Efficient PPA-SiO2-catalyzed Synthesis of β-enaminones Under Solvent-Free Conditions


 and
and
Abstract
:1. Introduction
2. Results and Discussion


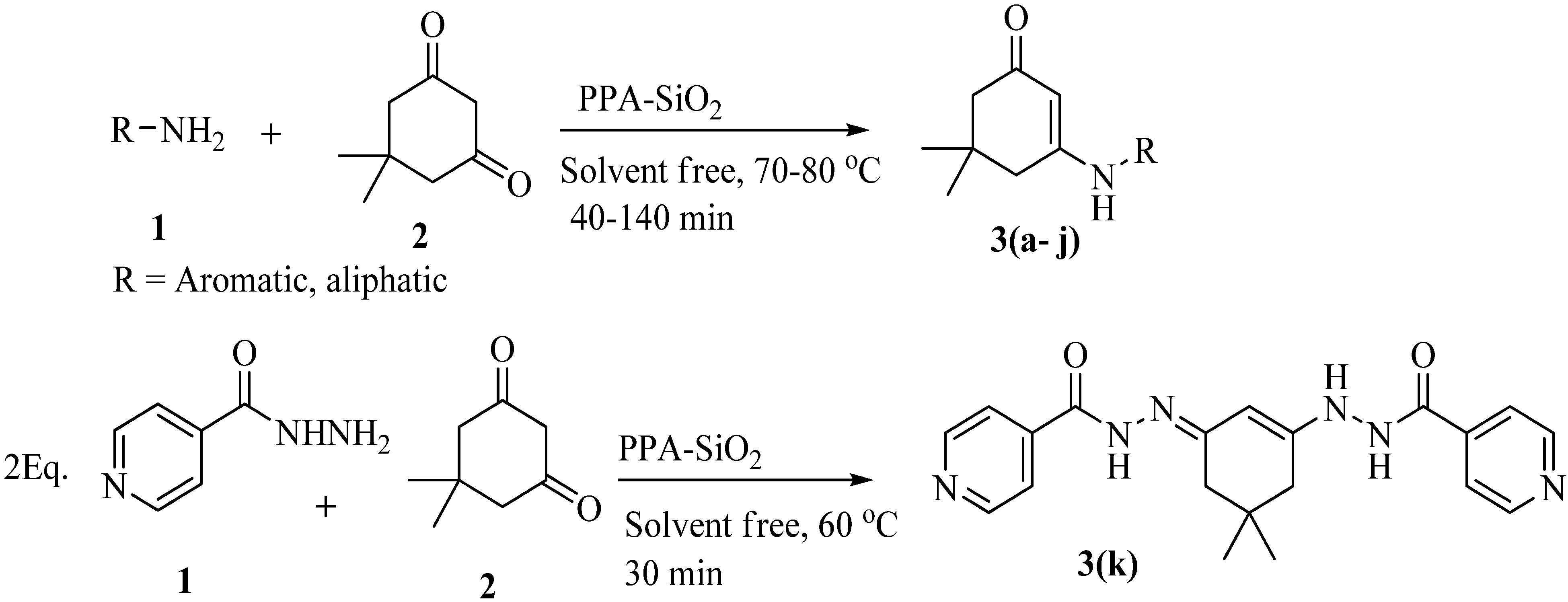{kind=link}
{kind=link}
| Entry a | Solvent | Catalyst | Temperature (°C) | Time (min) | Yield b (%) |
|---|---|---|---|---|---|
| 1. | Ethanol | − | Reflux | 4h | 30 |
| 2. | Water | PPA-SiO2 | 80 | 90 | Traces |
| 3. | Methanol | PPA-SiO2 | Reflux | 130 | 70 |
| 4. | Ethanol | PPA-SiO2 | Reflux | 140 | 71 |
| 5. | CH3CN | PPA-SiO2 | Reflux | 140 | 48 |
| 6. | Chloroform | PPA-SiO2 | Reflux | 130 | 56 |
| 7. | THF | PPA-SiO2 | Reflux | 140 | Traces |
| 8. | Solvent free | PPA-SiO2 | 70 | 40 | 88 |
2.1. Effect of Loading Catalyst
| Entry a | Catalyst (mg) | Time (min) | Yield b (%) |
|---|---|---|---|
| 1. | 50 | 60 | 50 |
| 2. | 80 | 50 | 64 |
| 3. | 110 | 40 | 89 |
| 4. | 130 | 45 | 86 |
| 5. | 150 | 45 | 86 |
2.2. Amines Substrate Scope

| Entry a | R | Time (min) | Product | Yield b (%) | Observed mp (°C) | Lit. mp (°C) |
|---|---|---|---|---|---|---|
| 1. | 1,2-Phenylene -dimethanamine | 40 |  | 89 | 187–188 | 186 [30] |
| 2. | C6H5- | 40 |  | 68 | 152–154 | − |
| 3. | 4-OCH3C6H5- | 45 |  | 74 | 201–202 | 199 [34] |
| 4. | 4-CH3C6H5- | 40 |  | 74 | 201–202 | 199 [34] |
| 5. | 4-BrC6H5- | 40 |  | 88 | 215–217 | 219 [32] |
| 6. | 4-NO2C6H5 | 50 |  | 67 | 194–195 | 191 [32] |
| 7. | (1S,2S)-(+)1-Amino-1-phenyl-1,3-proanediol | 60 |  | 71 | 103–105 | − |
| 8. | D-Threo-2-amino-1-(4-nitrophenyl)-1,3-propanediol | 60 |  | 70 | 176–178 | − |
| 9. | 4-(methyl)cyclohexane carboxylic acid | 140 |  | 48 | 270–272 | − |
| 10. | 2-(1-(methyl)cyclohexyl)acetic acid | 145 |  | 50 | 155–158 | − |
| 11. | Isonicotinic acid Hydrazide | 30 |  | 90 | 181–183 | − |
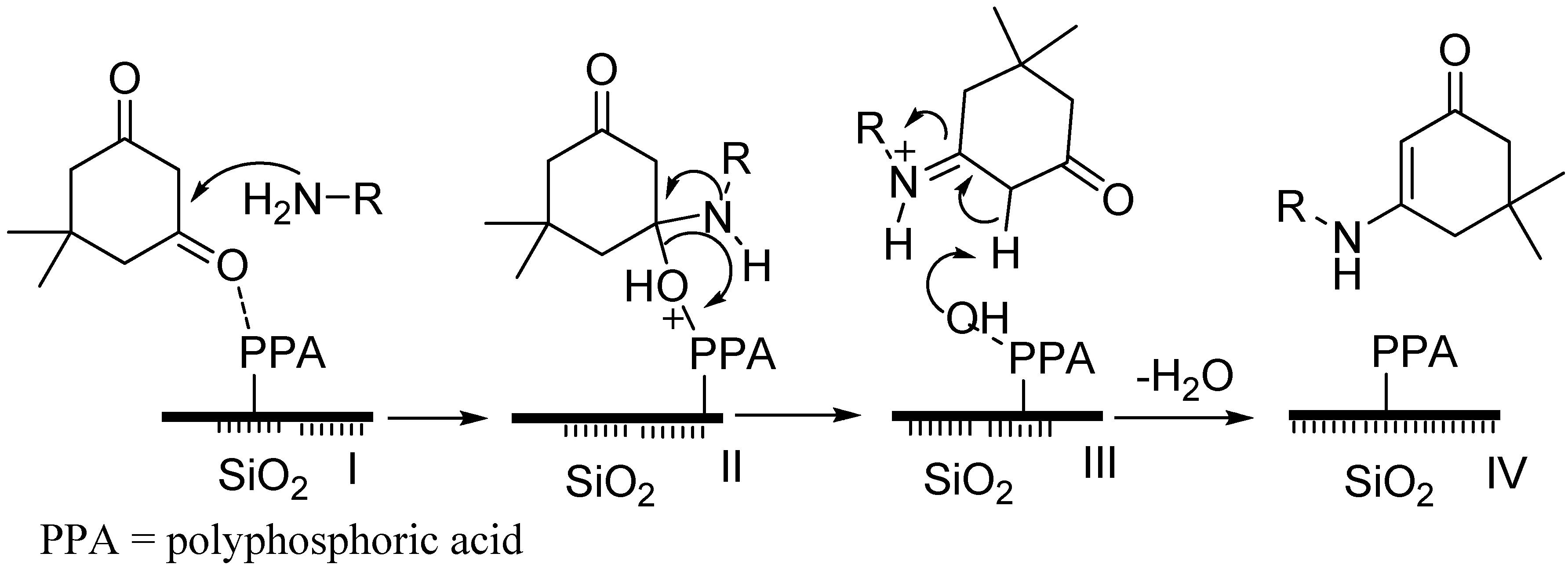
2.3. Proposed Mechanism

3. Experimental
3.1. General
3.2. Preparation of PPA-SiO2 Catalyst
3.3. Typical Procedure for PPA-SiO2 Catalyzed Synthesis of β-Enaminones
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Braibante, M.F.F.; Mara, E.F.; Braibante, H.T.S.; Morel, A.F.; Costa, C.C.; Lima, M.G. The solvent-free preparation of β-amino esters α,β-unsaturated ketones and esters with domestic microwave oven. J. Braz. Chem. Soc. 2006, 17, 184–188. [Google Scholar]
