Antioxidants Improve the Phenotypes of Dilated Cardiomyopathy and Muscle Fatigue in Mitochondrial Superoxide Dismutase-Deficient Mice


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
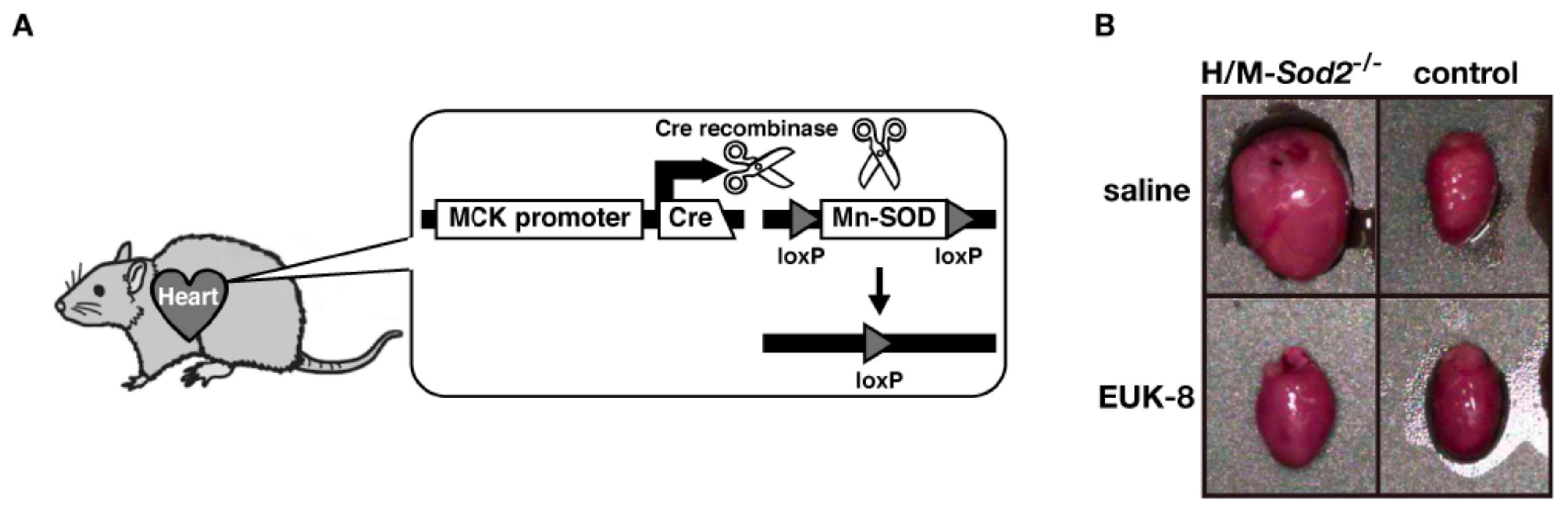
2. Heart and Muscle-Specific Mn-SOD-Deficient Mice
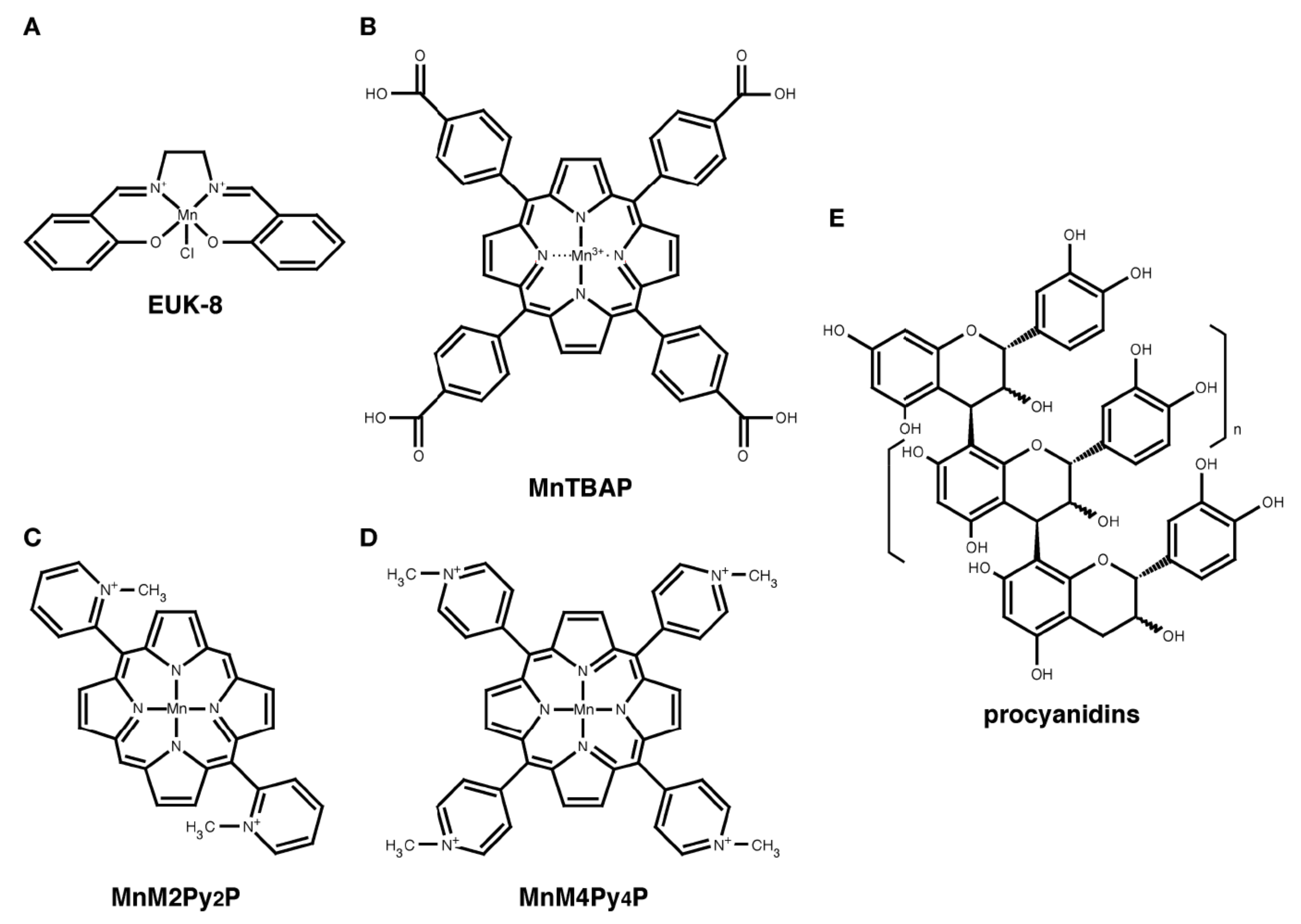
3. EUK-8
4. MnTBAP
5. Manganese Porphyrins
6. Procyanidins
7. Antioxidants for Human DCM
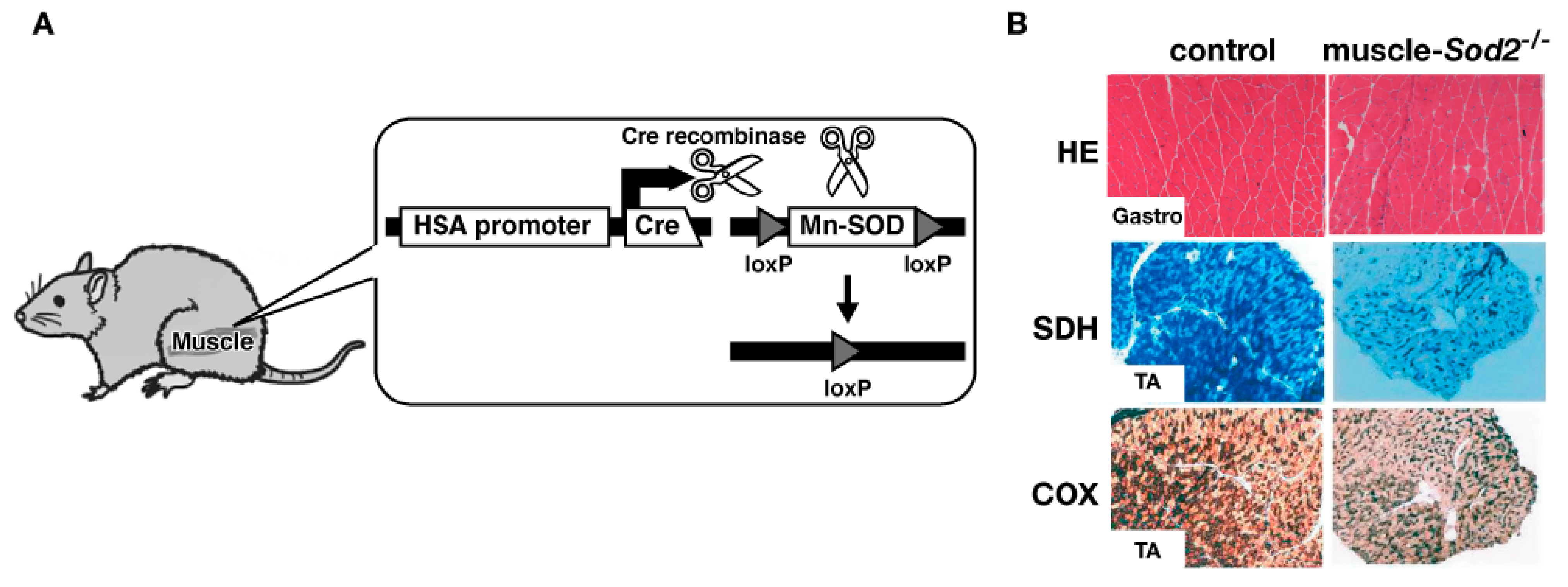
8. Effects of EUK-8 on Muscle
9. Conclusions
Acknowledgments
References
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [PubMed]
- Weisiger, R.A.; Fridovich, I. Mitochondrial superoxide dismutase. Site of synthesis and intramitochondrial localization. J. Biol. Chem. 1973, 248, 4793–4796. [Google Scholar] [PubMed]
- Marklund, S.L. Human copper-containing superoxide dismutase of high molecular weight. Proc. Natl. Acad. Sci. USA 1982, 79, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 2010, 1797, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Schneider, J.A.; Day, B.J.; Hinerfeld, D.; Coskun, P.; Mirra, S.S.; Crapo, J.D.; Wallace, D.C. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat. Genet. 1998, 18, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Coskun, P.; Patel, M.; Tuinstra, R.; Cottrell, B.; Jun, A.S.; Zastawny, T.H.; Dizdaroglu, M.; Goodman, S.I.; Huang, T.T.; et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc. Natl. Acad. Sci. USA 1999, 96, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Alameddine, F.M.; Zafari, A.M. Genetic polymorphisms and oxidative stress in heart failure. Congest. Heart Fail. 2002, 8, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Givertz, M.M.; Colucci, W.S. New targets for heart-failure therapy: Endothelin, inflammatory cytokines, and oxidative stress. Lancet 1998, 352, SI34–SI38. [Google Scholar] [CrossRef]
- Ikegami, T.; Suzuki, Y.; Shimizu, T.; Isono, K.; Koseki, H.; Shirasawa, T. Model mice for tissue-specific deletion of the manganese superoxidedismutase (MnSOD) gene. Biochem. Biophys. Res. Commun. 2002, 296, 729–736. [Google Scholar] [CrossRef]
