A Survey of Marine Natural Compounds and Their Derivatives with Anti-Cancer Activity Reported in 2011



Abstract
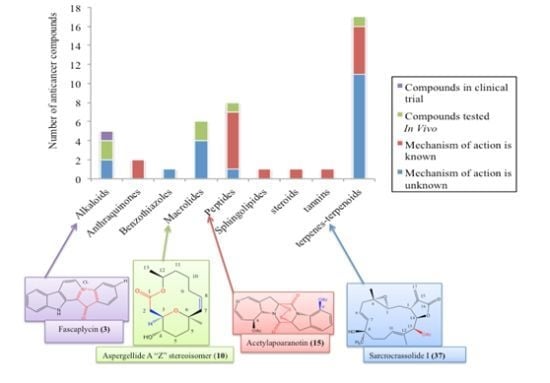
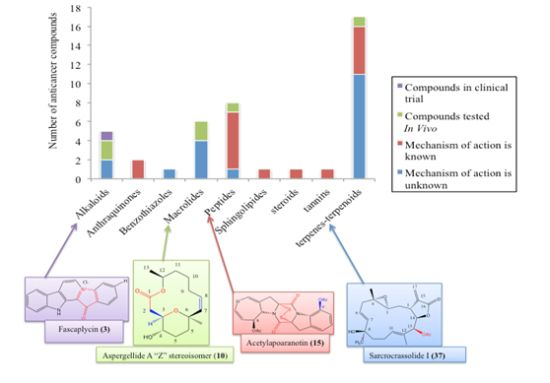
:
{kind=link}