- Negri, G.; Kascheres, C.; Kascheres, A.J. Recent developments in the chemistry of enaminones. J. Heterocycl. Chem. 2004, 41, 461–491. [Google Scholar] [CrossRef]
- Michael, J.P.; Koning, C.B.; Hosken, G.D.; Stanbury, T.V. Reformatsky reactions with N-arylpyrrolidine-2-thiones: Synthesis of tricyclic analogues of quinolone antibacterial agents. Tetrahedron 2001, 57, 9635–9648. [Google Scholar] [CrossRef]
- Eddingtona, N.D.; Coxa, S.D.; Robertsb, R.R.; Stablesc, J.P.; Powelld, C.B.; Scotte, R.K. Enaminones a versatile therapeutic pharmacophores. Further Advances. Curr. Med. Chem. 2000, 7, 417–436. [Google Scholar] [CrossRef]
- Savarin, C.G.; Murry, J.A.; Dormer, P.G. An expedient synthesis of highly functionalized naphthyridones and quinolines from a common N-aryl pyridinone template. Org. Lett. 2002, 4, 2071–2074. [Google Scholar] [CrossRef]
- David, O.; Blot, J.; Bellec, C.; Fargeau-Bellassoued, M.C.; Haviari, G.; Celerier, J.-P.; Lhommet, G.; Gramain, J.-C.; Gardette, D. Enamino ester reduction: A short enantioselectiveroute to pyrrolizidine and indolizidine alkaloids: Synthesis of (+)-laburnine, (+)-tashiromine, and (−)-isoretronecanol. J. Org. Chem. 1999, 64, 3122–3131. [Google Scholar] [CrossRef]
- Yun, X.L.; Bi, W.-Y.; Huang, J.H.; Liu, Y.; Zhang-Negrerie, D.; Du, Y.-F.; Zhao, K. Synthesis of N-substituted carbazolones from a-iodo enaminones via Pd(0)-catalyzed intermolecular coupling under microwave irradiation. Tetrahedron Lett. 2012, 53, 5076–5080. [Google Scholar] [CrossRef]
- Beholz, L.G.; Benovsky, R.; Ward, D.L.; Bata, N.S.; Stille, J.R. Formation of dihydropyridone and pyridone-based peptide analogs through Aza-annulation of β-enamino ester and amide substrates with α-amido acrylate derivatives. J. Org. Chem. 1997, 62, 1033–1042. [Google Scholar] [CrossRef]
- Nakamura, I.; Yamamoto, Y. Transition-Metal-catalyzed reactions in heterocyclic synthesis. Chem. Rev. 2004, 104, 2127–2198. [Google Scholar] [CrossRef]
- Chaaban, I.; Greenhill, J.V.; Akhtar, P. Enaminones in the mannich reaction. Part 2. Further investigations of internal mannich reactions. J. Chem. Soc. Perkin Trans. 1 1979. [Google Scholar] [CrossRef]
- Figueiredo, L.J.O.; Kascheres, C. Quinone Diazides and Enaminones as a Source of New Azo compounds with potential nonlinear optical properties. J. Org. Chem. 1997, 62, 1164–1167. [Google Scholar] [CrossRef]