- Brüning, J.C.; Michael, M.D.; Winnay, J.N.; Hayashi, T.; Hörsch, D.; Accili, D.; Goodyear, L.J.; Kahn, C.R. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 1998, 2, 559–569. [Google Scholar] [CrossRef]
- Nojiri, H.; Shimizu, T.; Funakoshi, M.; Yamaguchi, O.; Zhou, H.; Kawakami, S.; Ohta, Y.; Sami, M.; Tachibana, T.; Ishikawa, H.; et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. J. Biol. Chem. 2006, 281, 33789–33801. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, T.; Watanabe, K.; Ozawa, Y.; Nakashima, S.; Kanda, T.; Tagashira, M.; Sami, M.; Kaneko, T.; Tahara, S.; Nakaya, H.; et al. Apple polyphenols regulate mitochondrial superoxide generation and extend survival in a mouse model of dilated cardiomyopathy. Int. J. Life Sci. Med. Res. 2012, 2, 46–51. [Google Scholar] [CrossRef]
- Pucheu, S.; Boucher, F.; Sulpice, T.; Tresallet, N.; Bonhomme, Y.; Malfroy, B.; de Leiris, J. EUK-8 a synthetic catalytic scavenger of reactive oxygen species protects isolated iron-overloaded rat heart from functional and structural damage induced by ischemia/reperfusion. Cardiovasc. Drugs Ther. 1996, 10, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.; Marcus, C.B.; Huffman, K.; Kruk, H.; Malfroy, B.; Doctrow, S.R. Synthetic combined superoxide dismutase/catalase mimetics are protective as a delayed treatment in a rat stroke model: A key role for reactive oxygen species in ischemic brain injury. J. Pharmacol. Exp. Ther. 1998, 284, 215–221. [Google Scholar] [PubMed]
- Morten, K.J.; Ackrell, B.A.; Melov, S. Mitochondrial reactive oxygen species in mice lacking superoxide dismutase 2: Attenuation via antioxidant treatment. J. Biol. Chem. 2006, 281, 3354–3359. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Armstrong, S.J.; Arenas, I.A.; Pehowich, D.J.; Davidge, S.T. Cardioprotection by chronic estrogen or superoxide dismutase mimetic treatment in the aged female rat. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H165–H171. [Google Scholar] [CrossRef] [PubMed]