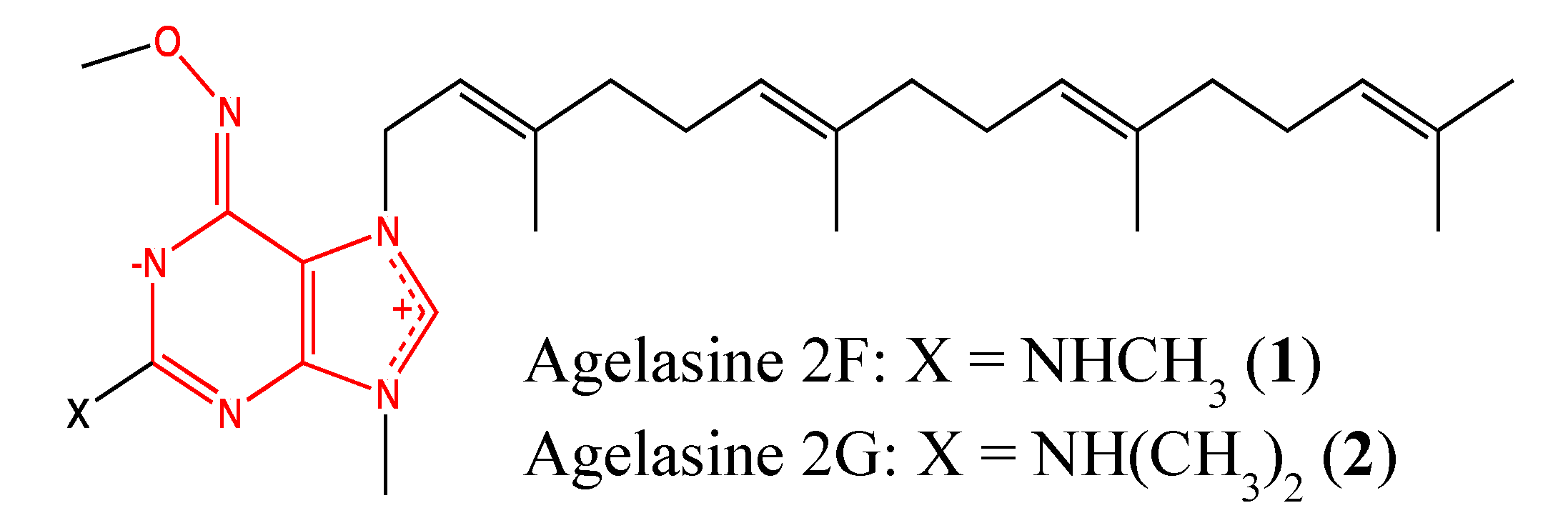{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
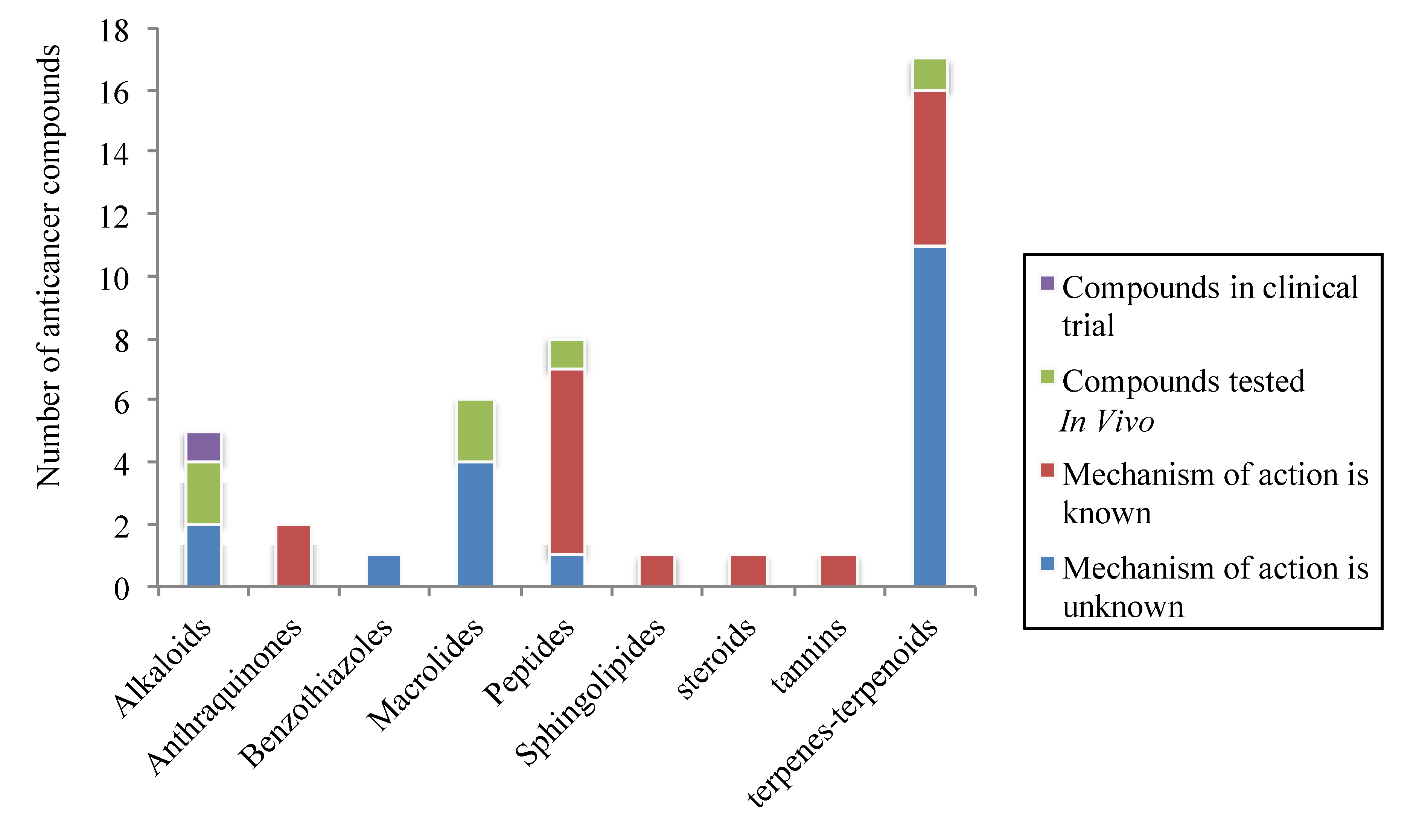
2. Promising Marine Anticancer Molecules Reported in 2011
2.1. Alkaloids
2.1.1. Agelasine analogs 2F and 2G
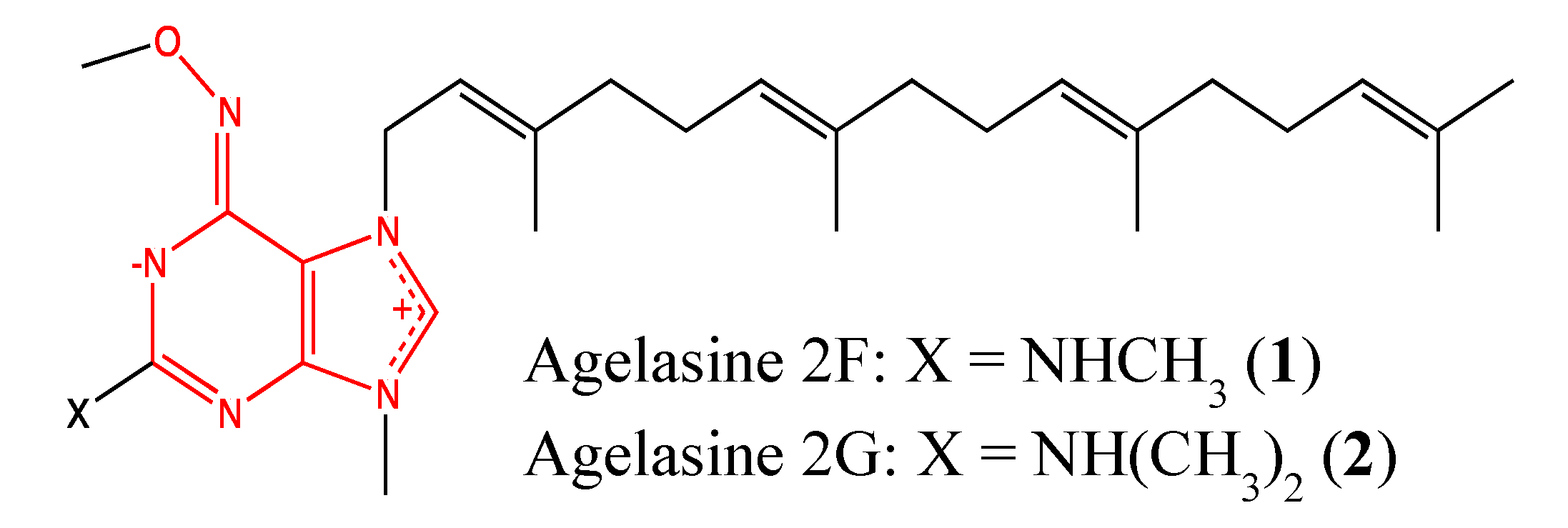
