Enthalpy/Entropy Contributions to Conformational KIEs: Theoretical Predictions and Comparison with Experiment

Abstract
:1. Introduction
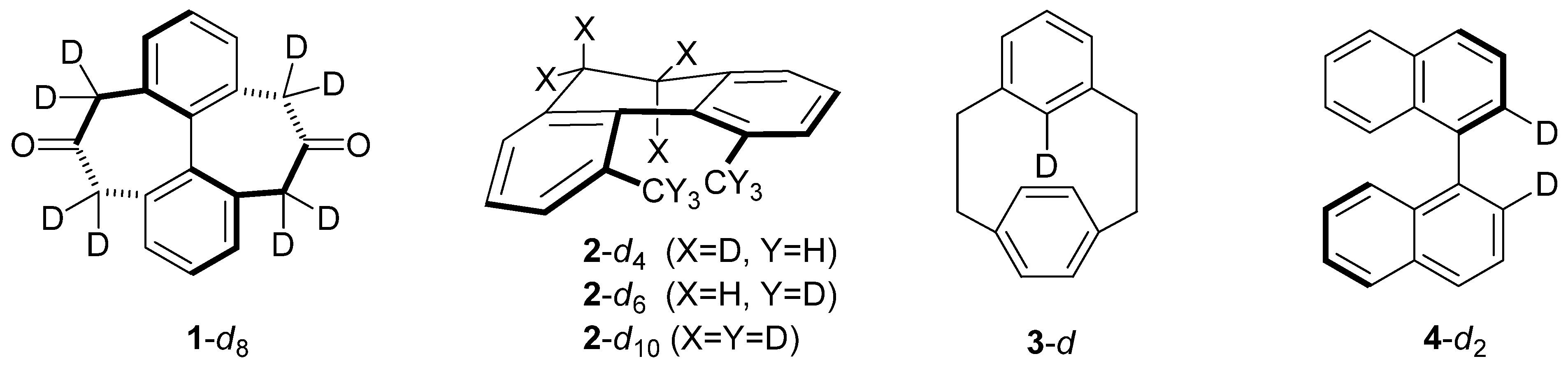
2. Results and Discussion
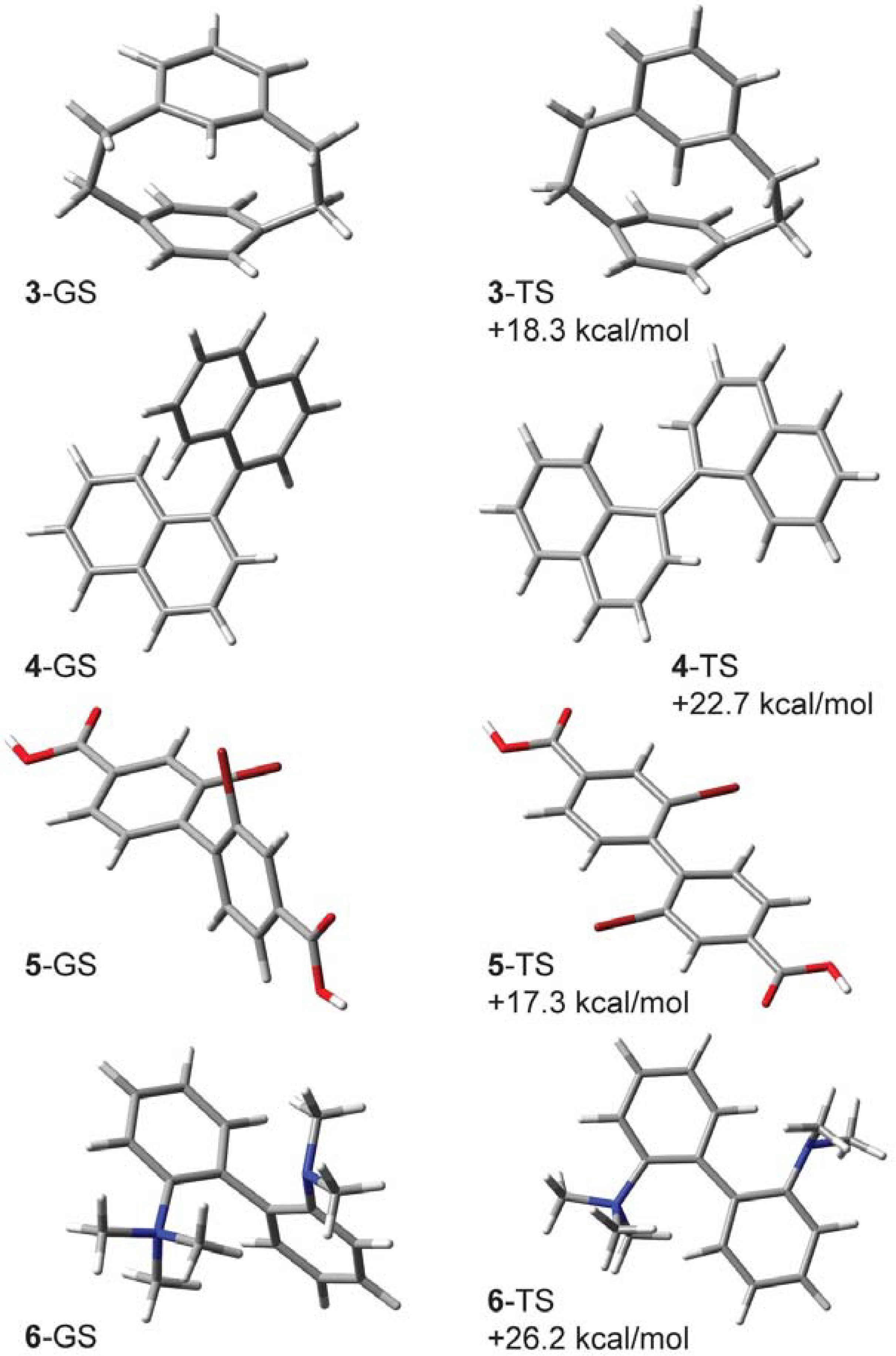
2.1. [2.2]-Metaparacyclophane-d (3-d)

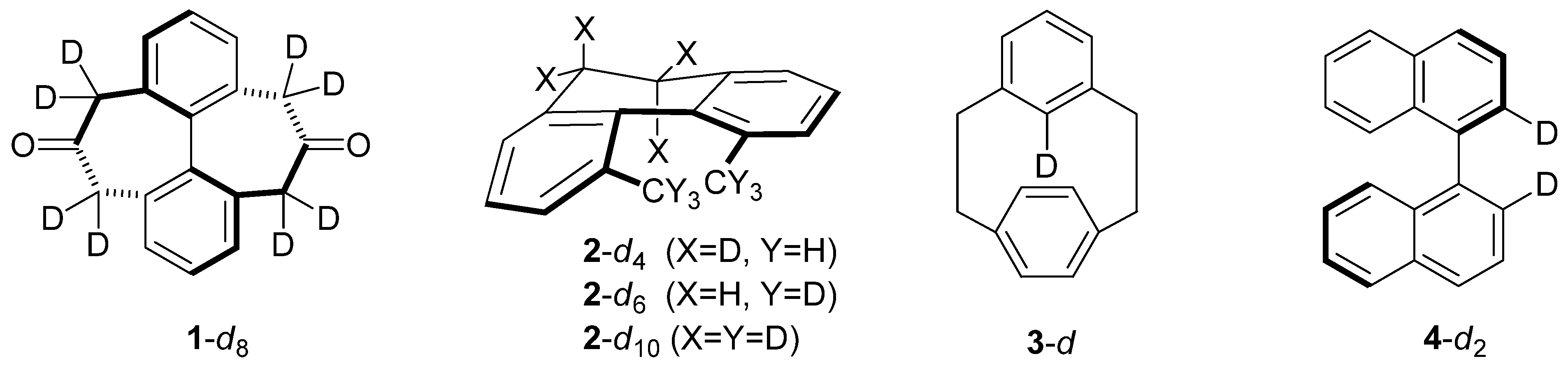{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| kH/kD | 3 | 4 | 5 |
|---|---|---|---|
| Experimental | 0.833 ± 0.040 a | 0.877 ± 0.024 b | 0.840 ± 0.050 c |
| Bigeleisen-Mayer | 0.832 a | 0.849 b | 0.825 c |
| ZPE | 0.829 | 0.822 | 0.820 |
| EXC | 0.999 | 1.027 | 1.001 |
| MMI | 1.005 | 1.005 | 1.005 |
| ΔΔG‡ | 0.833 a | 0.849 b | 0.825 c |
| ΔΔH‡ | 0.829 | 0.813 | 0.810 |
| −TΔΔS‡ | 1.005 | 1.044 | 1.018 |
2.2. 1,1'-Binaphthyl-2,2'-d2(4-d2)
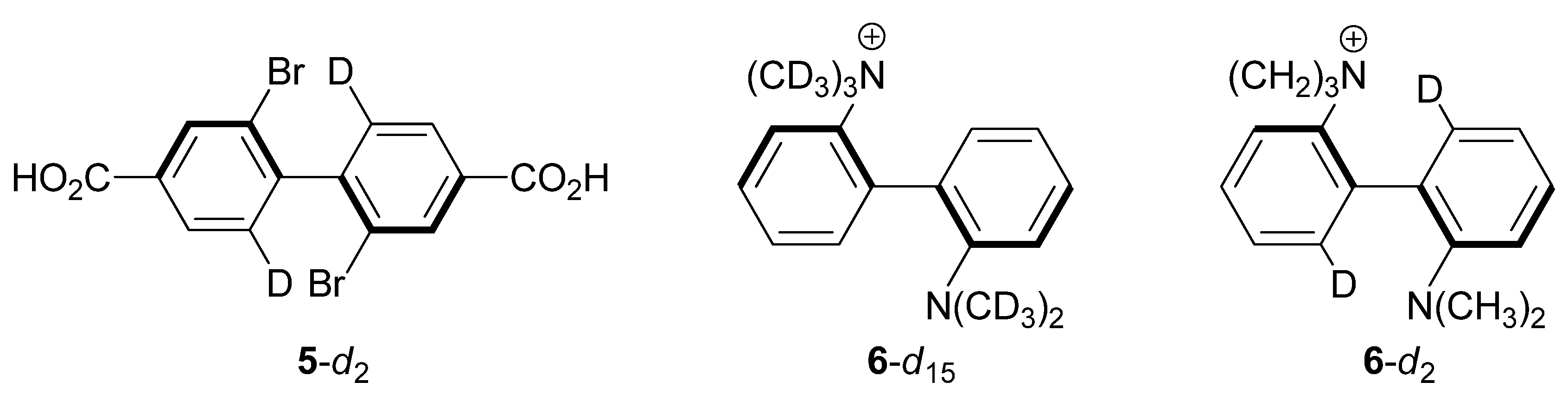
2.3. 2,2'-Dibromo-4,4'-dicarboxybiphenyl (5-d2)
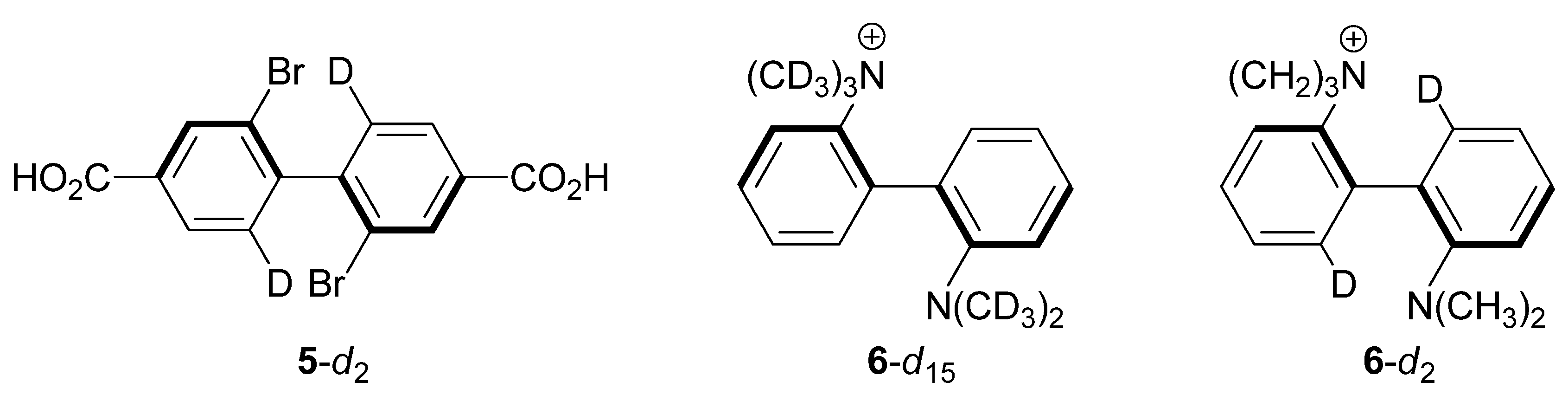
2.4. 2-(N,N,N-Trimethyl)-2'-(N,N-dimethyl)-diaminobiphenyl Cations 6-d15 and 6-d2
| kH/kD | 6-d15 | 6-d15 | 6-d2 |
|---|---|---|---|
| Experimental | 0.996 ± 0.007 a | 1.02 ± 0.01 b | 0.847 ± 0.027 c |
| Bigeleisen-Mayer | 1.041 a | 1.044 b | 0.829 c |