- Amin, J.K.; Xiao, L.; Pimental, D.R.; Pagano, P.J.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J. Mol. Cell Cardiol. 2001, 33, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, S.; Boucher, F.R.; Malfroy, B.; de Leiris, J.G. Free radicals in reperfusion-induced arrhythmias: Study with EUK 8, a novel nonprotein catalytic antioxidant. Free Radic. Biol. Med. 1996, 21, 945–954. [Google Scholar] [CrossRef]
- Melov, S.; Ravenscroft, J.; Malik, S.; Gill, M.S.; Walker, D.W.; Clayton, P.E.; Wallace, D.C.; Malfroy, B.; Doctrow, S.R.; Lithgow, G.J. Extension of life-span with superoxide dismutase/ catalase mimetics. Science 2000, 289, 1567–1569. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Doctrow, S.R.; Schneider, J.A.; Haberson, J.; Patel, M.; Coskun, P.E.; Huffman, K.; Wallace, D.C.; Malfroy, B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J. Neurosci. 2001, 21, 8348–8353. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, S.; Matsuda, A.; Sunagawa, T.; Noda, Y.; Kaneko, T.; Tahara, S.; Hiraumi, Y.; Adachi, S.; Matsui, H.; Ando, K.; et al. Antioxidant, EUK-8, prevents murine dilated cardiomyopathy. Circ. J. 2009, 73, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Makino, N.; Maeda, T.; Oyama, J.; Sasaki, M.; Higuchi, Y.; Mimori, K.; Shimizu, T. Antioxidant therapy attenuates myocardial telomerase activity reduction in superoxide dismutase-deficient mice. J. Mol. Cell. Cardiol. 2011, 50, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Day, B.J.; Shawen, S.; Liochev, S.I.; Crapo, J.D. A metalloporphyrin superoxide dismutase mimetic protects against paraquat-induced endothelial cell injury, in vitro. J. Pharmacol. Exp. Ther. 1995, 275, 1227–1232. [Google Scholar] [PubMed]
- Asayama, S.; Mizushima, K.; Nagaoka, S.; Kawakami, H. Design of metalloporphyrin-carbohydrate conjugates for a new superoxide dismutase mimic with cellular recognition. Bioconjug. Chem. 2004, 15, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Asayama, S.; Kawamura, E.; Nagaoka, S.; Kawakami, H. Design of manganese porphyrin modified with mitochondrial signal peptide for a new antioxidant. Mol. Pharm. 2006, 3, 468–470. [Google Scholar] [CrossRef] [PubMed]