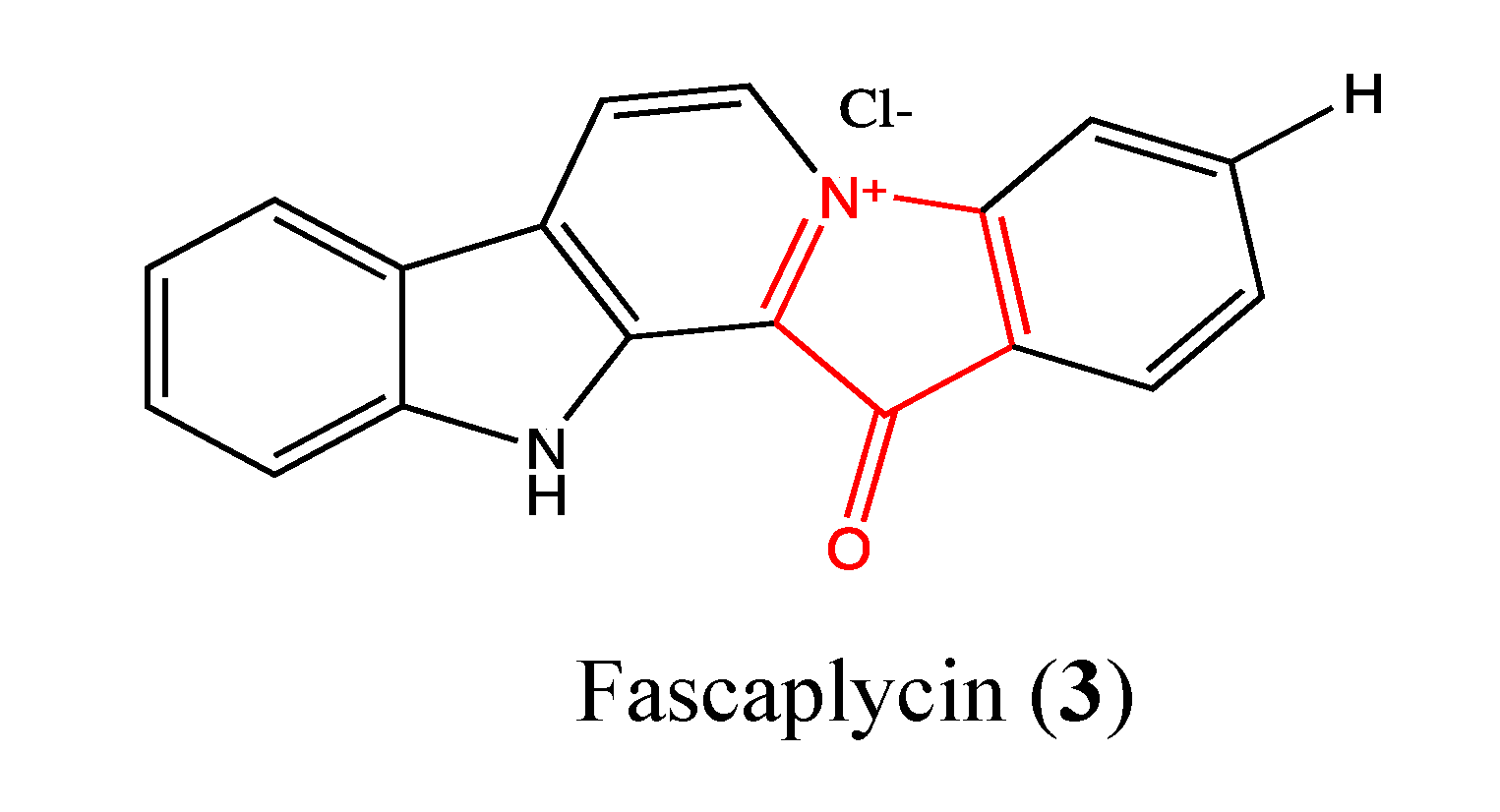
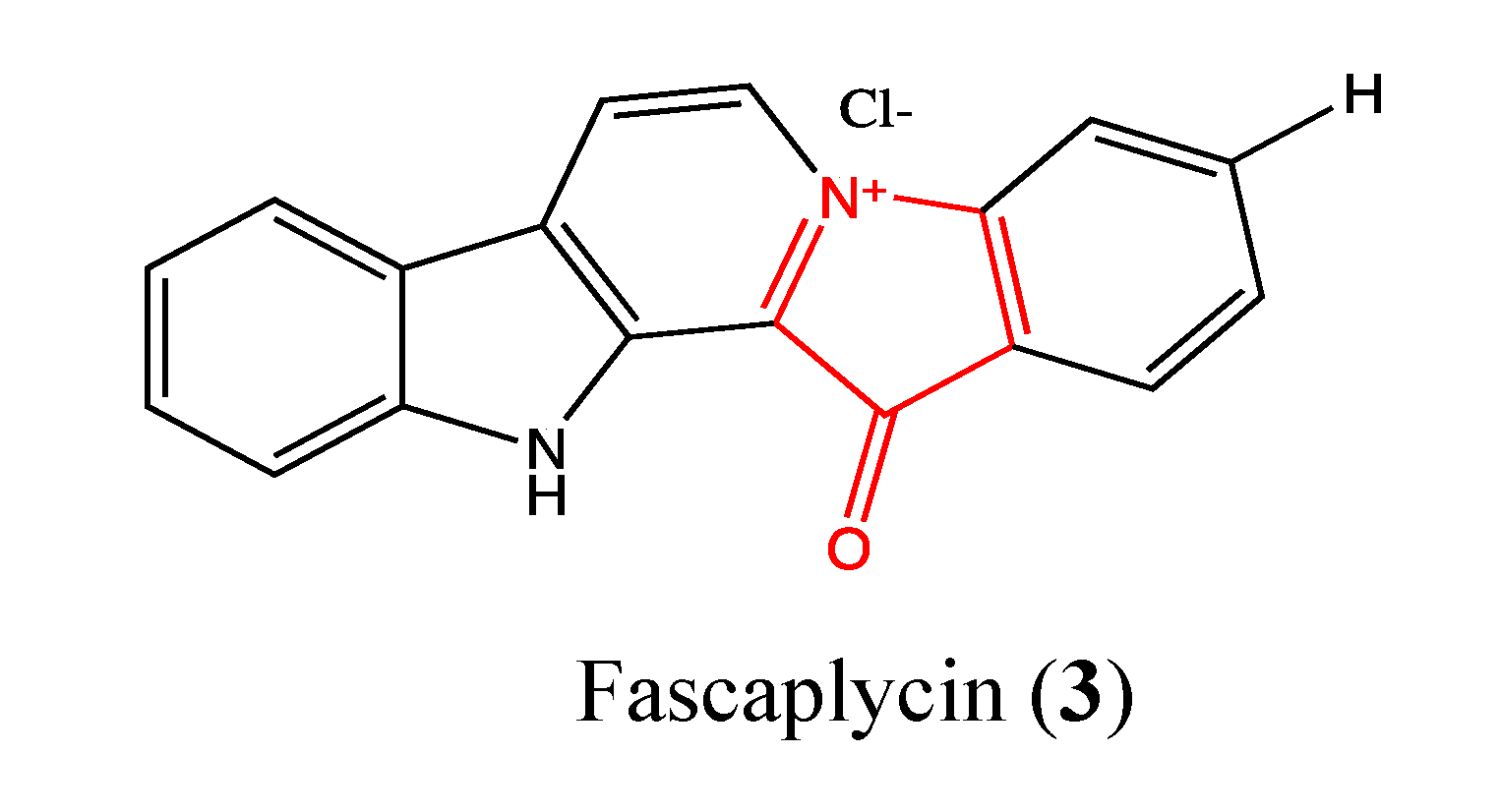
2.1.2. Fascaplysin
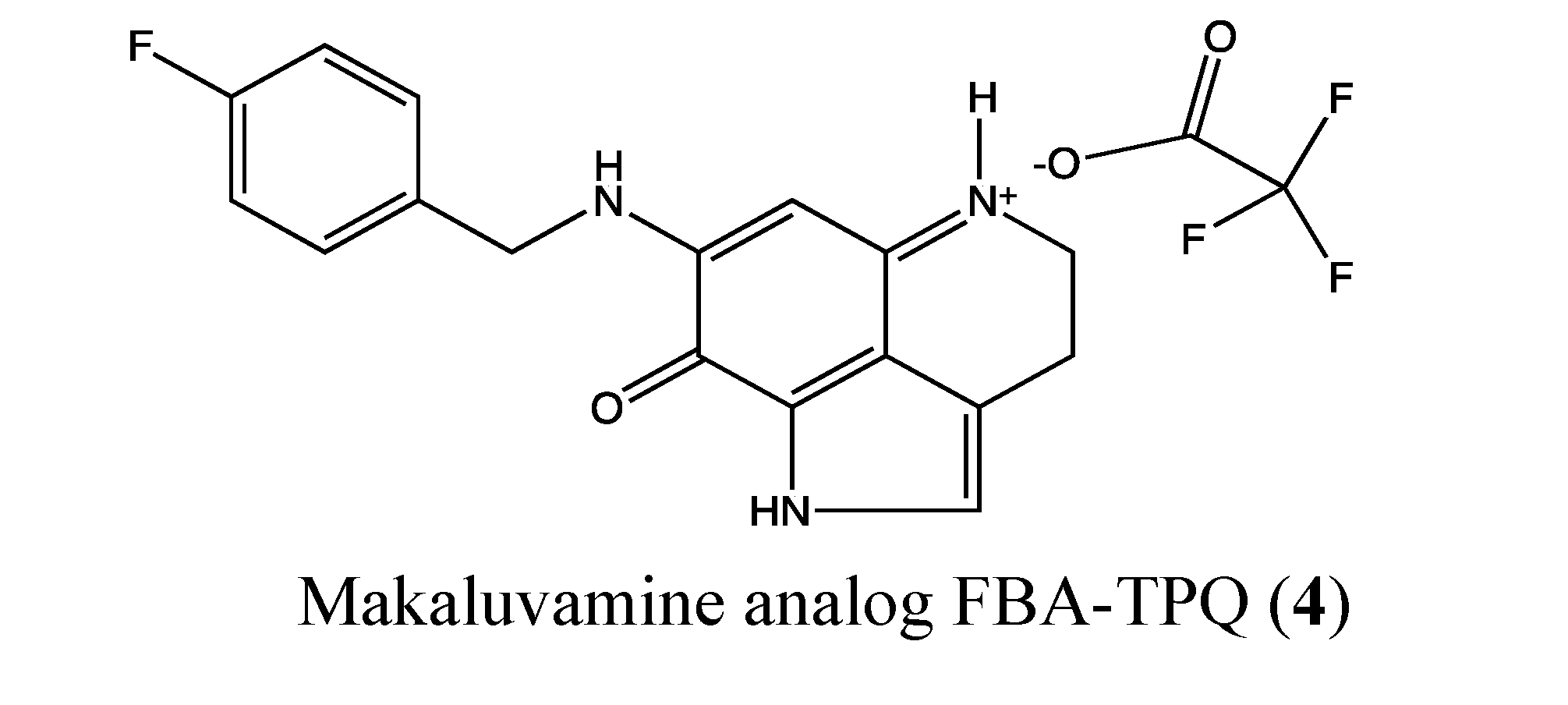
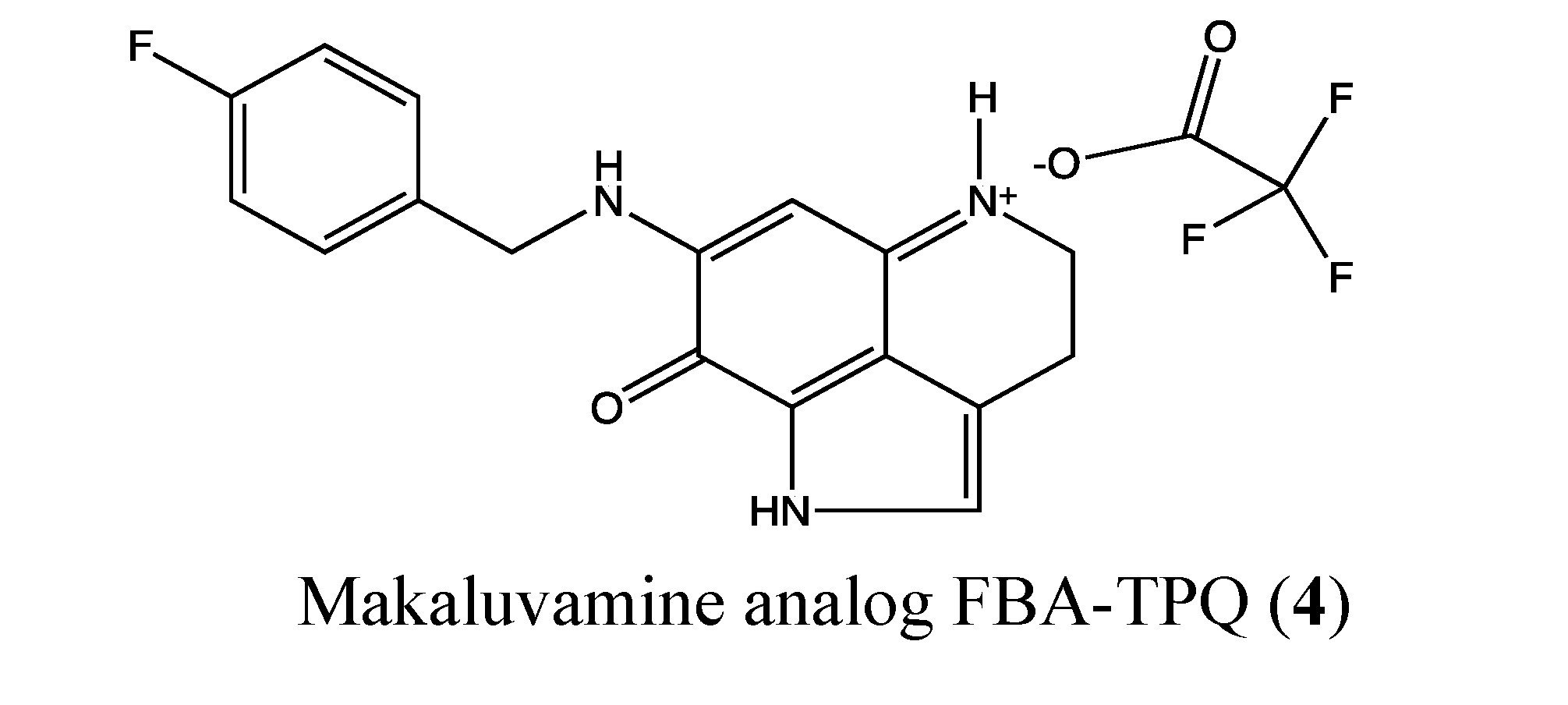
2.1.3. Makaluvamine Analog FBA-TPQ
