524. [Google Scholar] [CrossRef]
- Shirani, H.; Bergman, J.; Janosik, T. New syntheses of unsymmetrical thiepins and their selenium analogues. Tetrahedron 2009, 65, 8350–8353. [Google Scholar] [CrossRef]
- Harrowven, D.C.; Guy, I.L.; Howell, M.; Packham, G. The synthesis of a combretastatin A-4 based library and discovery of new cooperative ortho-effects in Wittig reactions leading to (Z)-stilbenes. Synlett 2006, 18, 2977–2980. [Google Scholar] [CrossRef]
- Radix, S.; Barret, R. Total synthesis of two natural phenanthrenes: Confusarin and a regioisomer. Tetrahedron 2007, 63, 12379–12387. [Google Scholar] [CrossRef]
- Serban, G.; Abe, H.; Takeuchi, Y.; Harayama, T. A new approach to the benzopyridoxepine core by metal mediated intramolecular biaryl ether formation. Heterocycles 2008, 75, 2949–2958. [Google Scholar] [CrossRef]
- Manske, R.H.F.; Ledingham, A.E. Synthesis and reactions of some dibenzoxepins. J. Am. Chem. Soc. 1950, 72, 4797–4799. [Google Scholar] [CrossRef]
- Ramana, C.V.; Mondal, M.A.; Puranik, V.G.; Gurjar, M.K. A carbohydrate based approach towards the synthesis of aspercyclide C. Tetrahedron Lett. 2007, 48, 7524–7527. [Google Scholar] [CrossRef]
- Maiti, D.; Buchwald, S.L. Cu-Catalyzed arylation of phenols: Synthesis of sterically hindered and heteroaryl diaryl ethers. J. Org. Chem. 2010, 75, 1791–1794. [Google Scholar] [CrossRef]
- Li, F.; Wang, Q.R.; Ding, Z.B.; Tao, F.G. Microwave-assisted synthesis of diaryl ethers without catalyst. Org. Lett. 2003, 5, 2169–2171. [Google Scholar] [CrossRef]
- McMurry, J.E. Titanium-induced dicarbonyl-coupling reactions. Acc. Chem. Res. 1983, 16, 405–411. [Google Scholar] [CrossRef]
- Dieguez, H.R.; Lopez, A.; Domingo, V.; Arteaga, J.F.; Dobado, J.A.; Herrador, M.M.; del Moral, J.F.Q.; Barrero, A.F. Weakening C-O bonds: Ti(III), a new reagent for alcohol deoxygenation and carbonyl coupling olefination. J. Am. Chem. Soc. 2010, 132, 254–259. [Google Scholar] [CrossRef]
- Stahl, M.; Pidun, U.; Frenking, G. On the mechanism of the McMurry reaction. Angew. Chem. Int. Ed. Engl. 1997, 36, 2234–2237. [Google Scholar] [CrossRef]
- Ephritikhine, M. A new look at the McMurry reaction. Chem. Commun. 1998. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, Z.X.; Xiao, Q.; Qu, P.Y.; Ge, R.; Zhang, Y.; Wang, J.B. Rhodium(II)-catalyzed cyclization of bis(N-tosylhydrazone)s: An efficient approach towards polycyclic aromatic compounds. Angew. Chem. Int. Ed. Engl. 2012, 51, 5714–5717. [Google Scholar] [CrossRef]
- Matsuda, T.; Sato, S. Synthesis of dibenzoheteropines of group 13-16 elements via ring-closing metathesis. J. Org. Chem. 2013, 78, 3329–3335. [Google Scholar] [CrossRef]
- Osuka, A.; Kobayashi, F.; Maruyama, K. Synthesis of dimeric and trimeric porphyrins based on intramolecular macrocyclization reactions. Chem. Lett. 1990, 19, 1521–1524. [Google Scholar]
- Yang, Z.; Wong, H.N.C.; Hon, P.M.; Chang, H.M.; Lee, C.M. A novel synthesis of the dibenz[b,f]oxepin ring-system: 10,11-dihydro-11-hydroxydibenz[b,f]oxepin-10(11H)-one. J. Org. Chem. 1992, 57, 4033–4034. [Google Scholar] [CrossRef]
- Zacharie, B.; Attardo, G.; Barriault, N.; Penney, C. Regioselective synthesis of 6-substituted 2-hydroxybenzaldehyde: Efficient synthesis of the immunomodulator tucaresol and related analogues. J. Chem. Soc. Perkin Trans. 1997. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moreno, D.R.R.; Giorgi, G.; Salas, C.O.; Tapia, R.A. New Short Strategy for the Synthesis of the Dibenz[b,f]oxepin Scaffold. Molecules 2013, 18, 14797-14806. https://doi.org/10.3390/molecules181214797
Moreno DRR, Giorgi G, Salas CO, Tapia RA. New Short Strategy for the Synthesis of the Dibenz[b,f]oxepin Scaffold. Molecules. 2013; 18(12):14797-14806. https://doi.org/10.3390/molecules181214797
Chicago/Turabian StyleMoreno, David R. R., Giorgio Giorgi, Cristian O. Salas, and Ricardo A. Tapia. 2013. "New Short Strategy for the Synthesis of the Dibenz[b,f]oxepin Scaffold" Molecules 18, no. 12: 14797-14806. https://doi.org/10.3390/molecules181214797