| ZPE | 0.966 | 0.968 | 0.798 |
| EXC | 1.089 | 1.090 | 1.034 |
| MMI | 0.989 | 0.989 | 1.005 |
| ΔΔG‡ | 1.041 a | 1.045 b | 0.828 c |
| ΔΔH‡ | 0.945 | 0.950 | 0.779 |
| −TΔΔS‡ | 1.101 | 1.100 | 1.063 |
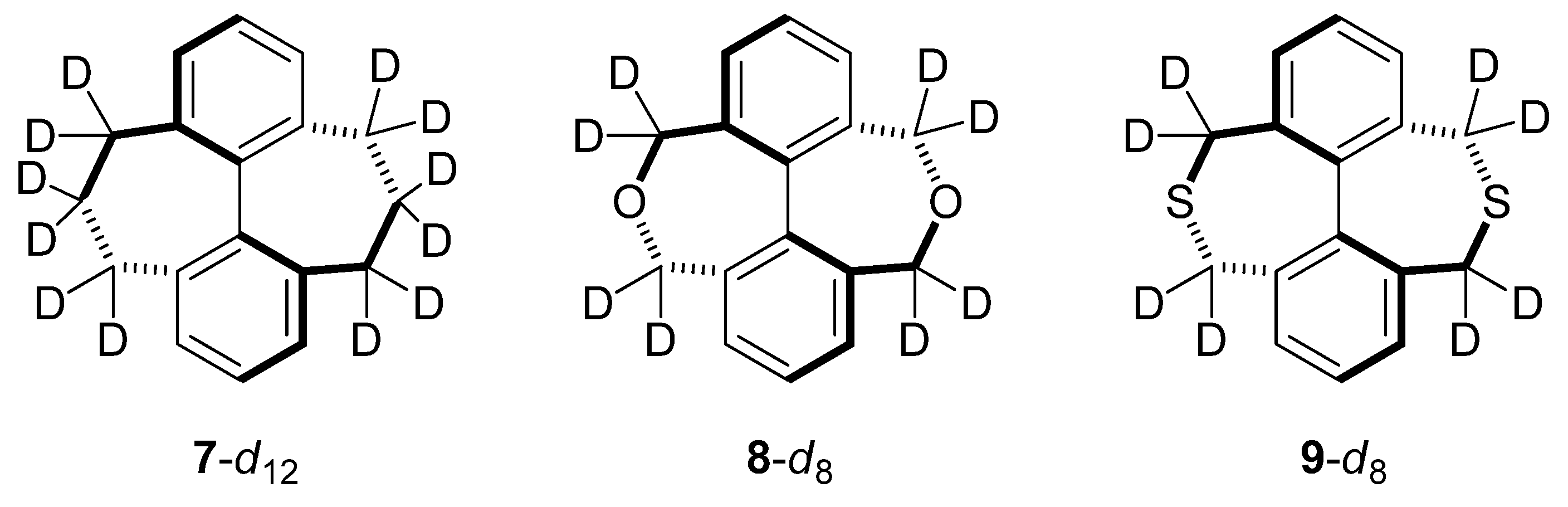
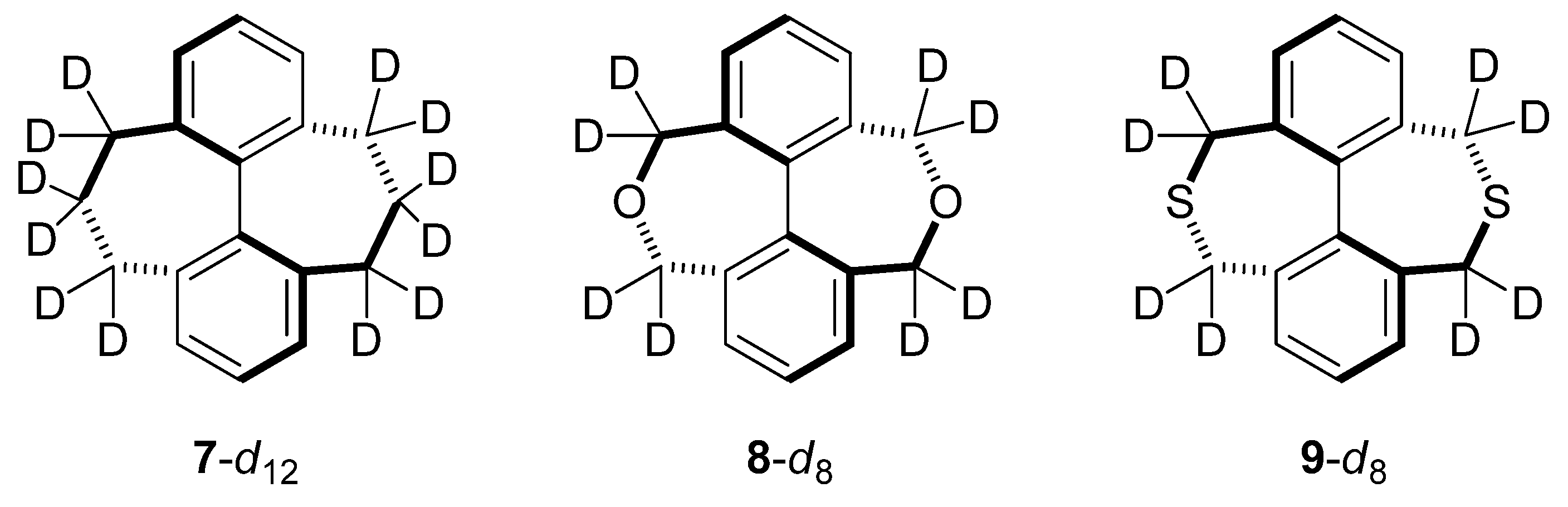
2.5. Compounds Structurally Similar to Mislow’s Doubly-Bridged Diketone 1
| kH/kD | 1-d8 | 7-d12 | 8-d8 | 9-d8 |
|---|---|---|---|---|
| Bigeleisen-Mayer | 1.075 | 1.172 | 1.147 | 1.057 |
| ZPE | 1.026 | 1.022 | 1.069 | 0.989 |
| EXC | 1.050 | 1.148 | 1.073 | 1.069 |
| MMI | 0.998 | 0.998 | 0.999 | 1.000 |
| ΔΔG‡ | 1.075 | 1.171 | 1.146 | 1.058 |
| ΔΔH‡ | 0.973 | 0.935 | 1.005 | 0.931 |
| −TΔΔS‡ | 1.105 | 1.252 | 1.140 | 1.136 |
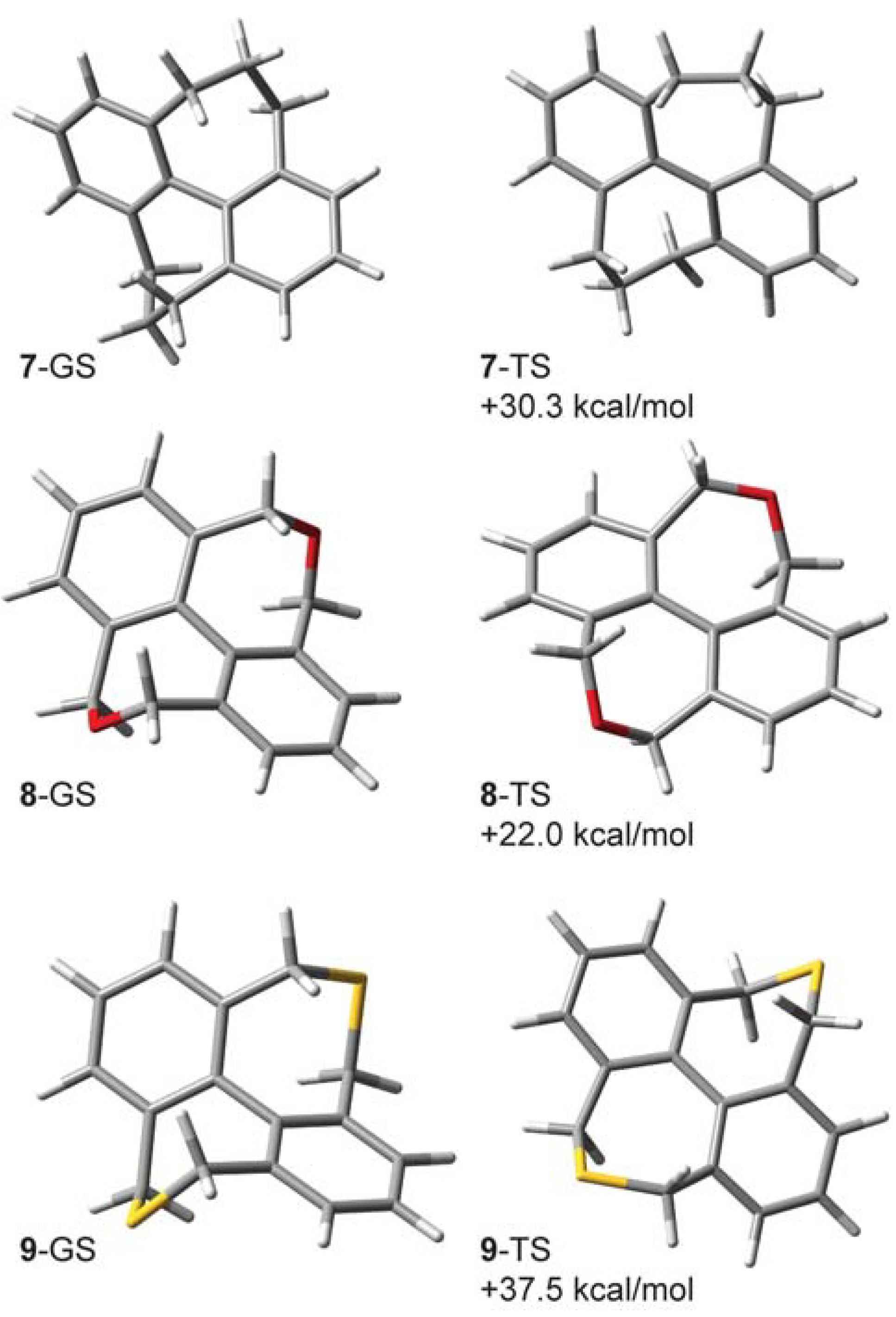
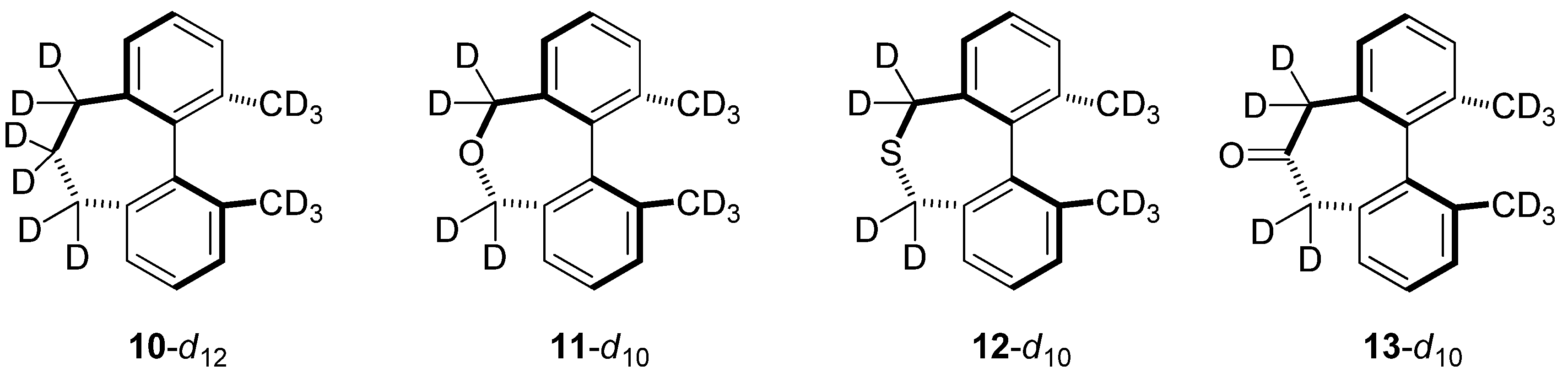
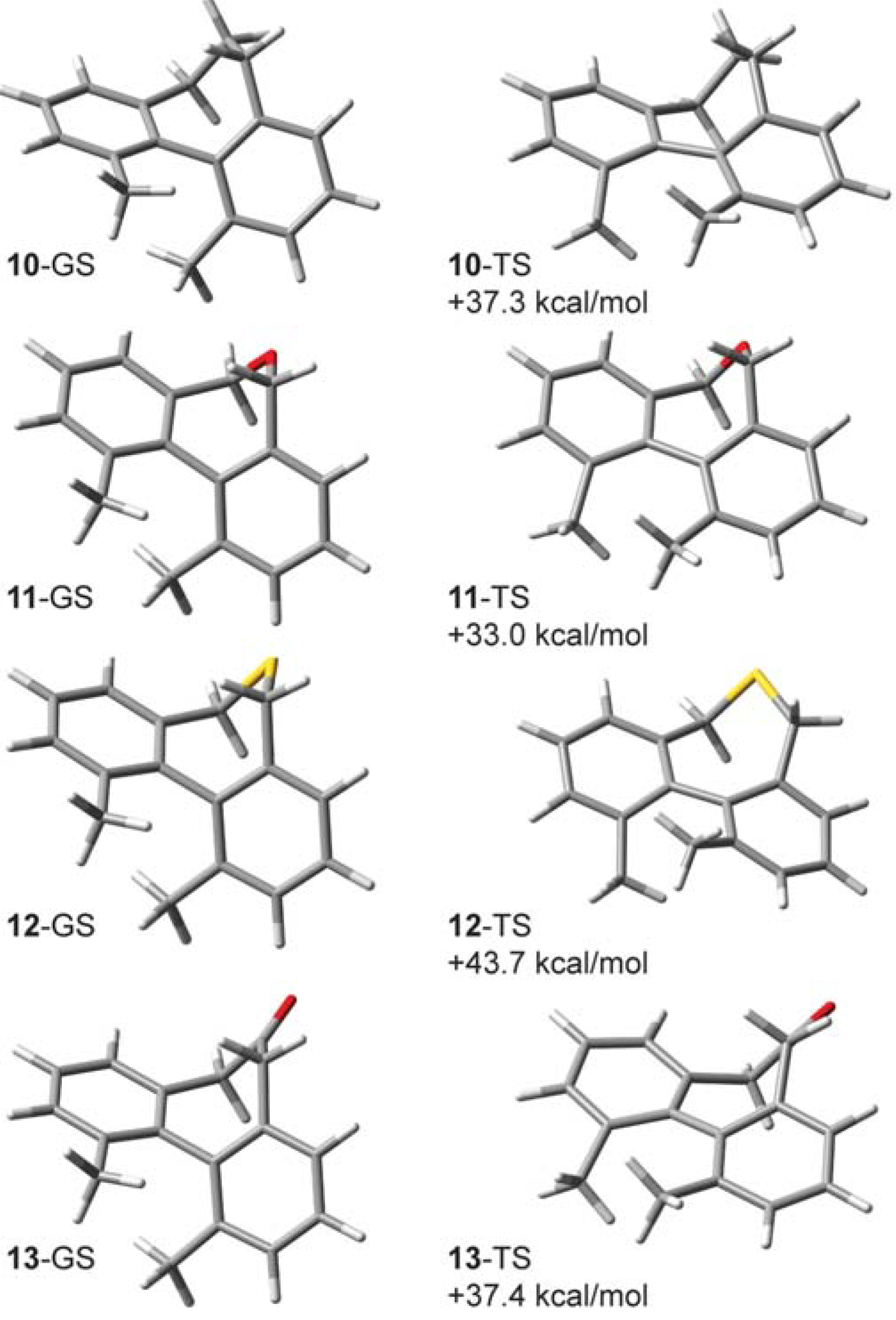
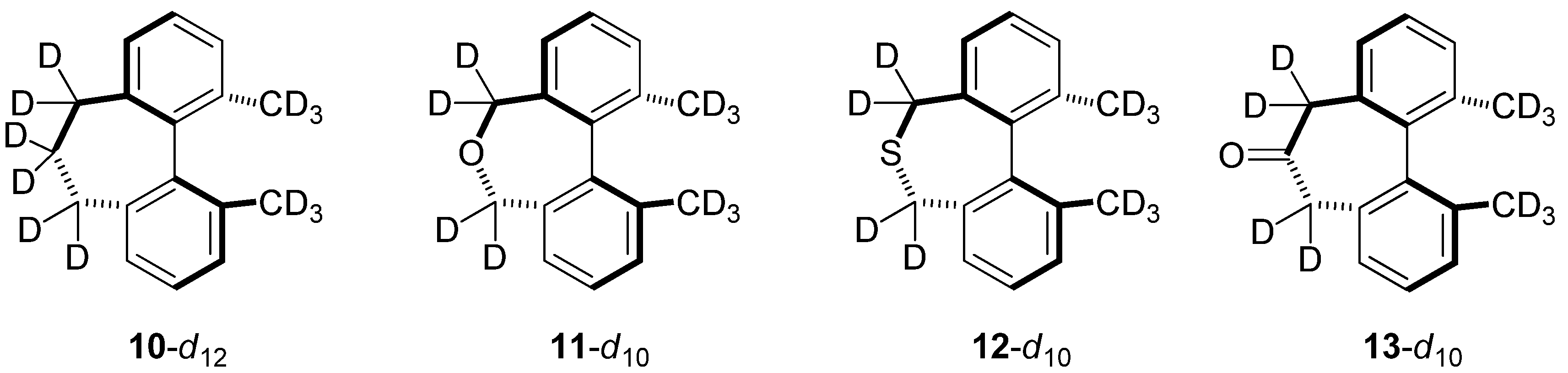
2.6. Structures Related to Mislow’s Singly Bridged Dihydrodimethylphenanthrene 2
| kH/kD | 2-d10 | 10-d12 | 11-d10 | 12-d10 | 13-d10 |
|---|---|---|---|---|---|
| Bigeleisen-Mayer | 0.846 | 0.869 | 0.868 | 0.959 | 0.867 |
| ZPE | 0.712 | 0.660 | 0.678 | 0.762 | 0.664 |
| EXC | 1.193 | 1.321 | 1.284 | 1.256 | 1.308 |