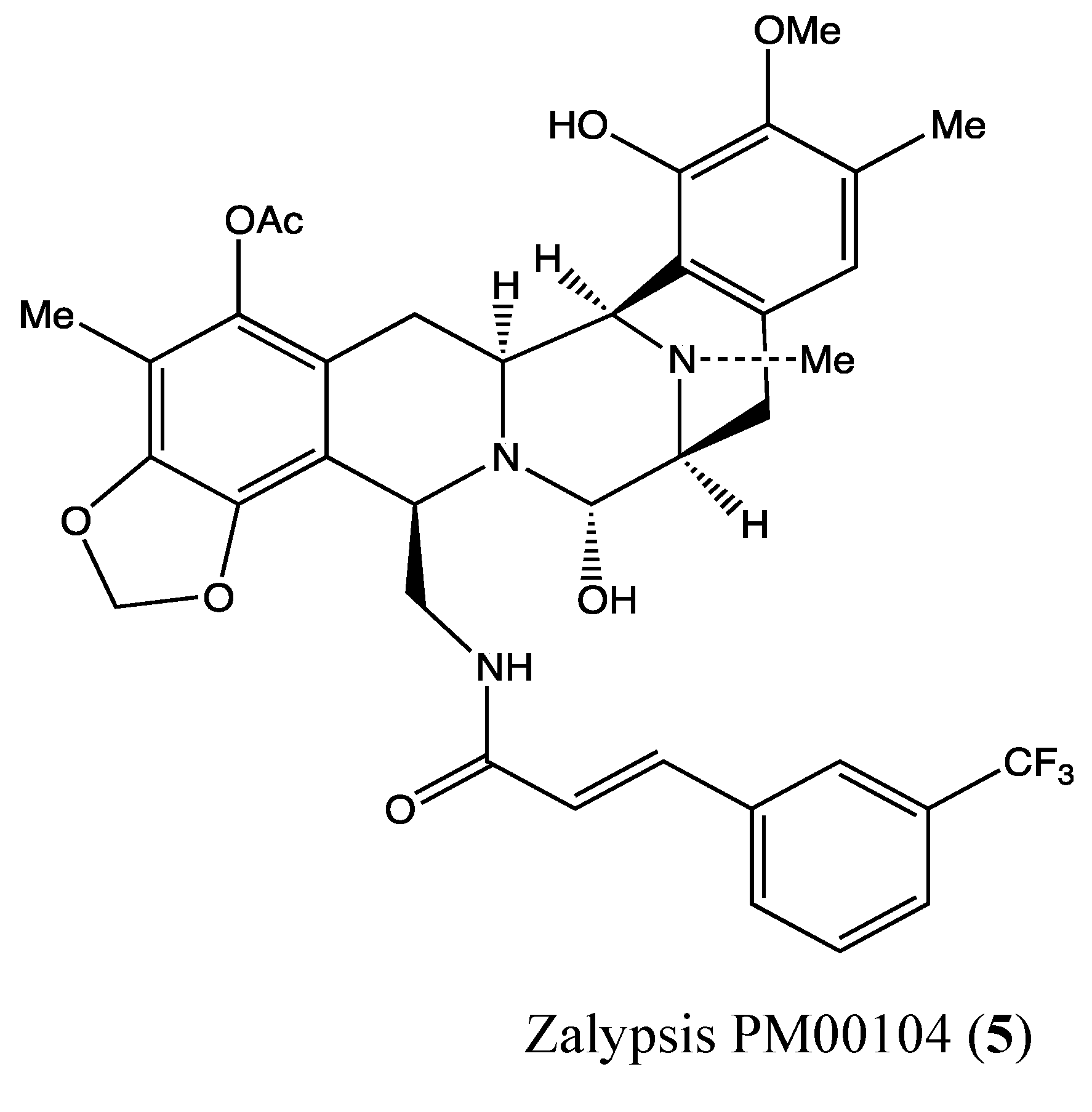
2.1.4. Zalypsis (PM00104)
2.2. Anthraquinones
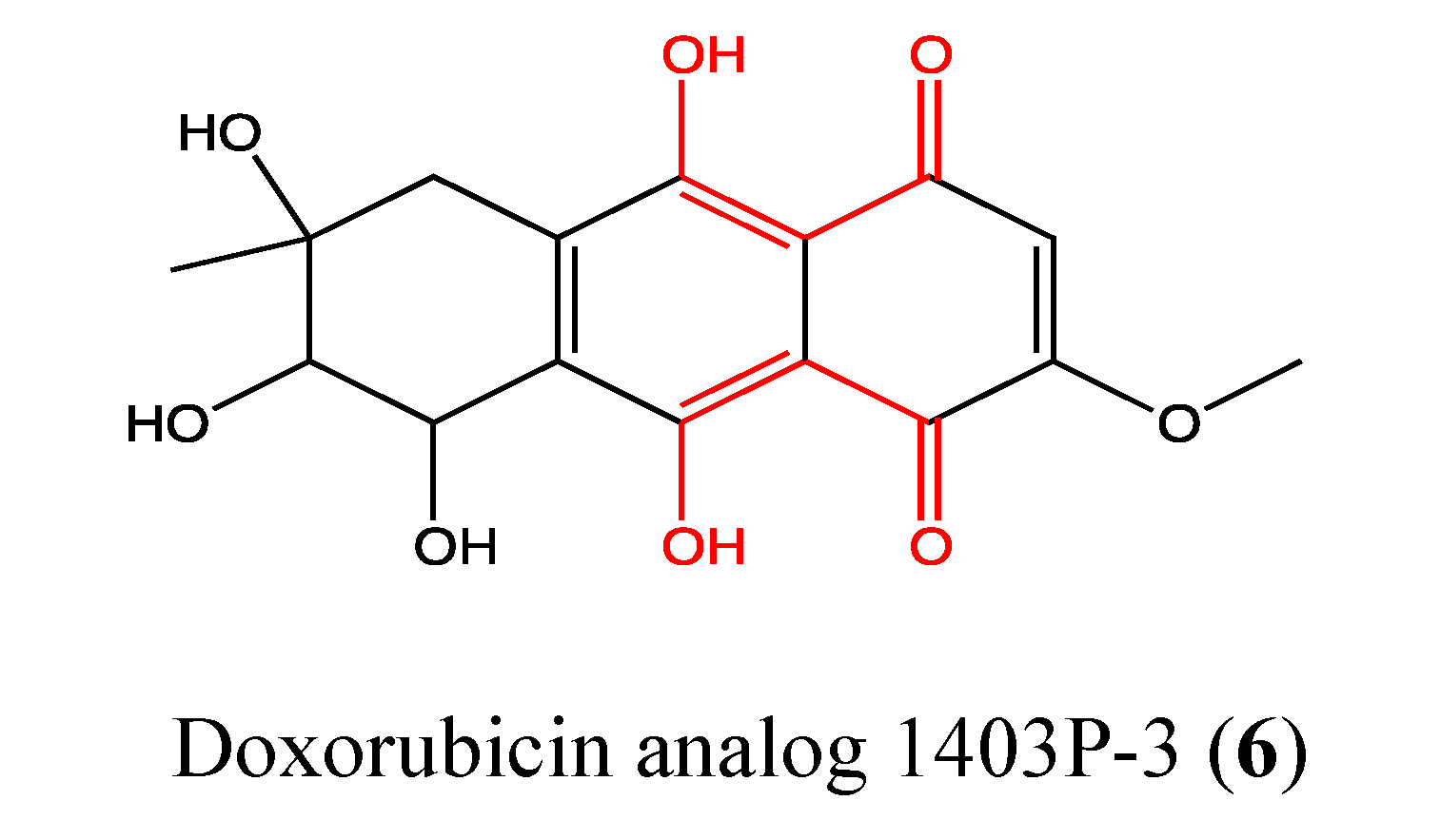
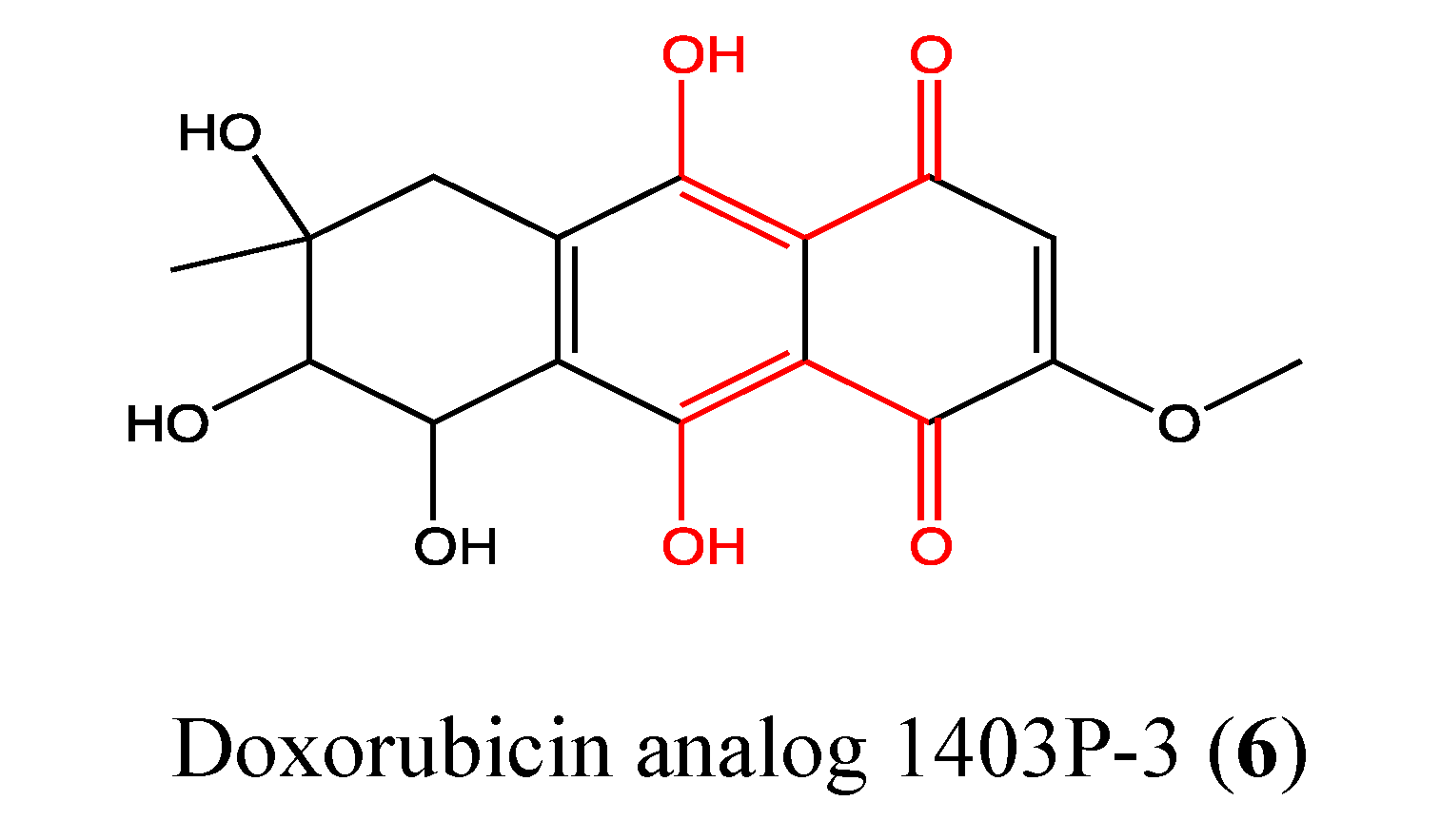
2.2.1. Doxorubicin Analogue 1403P-3
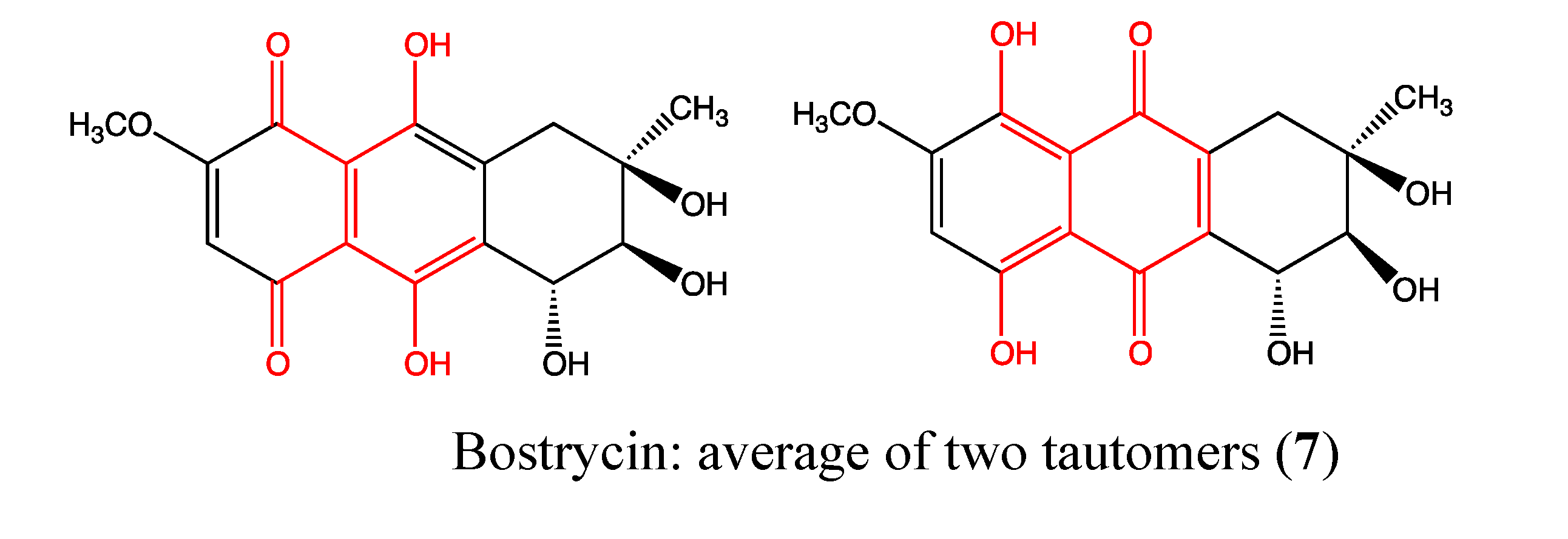