- Asayama, S.; Nakajima, T.; Kawakami, H. New water-soluble Mn-porphyrin with catalytic activity for superoxide dismutation and peroxynitrite decomposition. Metallomics 2011, 3, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Benov, L.; Spasojevic, I.; Fridovich, I. The ortho effect makes manganese(III) meso-tetrakis(N-methylpyridinium-2-yl)porphyrin a powerful and potentially useful superoxide dismutase mimic. J. Biol. Chem. 1998, 273, 24521–24528. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, N.; Asayama, S.; Noda, Y.; Shimizu, T.; Kawakami, H. Pharmaceutical effect of manganese porphyrins on manganese superoxide dismutase deficient mice. Mol. Pharm. 2012, 9, 2956–2959. [Google Scholar] [CrossRef] [PubMed]
- Tsao, R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010, 2, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Hertog, M.G.; Feskens, E.J.; Hollman, P.C.; Katan, M.B.; Kromhout, D. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet 1993, 342, 1007–1011. [Google Scholar] [CrossRef]
- Mursu, J.; Voutilainen, S.; Nurmi, T.; Tuomainen, T.P.; Kurl, S.; Salonen, J.T. Flavonoid intake and the risk of ischaemic stroke and CVD mortality in middle-aged Finnish men: The Kuopio Ischaemic Heart Disease Risk Factor Study. Br. J. Nutr. 2008, 100, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Chiba, M.; Kasai, K.; Nozaka, H.; Nakamura, T.; Shoji, T.; Kanda, T.; Ohtake, Y.; Sato, T. Apple procyanidins induce tumor cell apoptosis through mitochondrial pathway activation of caspase-3. Carcinogenesis 2008, 29, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Sato, Y.; Watanabe, T.; Nagaoka, M.H.; Yoshioka, Y.; Shoji, T.; Kanda, T.; Yamada, K.; Totsuka, M.; Teshima, R.; et al. Dietary unripe apple polyphenol inhibits the development of food allergies in murine models. FEBS Lett. 2005, 579, 4485–4491. [Google Scholar] [CrossRef] [PubMed]
- Osada, K.; Funayama, M.; Fuchi, S.; Sami, M.; Ohta, Y.; Kanda, T.; Ikeda, M. Effects of dietary procyanidins and tea polyphenols on adipose tissue mass and fatty acid metabolism in rats on a high fat diet. J. Oleo Sci. 2006, 55, 79–89. [Google Scholar] [CrossRef]