| MMI | 0.997 | 0.997 | 0.998 | 1.001 | 0.997 |
| ΔΔG‡ | 0.846 | 0.869 | 0.869 | 0.958 | 0.868 |
| ΔΔH‡ | 0.689 | 0.712 | 0.713 | 0.805 | 0.710 |
| −TΔΔS‡ | 1.227 | 1.221 | 1.219 | 1.191 | 1.219 |

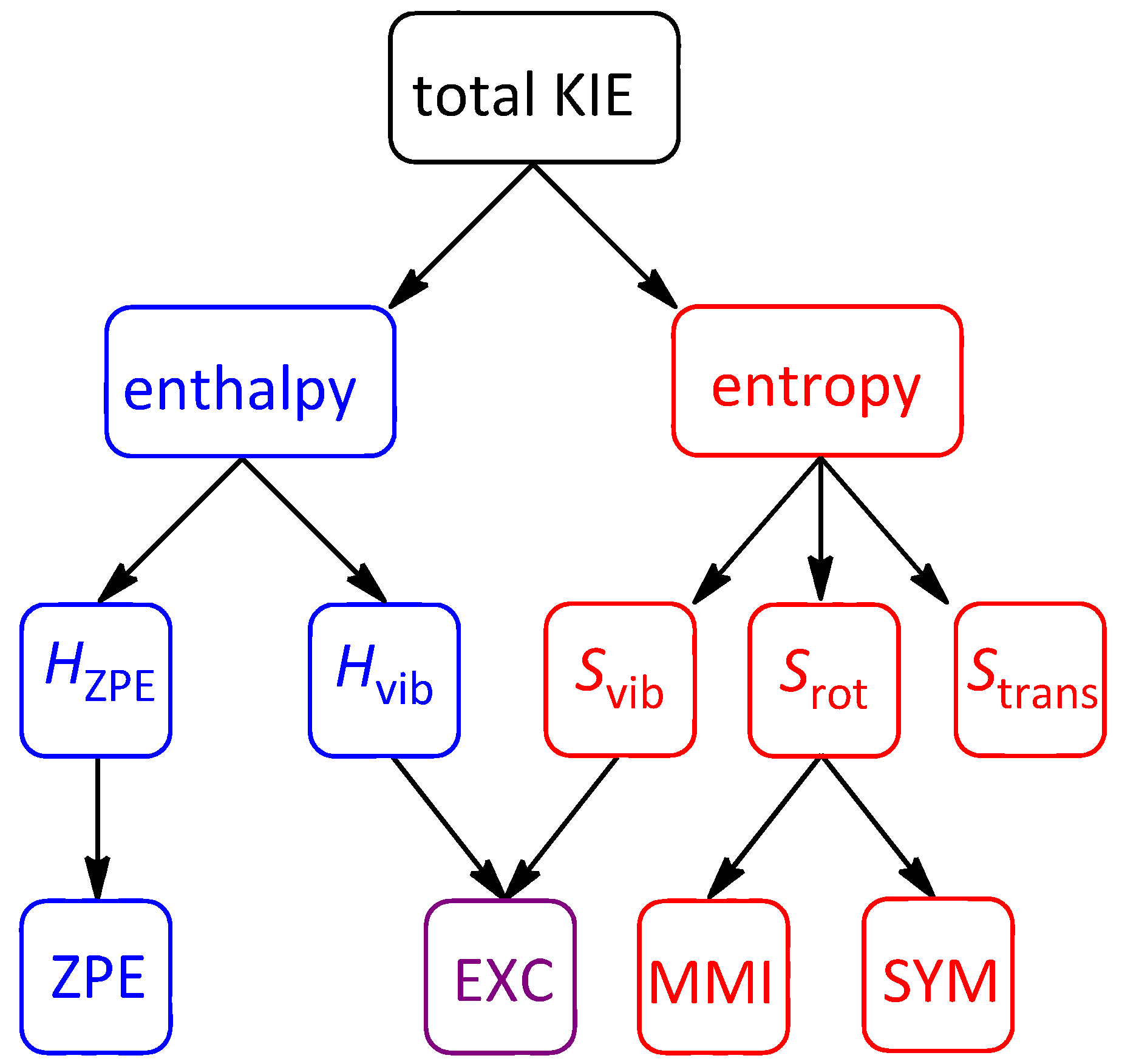
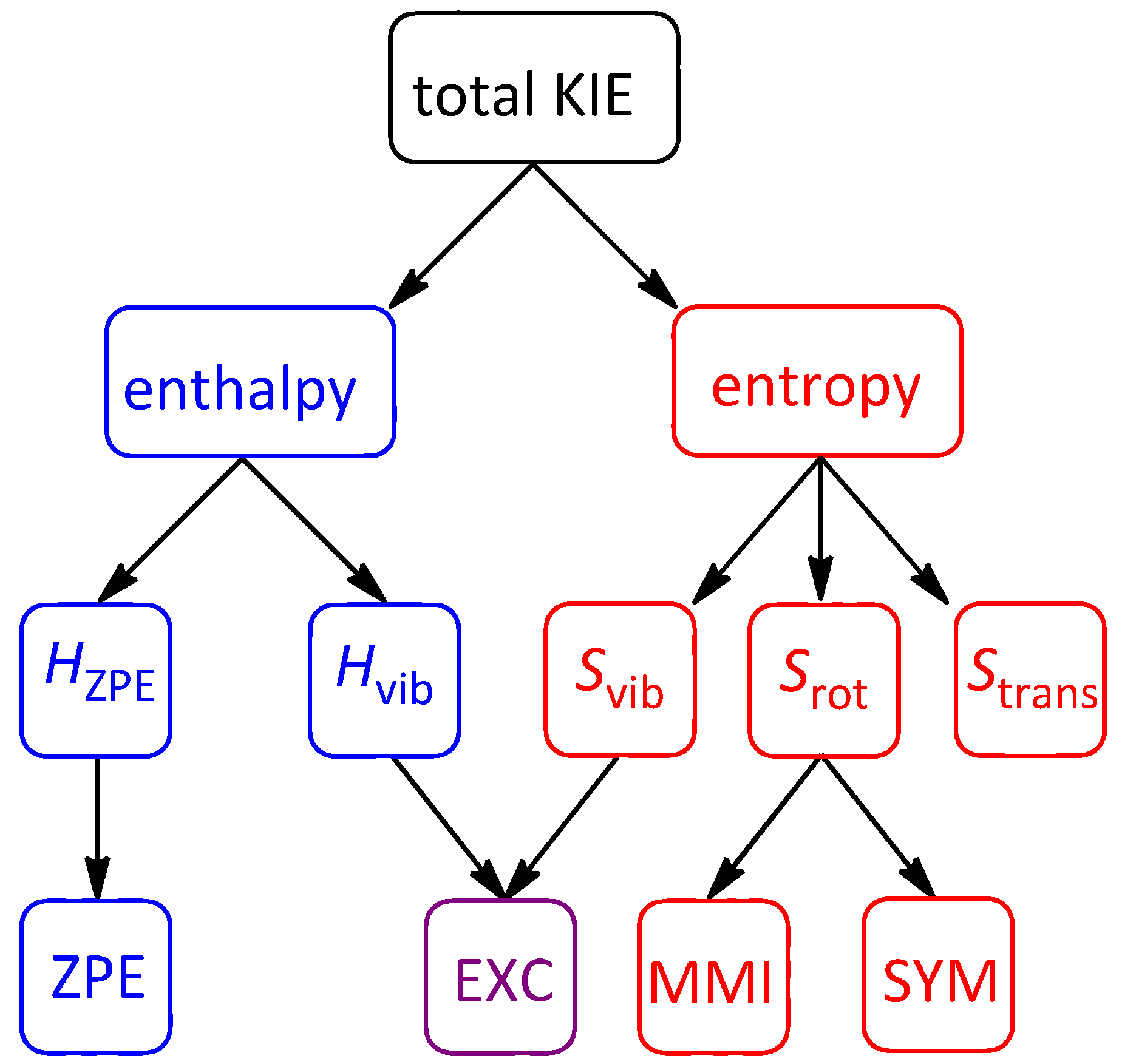
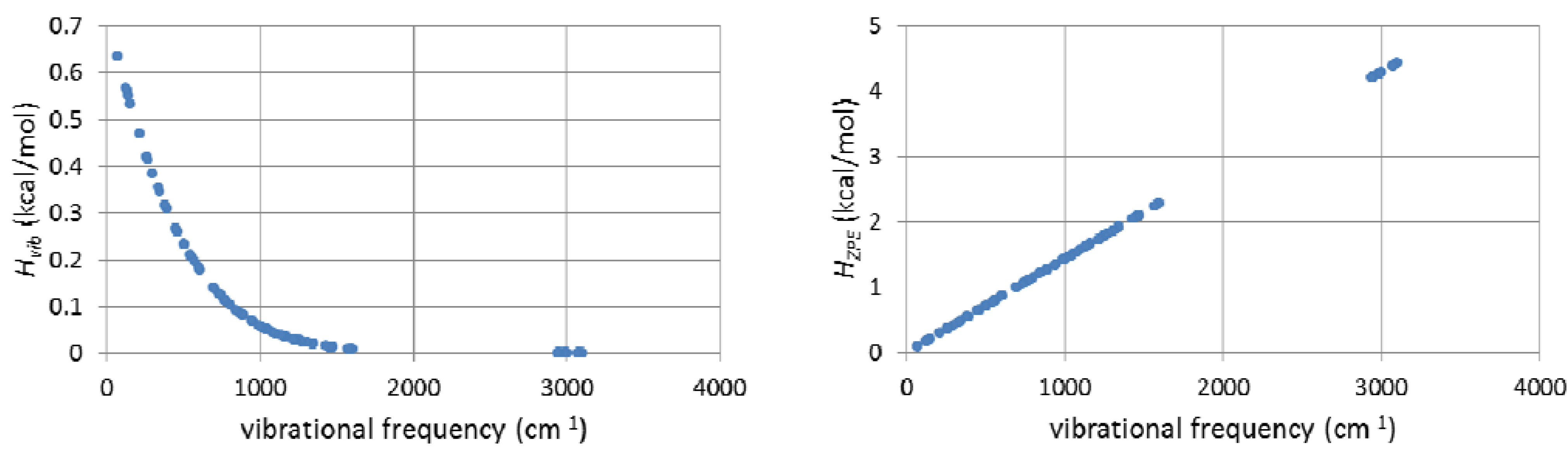
2.7. On the Interplay and Relative Magnitudes of ΔΔH‡ZPE and ΔΔH‡vib
| Term | 1-d8 | 7-d12 | 2-d6 | 2-d10 | 10-d12 |
|---|---|---|---|---|---|
| ΔΔH‡vib | −0.038 | −0.065 | −0.009 | −0.020 | +0.047 |
| ΔΔH‡ZPE | 0.018 | 0.016 | −0.176 | −0.213 | −0.260 |
| ΔΔH‡thermal | −0.020 | −0.049 | −0.185 | −0.233 | −0.213 |

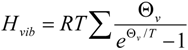
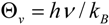
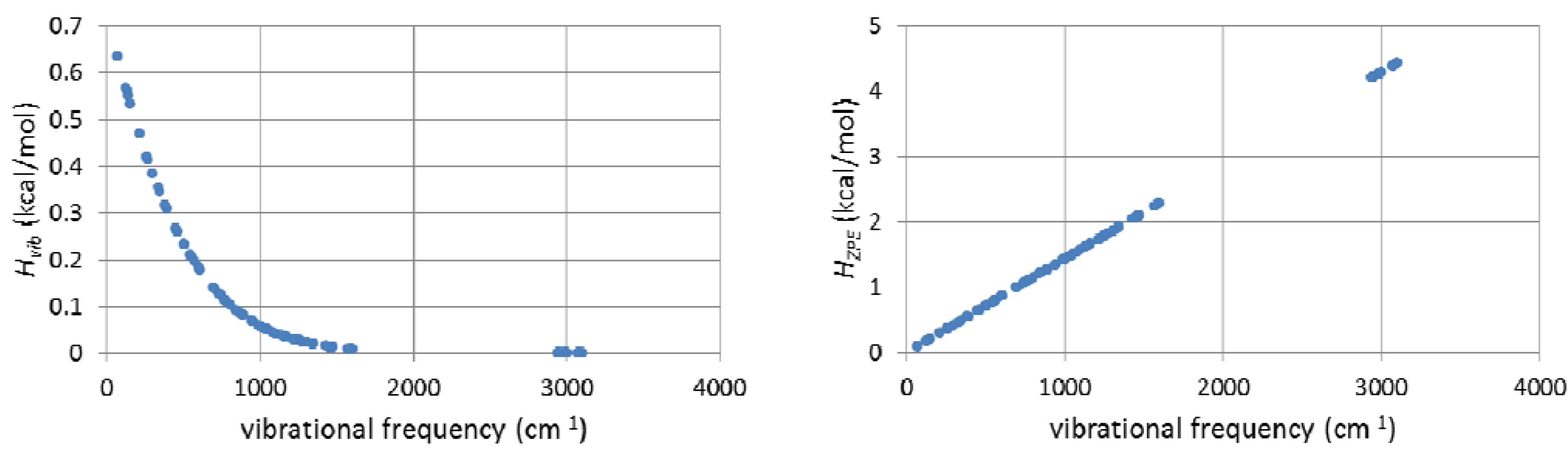
3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Perrin, C.L.; Thoburn, J.D.; Kresge, A.J. Secondary kinetic isotope effects in C–N rotation of amides. J. Am. Chem. Soc. 1992, 114, 8800–8807. [Google Scholar] [CrossRef]
- Olson, L.P.; Li, Y.; Houk, K.N.; Kresge, A.J.; Schaad, L.J. Theoretical analysis of secondary kinetic isotope effects in C–N rotation of amides. J. Am. Chem. Soc. 1995, 117, 2992–2997. [Google Scholar]