- Aceña, J.L.; Arjona, O.; Mañas, R.; Plumet, J. Unexpected one-pot epoxy sulfone-enaminone transformation. Synthesis of 5α-carba-β-mannopyranosylamine. J. Org. Chem. 2000, 65, 2580–2582. [Google Scholar] [CrossRef]
- Cimarelli, C.; Palmieri, G. Stereoselective reduction of enantiopure β-enaminone esters byhydride: A convenient synthesis of both enantiopure b-amino esters. J. Org. Chem. 1996, 61, 5557–5563. [Google Scholar] [CrossRef]
- Potin, D.; Dumas, F.; Angelo, J.D. New chiral auxiliaries: Their use in the asymmetric hydrogenation of β-acetamidocrotonates. J. Am. Chem. Soc. 1990, 112, 3483–3486. [Google Scholar] [CrossRef]
- Palmieri, G.; Cimarelli, C. Chemo- and stereoselective reduction of enaminones for the preparation of biologically active compounds. ARKIVOC 2006, 2006, 104–126. [Google Scholar] [CrossRef]
- Jackson, P.L.; Hanson, C.D.; Farrell, A.K.; Butcher, R.J.; Stables, J.P.; Eddington, N.D.; Scott, R.K. Enaminones 12. An explanation of anticonvulsant activity and toxicity per Linus Pauling’s clathrate hypothesis . Eur. J. Med. Chem. 2012, 5, 42–51. [Google Scholar]
- Edafiogho, I.O.; Ananthalakshmi, K.V.; Kombian, S.B. Anticonvulsant evaluation and mechanism of action of benzylamino enaminones. Bioorg. Med. Chem. 2006, 14, 5266–5272. [Google Scholar] [CrossRef]
- Bruno, O.; Schenone, S.; Ranise, A.; Bondavalli, F.; Filippelli, W. Anti-inflammatory agents: New series of N-substituted amino acids with complex pyrimidine structures endowed with antiphlogistic activity. Farmaco 1999, 54, 95–100. [Google Scholar] [CrossRef]
- Farag, A.M.; Mayhoub, A.S.; Barakat, S.E.; Bayomi, A.H. Regioselective synthesis and antitumor screening of some novel N-phenylpyrazole derivatives. Bioorg. Med. Chem. 2008, 16, 881–889. [Google Scholar] [CrossRef]
- Boger, D.L.; Ishizaki, T.; Wysocki, J.R.J.; Munk, S.A.; Kitos, P.A.; Suntornwat, O. Total synthesis and evaluation of (±)-N-(tert-butoxycarbonyl)-CBI, (±)-CBI-CDPI1, and (±)-CBICDPI2: CC-1065 functional agents incorporating the equivalent 1,2,9,9a-tetrahydrocyclopropa[1,2-c]benz[1,2-e]indol-4-one (CBI) left-hand subunit. J. Am. Chem. Soc. 1989, 111, 6461–6463. [Google Scholar] [CrossRef]