- Ohnishi-Kameyama, M.; Yanagida, A.; Kanda, T.; Nagata, T. Identification of catechin oligomers from apple (Malus pumila cv. Fuji) in matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and fast-atom bombardment mass spectrometry. Rapid Commun. Mass Spectrom. 1997, 11, 31–36. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; Scalbert, A. Proanthocyanidins and tannin-like compounds: Nature, occurrence, dietary intake and effects on nutrition and health. J. Sci. Food Agric. 2000, 80, 1094–1117. [Google Scholar] [CrossRef]
- Sunagawa, T.; Shimizu, T.; Kanda, T.; Tagashira, M.; Sami, M.; Shirasawa, T. Procyanidins from apples (Malus pumila Mill.) extend the lifespan of Caenorhabditis elegans. Planta Med. 2011, 77, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Senoo-Matsuda, N.; Yasuda, K.; Tsuda, M.; Ohkubo, T.; Yoshimura, S.; Nakazawa, H.; Hartman, P.S.; Ishii, N. A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. J. Biol. Chem. 2001, 276, 41553–41558. [Google Scholar] [CrossRef] [PubMed]
- Leontowicz, H.; Gorinstein, S.; Lojek, A.; Leontowicz, M.; Cíz, M.; Soliva-Fortuny, R.; Park, Y.S.; Jung, S.T.; Trakhtenberg, S.; Martin-Belloso, O. Comparative content of some bioactive compounds in apples, peaches and pears and their influence on lipids and antioxidant capacity in rats. J. Nutr. Biochem. 2002, 13, 603–610. [Google Scholar] [CrossRef]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.A.; Shukitt-Hale, B.; Kalt, W.; Ingram, D.K.; Joseph, J.A.; Wolkow, C.A. Blueberry polyphenols increase lifespan and thermotolerance in Caenorhabditis elegans. Aging Cell 2006, 5, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L.; Hatefi, Y.; Lester, R.L.; Widmer, C. Isolation of a quinone from beef heart mitochondria. Biochim. Biophys. Acta 1957, 25, 220–221. [Google Scholar] [CrossRef]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Manzoli, U.; Rossi, E.; Littarru, G.P.; Frustaci, A.; Lippa, S.; Oradei, A.; Aureli, V. Coenzyme Q10 in dilated cardiomyopathy. Int. J. Tissue React. 1990, 12, 173–178. [Google Scholar] [PubMed]