- Parkin, G. Applications of deuterium isotope effects for probing aspects of reactions involving oxidative addition and reductive elimination of H–H and C–Hh bonds. J. Labelled Compd. Rad. 2007, 50, 1088–1114. [Google Scholar] [CrossRef]
- O'Leary, D.J.; Hickstein, D.D.; Hansen, B.K.; Hansen, P.E. Theoretical and NMR studies of deuterium isotopic perturbation of hydrogen bonding in symmetrical dihydroxy compounds. J. Org. Chem. 2010, 75, 1331–1342. [Google Scholar]
- O’Leary, D.J.; Rablen, P.R.; Meyer, M.P. On the origin of conformational kinetic isotope effects. Angew Chem. Int. Ed. 2011, 50, 2564–2567. [Google Scholar] [CrossRef]
- Bartell, L.S. Role of non-bonded repulsions in secondary isotope effects. 1. Alpha and beta substitution effects. J. Am. Chem. Soc. 1961, 83, 3567–3571. [Google Scholar] [CrossRef]
- Bigeleisen, J.; Mayer, M.G. Calculation of equilibrium constants for isotopic exchange reactions. J. Chem. Phys. 1947, 15, 261–267. [Google Scholar] [CrossRef]
- Saunders, M.; Laidig, K.E.; Wolfsberg, M. Theoretical calculation of equilibrium isotope effects using abinitio force-constants: Application to NMR isotopic perturbation studies. J. Am. Chem. Soc. 1989, 111, 8989–8994. [Google Scholar] [CrossRef]