- Haycock-Lewandowski, S.J.; Wilder, A.; Ahman, J. Development of a bulk enabling route to Maraviroc (UK-427,857), a CCR-5 receptor antagonist. J. Org. Process Res. Dev. 2008, 12, 1094–1103. [Google Scholar] [CrossRef]
- Baraldi, G.; Simoni, D.; Manfredini, S. An improved preparation of enaminones from 1,3-diketones and ammonium acetate or amine acetates. Synthesis 1983, 902–903. [Google Scholar] [CrossRef]
- Rechsteiner, B.; Boullet, T.F.; Hamelin, J. Synthesis in dry media coupled with microwave irradiation: Application to the preparation of enaminoketones. Tetrahedron Lett. 1993, 34, 5071–5074. [Google Scholar] [CrossRef]
- Arcadi, A.; Bianchi, G.; Giuseppe, D.S.; Marinelli, F. Gold catalysis in the reactions of 1,3-dicarbonyls with nucleophiles. Green Chem. 2003, 5, 64–67. [Google Scholar] [CrossRef]
- Gogoi, S.; Bhuyan, R.; Barua, N.C. Iodine-catalyzed conversion of b-dicarbonyl compounds into β-enaminones within a minute under solvent-free conditions. Synth. Commun. 2005, 35, 2811–2818. [Google Scholar] [CrossRef]
- Das, B.; Venkateswarlu, K.; Majhi, A.; Reddy, M.R.; Reddy, K.N. Highly efficient, mild, and chemo- and stereoselective synthesis of enaminones and enamino esters using silica-supported perchloric acid under solvent-free conditions. J. Mol. Catal. A Chem. 2006, 246, 276–281. [Google Scholar] [CrossRef]
- Gholap, A.R.; Chakor, N.S.; Daniel, T.; Lahoti, R.J.; Srinivasan, K.V. A remarkablyrapid regioselective synthesis of β-enaminones using silica chloride in a heterogeneous as well as an ionic liquid in a homogeneous medium at room temperature. J. Mol. Catal. A Chem. 2006, 245, 37–46. [Google Scholar] [CrossRef]
- Eshghi, H.; Mohammad, S.S.; Safaei, E.; Vakili, M.; Farhadipour, A.; Bayat-Mokhtari, M. Silica supported Fe(HSO4)3 as an efficient, heterogeneous and recyclable catalyst for synthesis of β-enaminones and β-enaminone esters. J. Mol. Catal. A Chem. 2012, 430–436. [Google Scholar]
- Calle, M.; Calvo, L.A.; Gonzalez-Ortega, A.; Gonzalez-Nogal, A.M. Silylated β-enaminones as precursors in the regioselective synthesis of silyl pyrazoles. Tetrahedron 2006, 62, 611–618. [Google Scholar] [CrossRef]
- Kang, J.; Liang, F.; Sun, S.G.; Liu, Q.; Bi, X.H. Copper-mediated C–N bond formationviadirect aminolysis of dithioacetals. Org. Lett. 2006, 8, 2547–2550. [Google Scholar] [CrossRef]