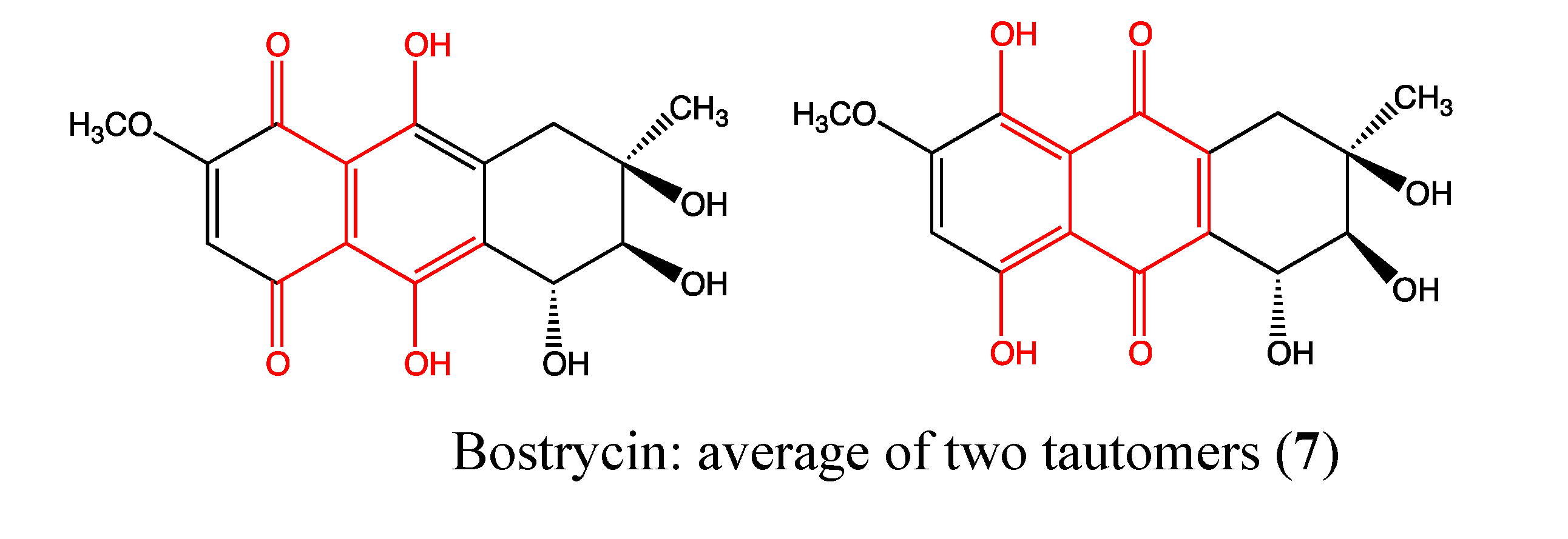
2.2.2. Bostrycin
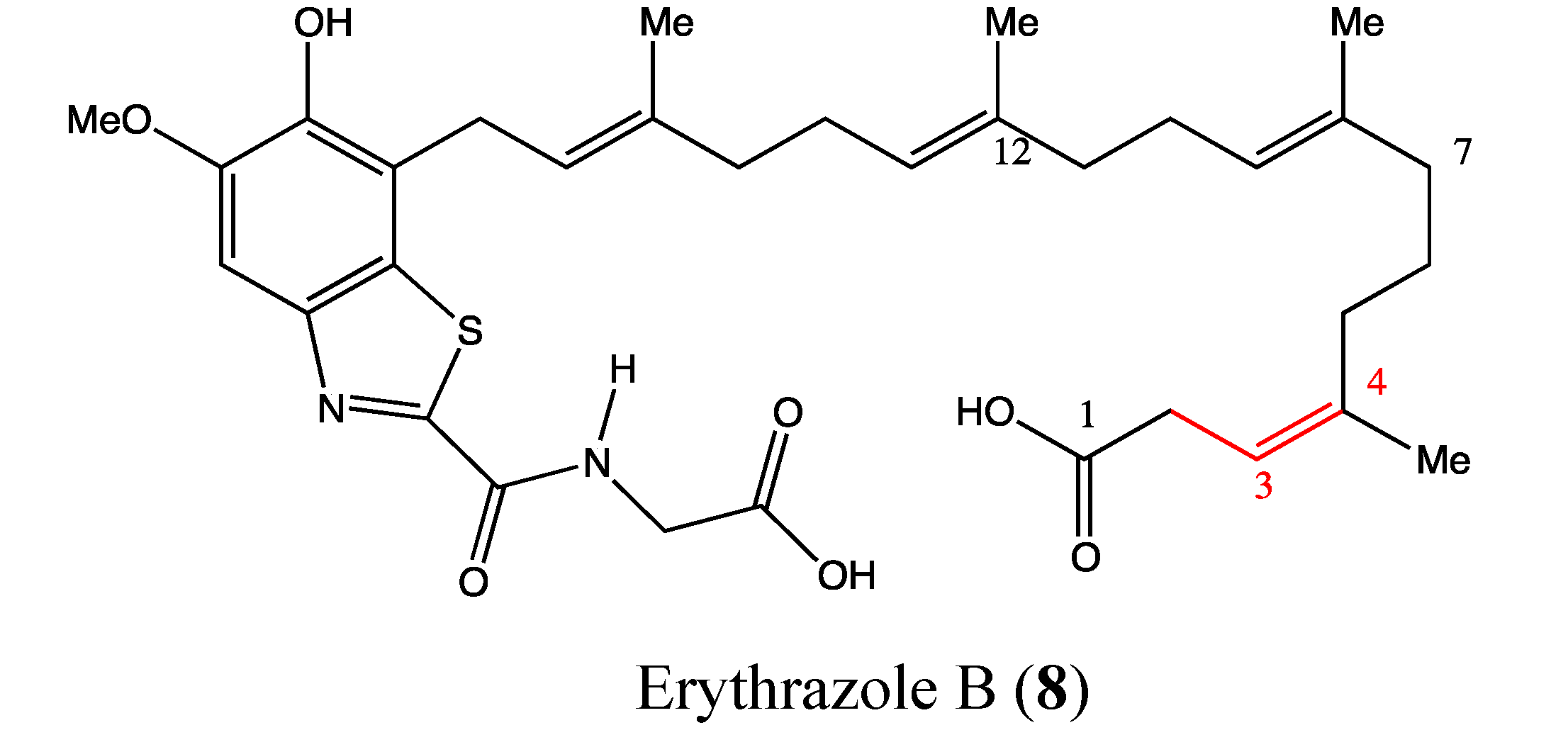
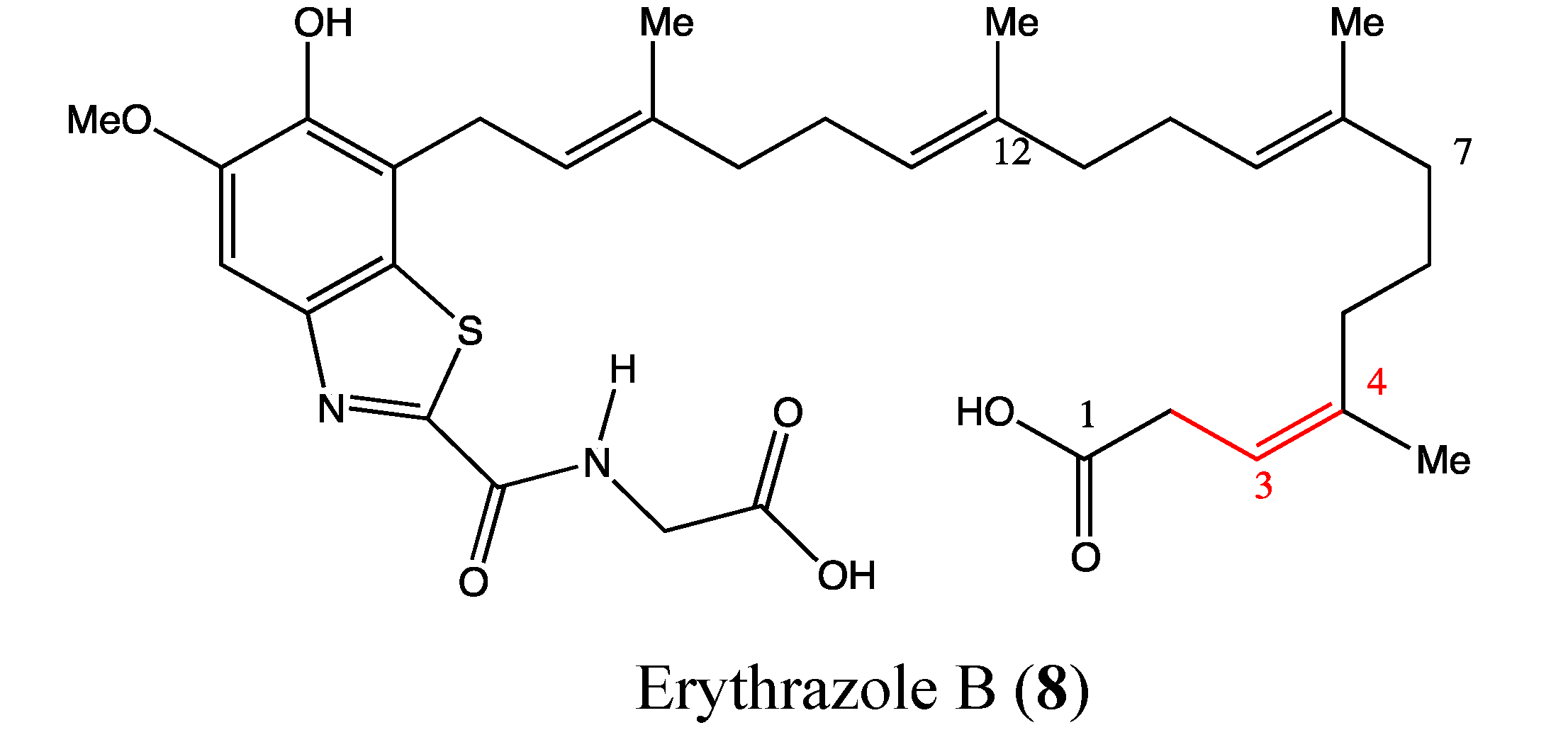
2.3. Benzothiazoles
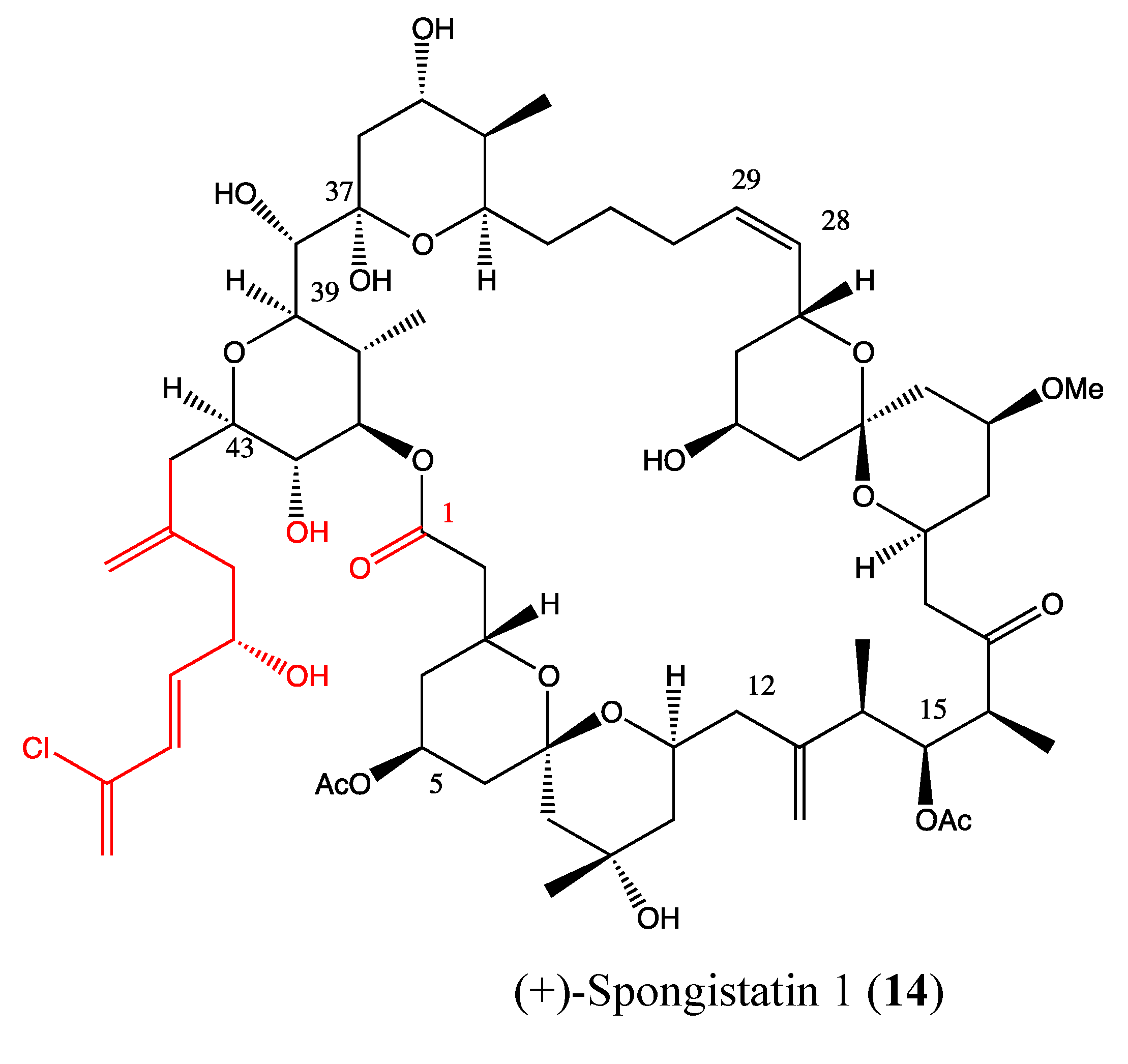
2.4. Macrolides
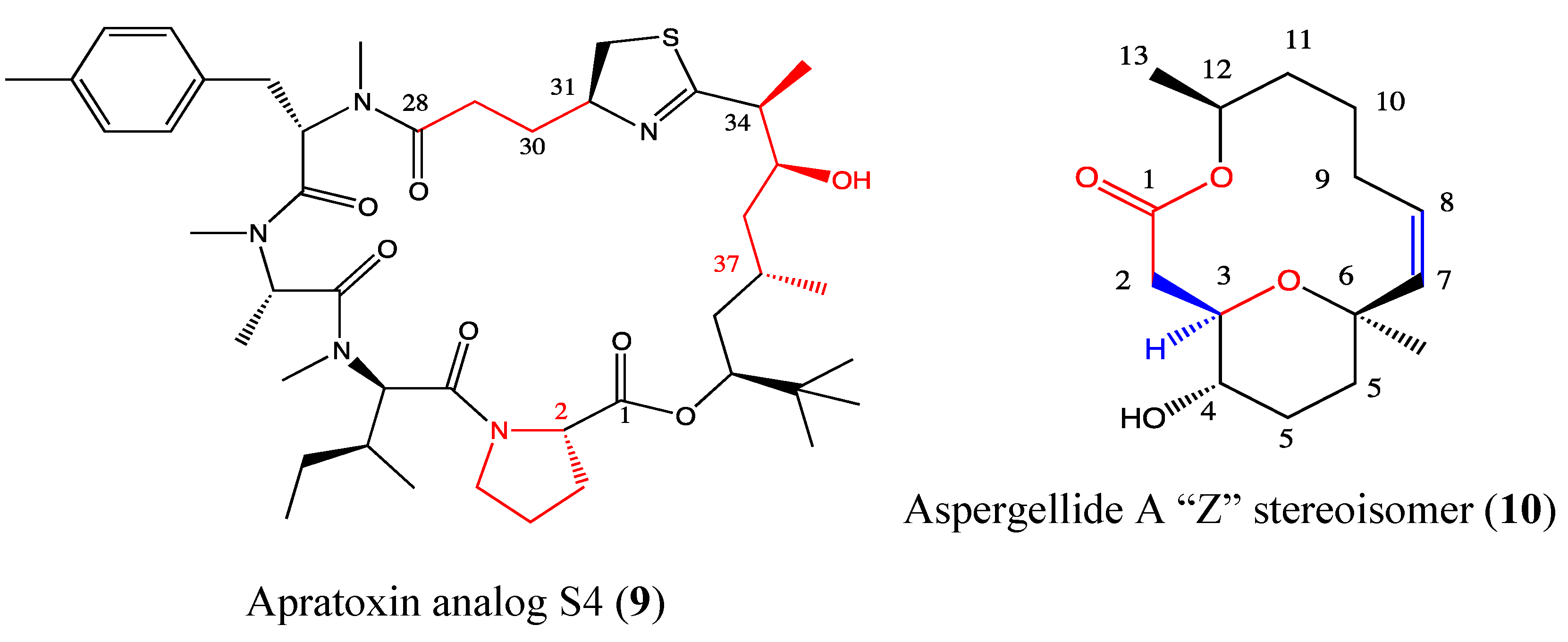