- Mislow, K.; Simon, E.; Hopps, H.B. A secondary kinetic isotope effect in conformational racemization. Tetrahedron Lett. 1962, 1011–1014. [Google Scholar] [CrossRef]
- Mislow, K.; Simon, E.; Glass, M.A.W.; Wahl, G.H.; Hopps, H.B. Stereochemistry of doubly bridged biphenyls: Synthesis, spectral properties, and optical stability. J. Am. Chem. Soc. 1964, 86, 1710–1733. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Ibberson, R.M. Is deuterium always smaller than protium? Angew. Chem. Int. Ed. 2008, 47, 4208–4210. [Google Scholar] [CrossRef]
- Mislow, K.; Graeve, R.; Gordon, A.J.; Wahl, G.H. A note on steric isotope effects. Conformational kinetic isotope effects in the racemization of 9,10-dihydro-4,5-dimethylphenanthrene. J. Am. Chem. Soc. 1963, 85, 1199–1200. [Google Scholar]
- Mislow, K.; Wahl, G.H.; Gordon, A.J.; Graeve, R. Conformational kinetic isotope effects in racemization of 9,10-dihydro-4,5-dimethylphenanthrene. J. Am. Chem. Soc. 1964, 86, 1733–1741. [Google Scholar] [CrossRef]