- Seki, H.; Georg, G.I. Synthesis of amino acid-derived enaminones via Wolff rearrangement using vinylogous Amides as carbon nucleophiles. J. Am. Chem. Soc. 2010, 10, 15512–15513. [Google Scholar] [CrossRef]
- Kantevari, S.; Bantu, R.; Nagarapu, L. HClO4–SiO2 and PPA–SiO2 catalyzed efficient one-pot Knoevenagel condensation, Michael addition and cyclodehydration of dimedone and aldehydes in acetonitrile, aqueous and solvent free conditions: Scope and limitations. J. Mol. Catal. A Chem. 2007, 269, 53–57. [Google Scholar] [CrossRef]
- Hasaninejad, A.; Zare, A.K.; Reza, M.M.; Shekouhy, M.; Rezamossavi-Zare, A. A green solventless protocol for the synthesis of β-enaminones and β-enaminone esters using silica sulfuric acid as a highly efficient, heterogeneous and reusable catalyst. E J. Chem. 2010, 7, 1546–1554. [Google Scholar] [CrossRef]
- Karimi-Jaberi, K.; Takmilifard, Z. Efficient synthesis of β-enaminones and β-enaininoester using Tris(hydrogensulfateo)boron or Trichloroacetic acid as catalysts. Eur. Chem. Bull. 2013, 2, 211–213. [Google Scholar]
- Nisar, M.; Ali, I.; Shah, M.R.; Badshah, A.; Qayum, M.; Khan, H.; Khan, I.; Ali, S. Amberlite IR-120H as a recyclable catalyst for the synthesis of 1,8-dioxo-octahydroxanthene analogs and their evaluation as potential leishmanicidal agents. RSC Adv. 2013, 3, 21753–21758. [Google Scholar]
- Sample Availability: Samples of the novel compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nisar, M.; Ali, I.; Shah, M.R.; Qayum, M.; Zia-Ul-Haq, M.; Rashid, U.; Islam, M.S. Efficient PPA-SiO2-catalyzed Synthesis of β-enaminones Under Solvent-Free Conditions. Molecules 2013, 18, 15182-15192. https://doi.org/10.3390/molecules181215182
Nisar M, Ali I, Shah MR, Qayum M, Zia-Ul-Haq M, Rashid U, Islam MS. Efficient PPA-SiO2-catalyzed Synthesis of β-enaminones Under Solvent-Free Conditions. Molecules. 2013; 18(12):15182-15192. https://doi.org/10.3390/molecules181215182
Chicago/Turabian StyleNisar, Muhammad, Ihsan Ali, Muhammad Raza Shah, Mughal Qayum, Muhammad Zia-Ul-Haq, Umer Rashid, and Md. Saiful Islam. 2013. "Efficient PPA-SiO2-catalyzed Synthesis of β-enaminones Under Solvent-Free Conditions" Molecules 18, no. 12: 15182-15192. https://doi.org/10.3390/molecules181215182