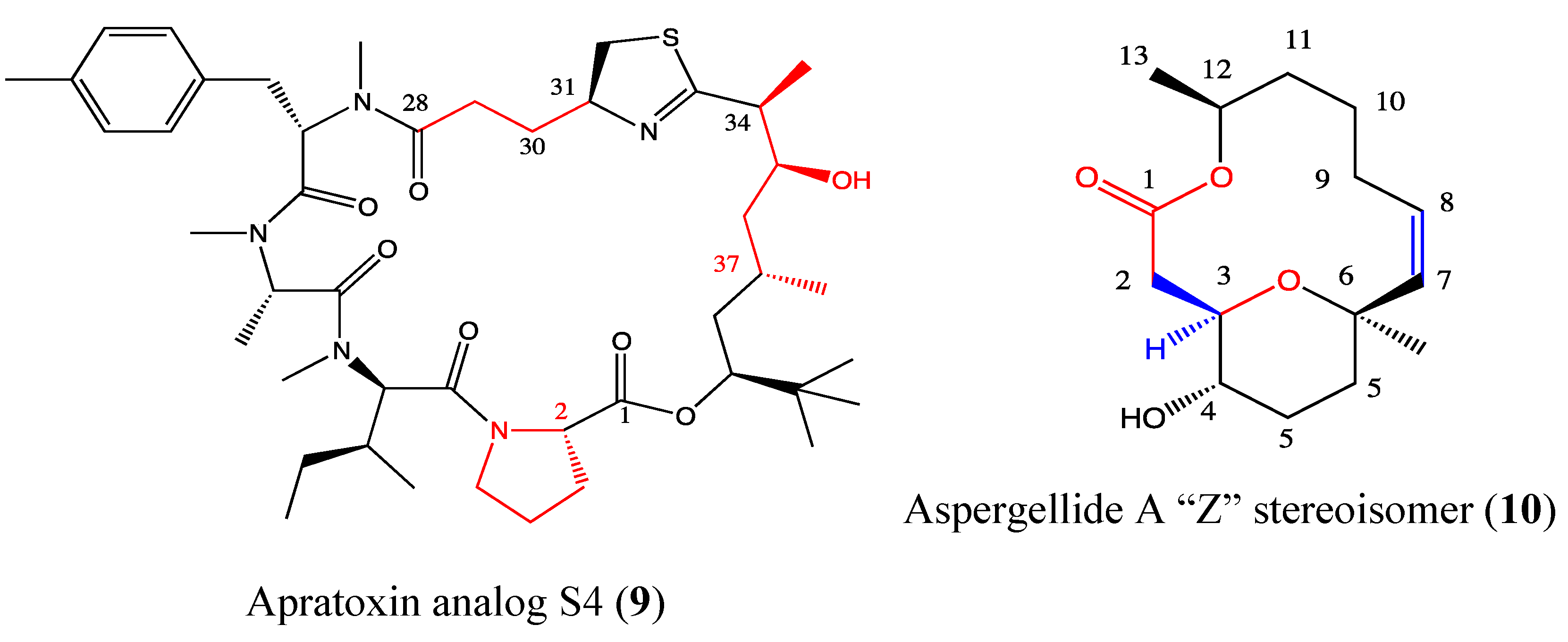
2.4.1. Apratoxin Analog S4-1e
2.4.2. Aspergillide A Analog
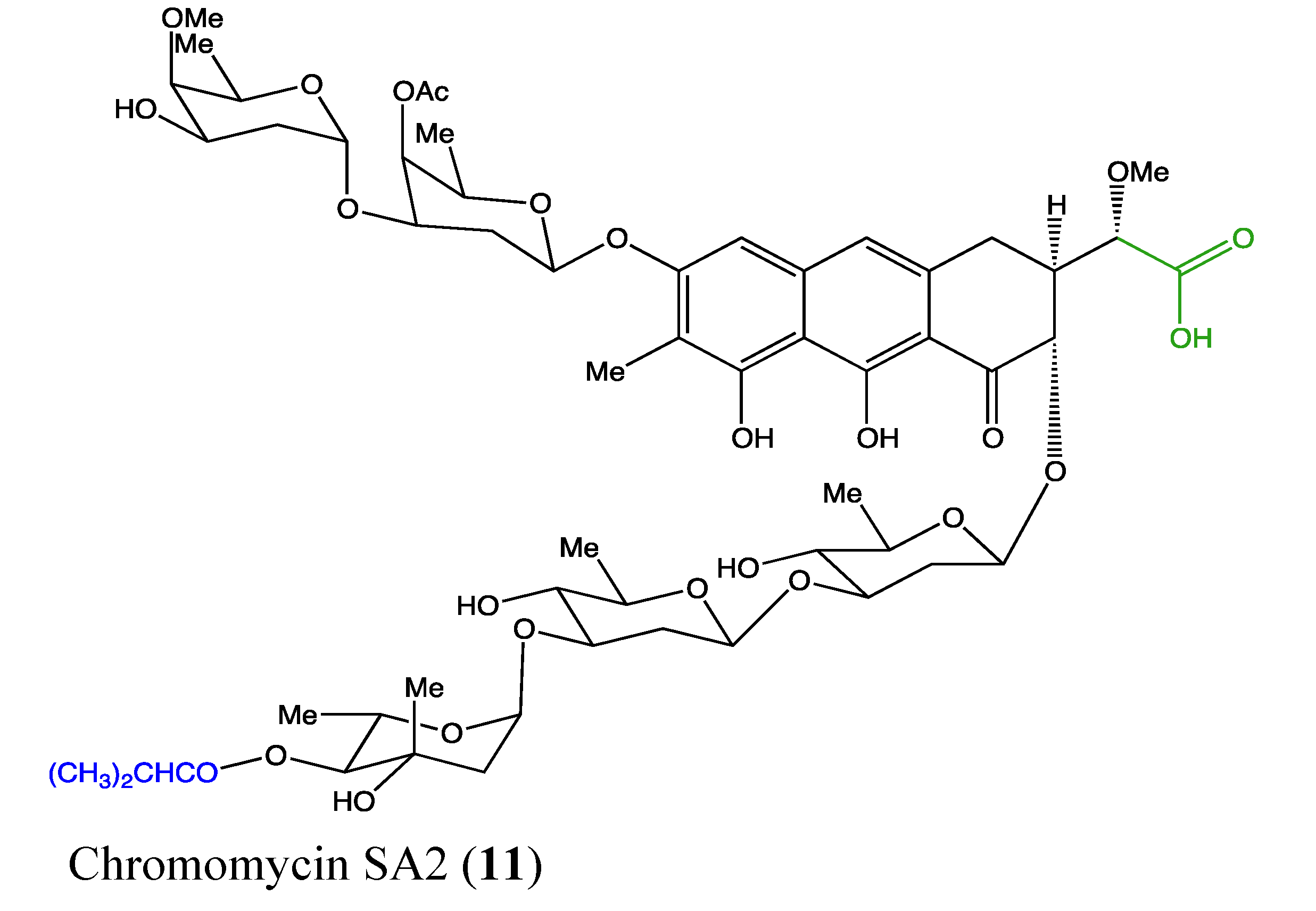
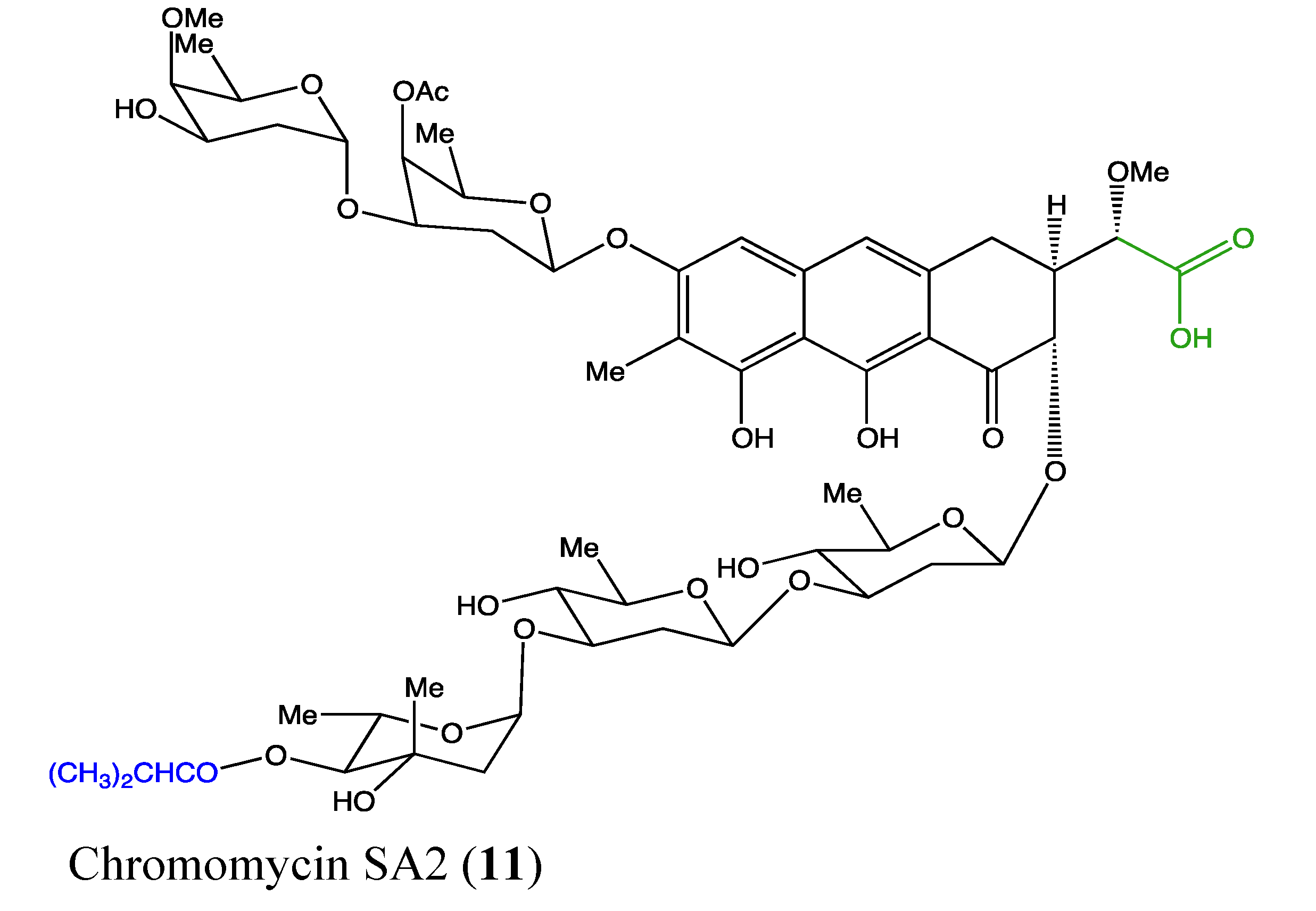
2.4.3. Chromomycin SA2