- Hayama, T.; Baldridge, K.K.; Wu, Y.T.; Linden, A.; Siegel, J.S. Steric isotope effects gauged by the bowl-inversion barrier in selectively deuterated pentaarylcorannulenes. J. Am. Chem. Soc. 2008, 130, 1583–1591. [Google Scholar]
- Mugridge, J.S.; Bergman, R.G.; Raymond, K.N. Does size really matter? The steric isotope effect in a supramolecular host-guest exchange reaction. Angew. Chem. Int. Ed. 2010, 49, 3635–3637. [Google Scholar] [CrossRef]
- West, J.D.; Stafford, S.E.; Meyer, M.P. A mechanistic probe for asymmetric reactions: Deuterium isotope effects at enantiotopic groups. J. Am. Chem. Soc. 2008, 130, 7816–7817. [Google Scholar] [CrossRef]
- Meyer, M.P. Nonbonding interactions and stereoselection in the Corey-Bakshi-Shibata reduction. Org. Lett. 2009, 11, 4338–4341. [Google Scholar] [CrossRef]
- Giagou, T.; Meyer, M.P. Kinetic isotope effects in asymmetric reactions. Chem.-Eur. J. 2010, 16, 10616–10628. [Google Scholar] [CrossRef]
- Sherrod, S.A.; Boekelheide, V. Unusually large conformational kinetic isotope-effect in [2.2]metaparacyclophane. J. Am. Chem. Soc. 1972, 94, 5513–5515. [Google Scholar] [CrossRef]