- Yue, T.L.; Cheng, H.Y.; Lysko, P.G.; McKenna, P.J.; Feuerstein, R.; Gu, J.L.; Lysko, K.A.; Davis, L.L.; Feuerstein, G. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J. Pharmacol. Exp. Ther. 1992, 263, 92–98. [Google Scholar] [PubMed]
- Packer, M.; Bristow, M.R.; Cohn, J.N.; Colucci, W.S.; Fowler, M.B.; Gilbert, E.M.; Shusterman, N.H. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N. Engl. J. Med. 1996, 334, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Karne, R.; Ghanim, H.; Hamouda, W.; Aljada, A.; Magsino, C.H., Jr. Carvedilol inhibits reactive oxygen species generation by leukocytes and oxidative damage to amino acids. Circulation 2000, 101, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, H.; Horie, T.; Ishikawa, S.; Tsuda, C.; Kawakami, S.; Noda, Y.; Kaneko, T.; Tahara, S.; Tachibana, T.; Okabe, M.; et al. Oxidative stress in skeletal muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radic. Biol. Med. 2010, 48, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Miniou, P.; Tiziano, D.; Frugier, T.; Roblot, N.; Le Meur, M.; Melki, J. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 1999, 27, e27. [Google Scholar] [CrossRef] [PubMed]



© 2013 by the authors. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Koyama, H.; Nojiri, H.; Kawakami, S.; Sunagawa, T.; Shirasawa, T.; Shimizu, T. Antioxidants Improve the Phenotypes of Dilated Cardiomyopathy and Muscle Fatigue in Mitochondrial Superoxide Dismutase-Deficient Mice. Molecules 2013, 18, 1383-1393. https://doi.org/10.3390/molecules18021383
Koyama H, Nojiri H, Kawakami S, Sunagawa T, Shirasawa T, Shimizu T. Antioxidants Improve the Phenotypes of Dilated Cardiomyopathy and Muscle Fatigue in Mitochondrial Superoxide Dismutase-Deficient Mice. Molecules. 2013; 18(2):1383-1393. https://doi.org/10.3390/molecules18021383
Chicago/Turabian StyleKoyama, Hirofumi, Hidetoshi Nojiri, Satoru Kawakami, Tadahiro Sunagawa, Takuji Shirasawa, and Takahiko Shimizu. 2013. "Antioxidants Improve the Phenotypes of Dilated Cardiomyopathy and Muscle Fatigue in Mitochondrial Superoxide Dismutase-Deficient Mice" Molecules 18, no. 2: 1383-1393. https://doi.org/10.3390/molecules18021383