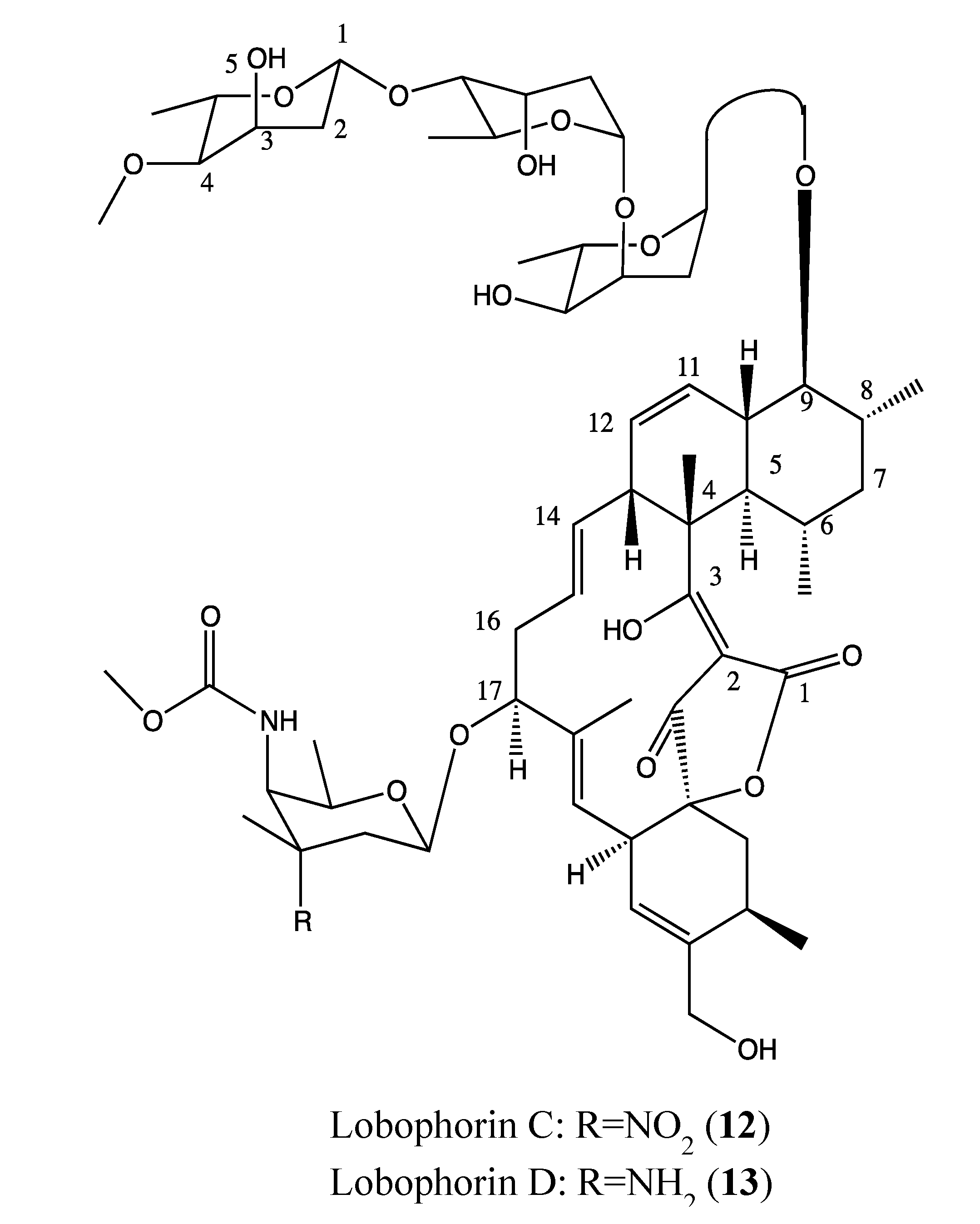
2.4.4. Lobophorin C and D


2.5. Peptides
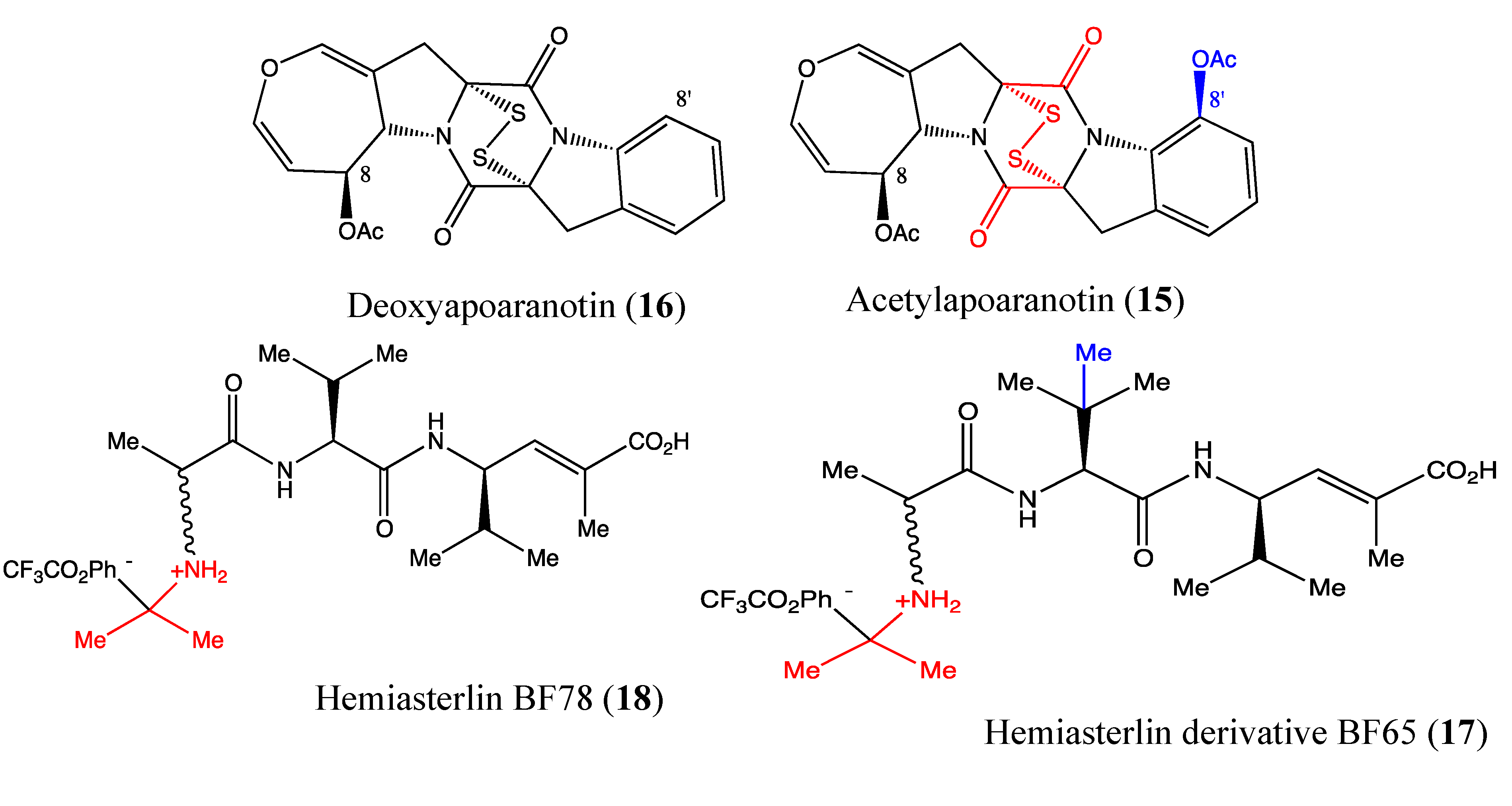
2.5.1. Acetylapoaranotin
2.5.2. Hemiasterlin Derivatives BF65 and BF78
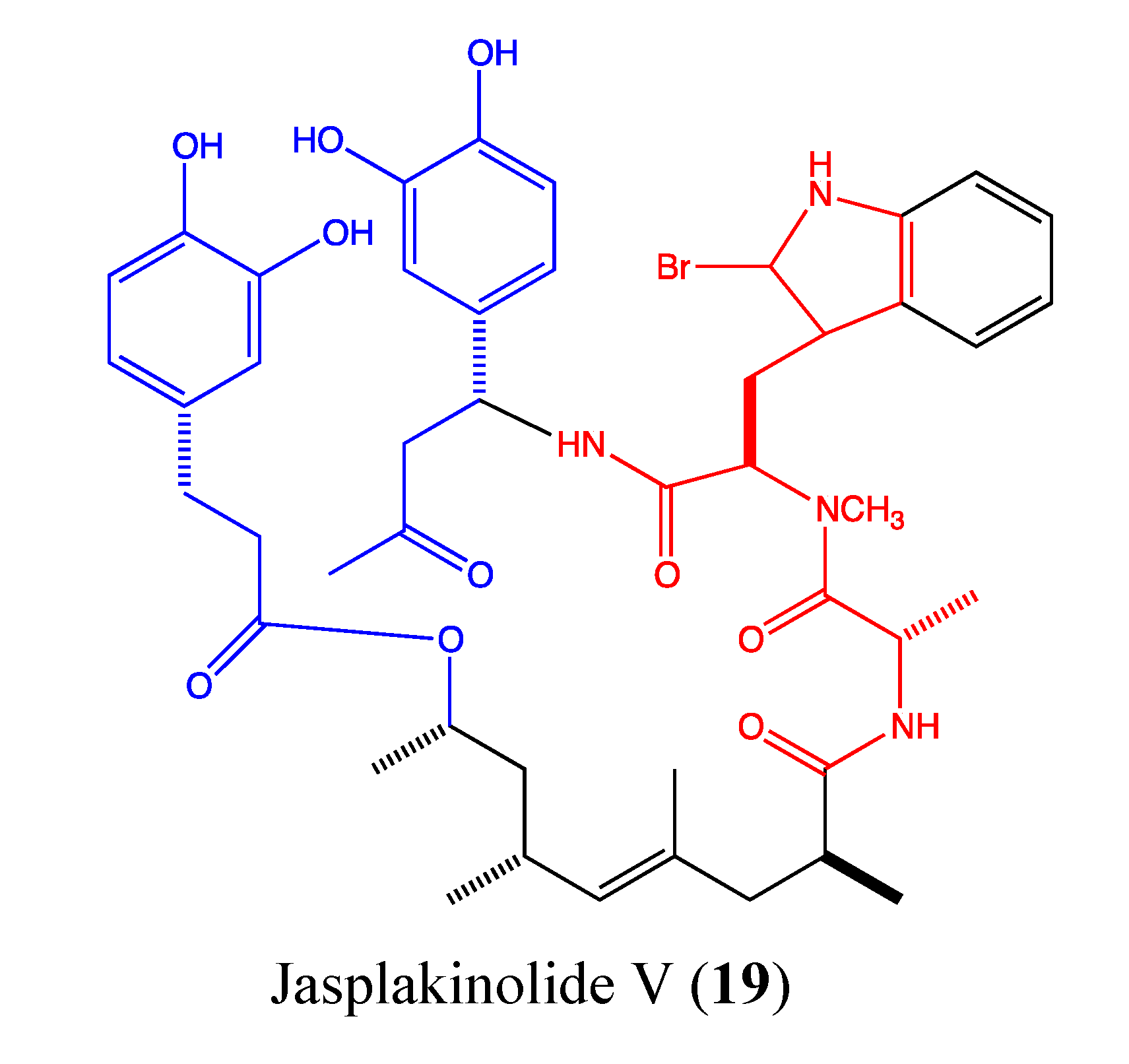
2.5.3. Jasplakinolide V