- Sherrod, S.A.; Costa, R.L.D.; Barnes, R.A.; Boekelheide, V. Study of steric effects in [2.2]metaparacyclophanes: Steric isotope-effect, remote substituent effects on a steric barrier, and other steric phenomena. J. Am. Chem. Soc. 1974, 96, 1565–1577. [Google Scholar]
- Irikura, K.K.; Johnson, R.D.; Kacker, R.N.; Kessel, R. Uncertainties in scaling factors for ab initio vibrational zero-point energies. J. Chem. Phys. 2009, 130, 114102–114111. [Google Scholar] [CrossRef]
- Carter, R.E.; Dahlgren, L. Steric isotope effects. 3. The deuterium isotope effect in racemization of (−)-1,1'-binaphthyl-2,2'-d2. Acta Chem. Scand. 1969, 23, 504–514. [Google Scholar] [CrossRef]
- Melander, L.; Carter, R.E. Steric isotope effects. The isotope effect on racemization of 2,2'-dibromo-4,4'-dicarboxybiphenyl. J. Am. Chem. Soc. 1964, 86, 295–296. [Google Scholar]
- Hall, D.M.; Harris, M.M. Some racemisation data for compounds owing their optical activity to restricted rotation. J. Chem. Soc. 1960, 490–494. [Google Scholar] [CrossRef]
- Heitner, C.; Leffek, K.T. Deuterium isotope effect on racemization of a highly hindered biphenyl derivative. Can. J. Chem. 1966, 44, 2567–2570. [Google Scholar] [CrossRef]
- Carter, R.E.; Dahlgren, L. Steric isotope effects. 4. Deuterium isotope effect in racemization of (+)-2-(N,N,N-trimethyl)-2'-(N,N-dimethyl)-diamino-biphenyl-6,6'-d2 cation. Acta Chem. Scand. 1970, 24, 633–643. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Hirschi, J.S.; Takeya, T.; Hang, C.; Singleton, D.A. Transition-state geometry measurements from C-13 isotope effects. The experimental transition state for the epoxidation of alkenes with oxaziridines. J. Am. Chem. Soc. 2009, 131, 2397–2403. [Google Scholar]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fong, A.; Meyer, M.P.; O'Leary, D.J. Enthalpy/Entropy Contributions to Conformational KIEs: Theoretical Predictions and Comparison with Experiment. Molecules 2013, 18, 2281-2296. https://doi.org/10.3390/molecules18022281
Fong A, Meyer MP, O'Leary DJ. Enthalpy/Entropy Contributions to Conformational KIEs: Theoretical Predictions and Comparison with Experiment. Molecules. 2013; 18(2):2281-2296. https://doi.org/10.3390/molecules18022281
Chicago/Turabian StyleFong, Aaron, Matthew P. Meyer, and Daniel J. O'Leary. 2013. "Enthalpy/Entropy Contributions to Conformational KIEs: Theoretical Predictions and Comparison with Experiment" Molecules 18, no. 2: 2281-2296. https://doi.org/10.3390/molecules18022281