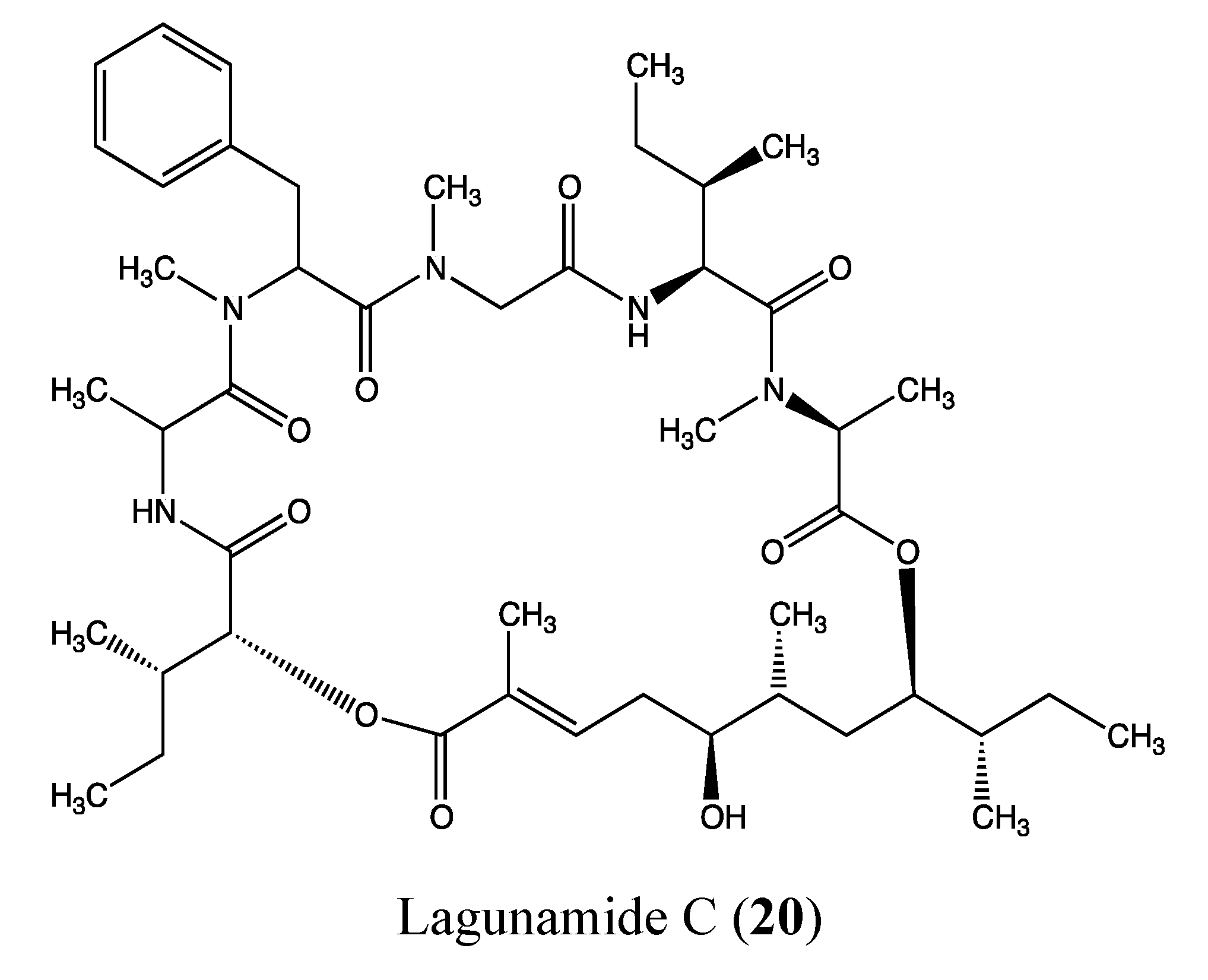
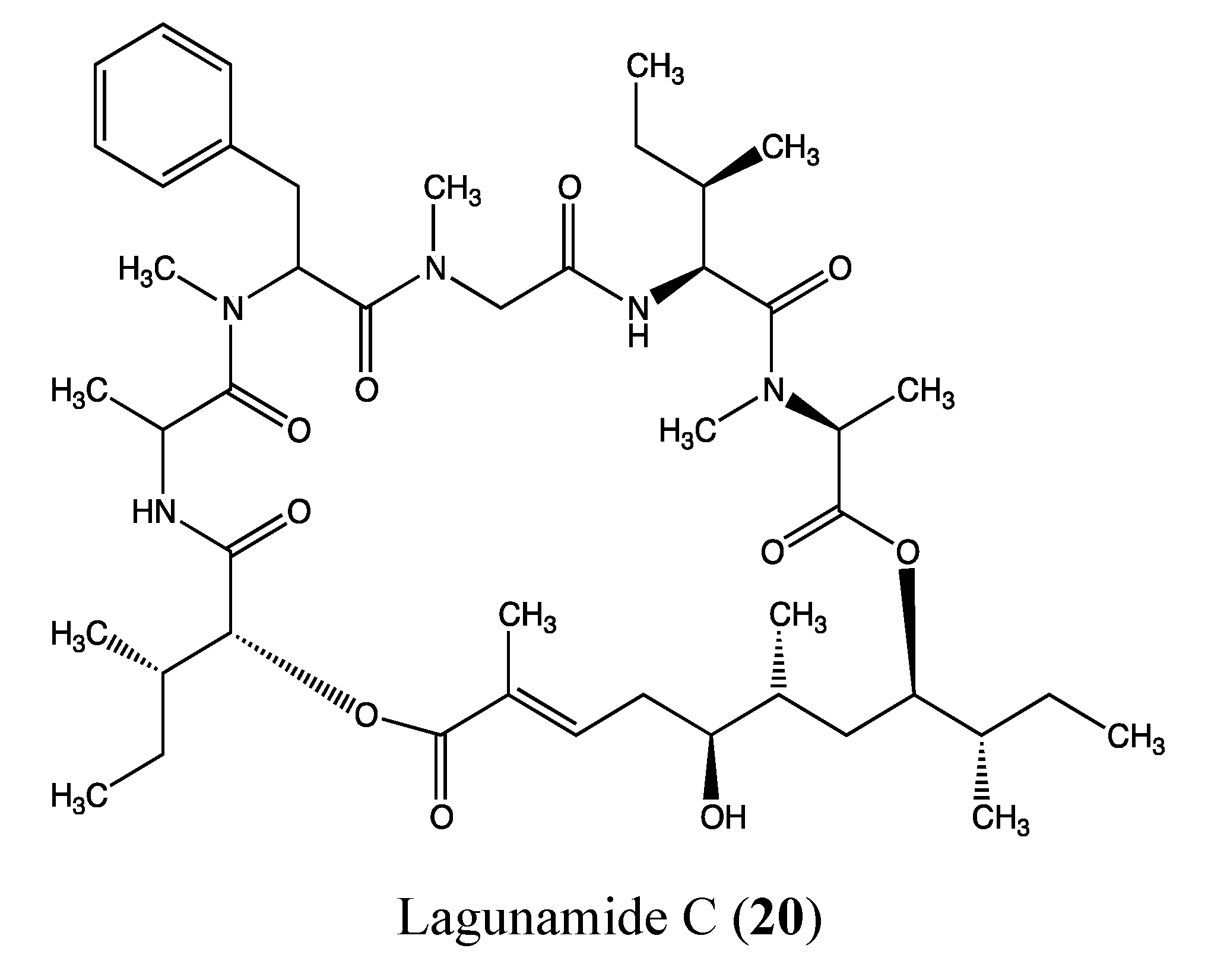
2.5.4. Lagunamide C
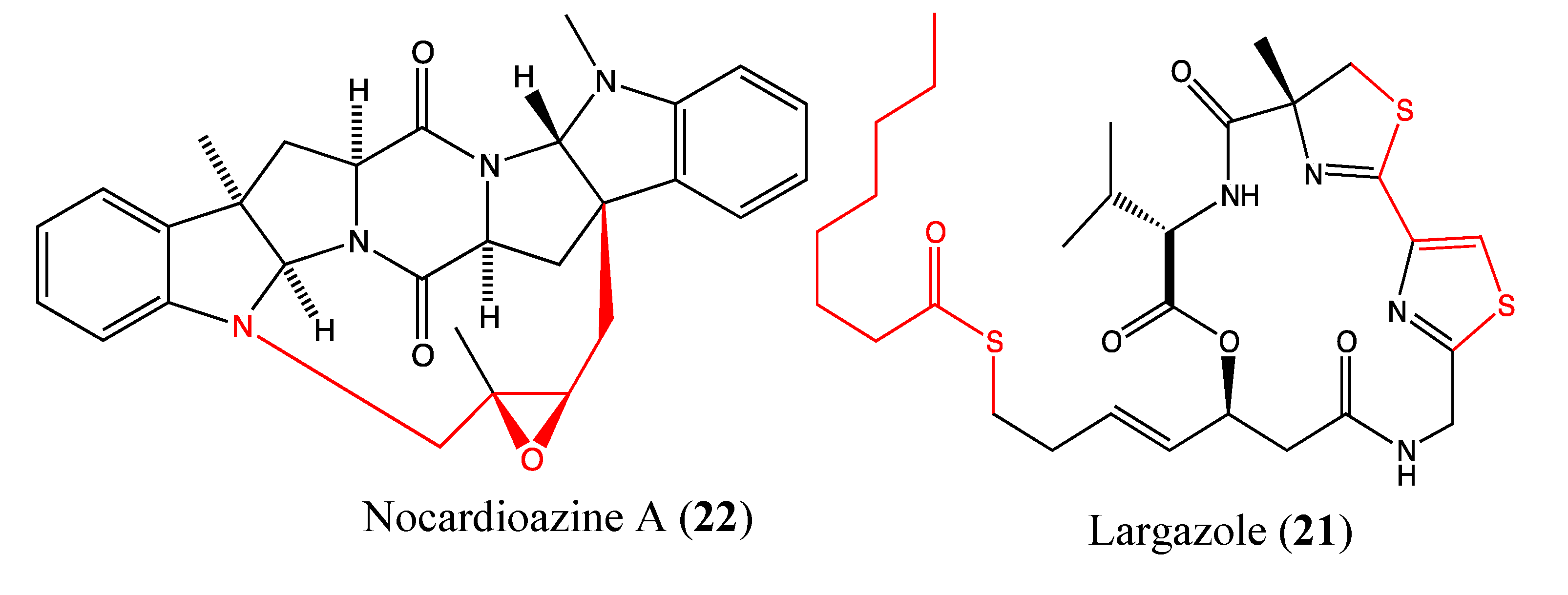
2.5.5. Largazole
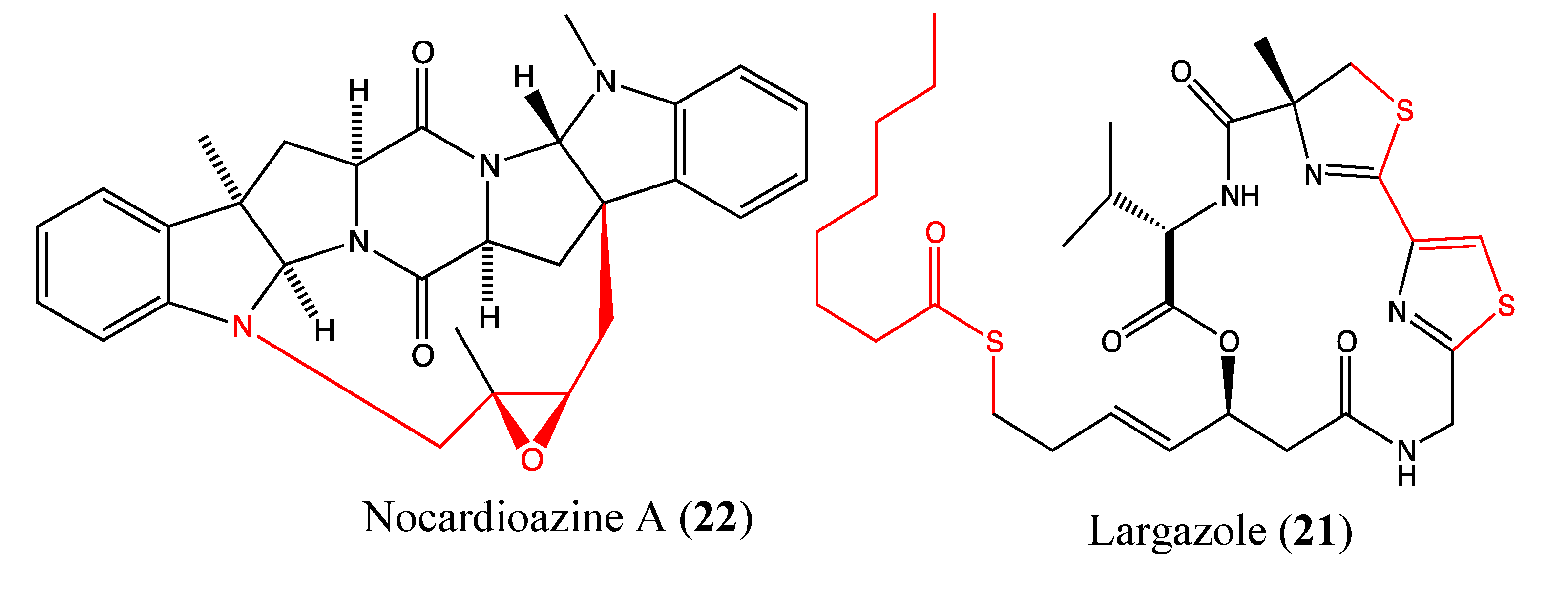
2.5.6. Nocardioazine A
2.5.7. Pardaxin
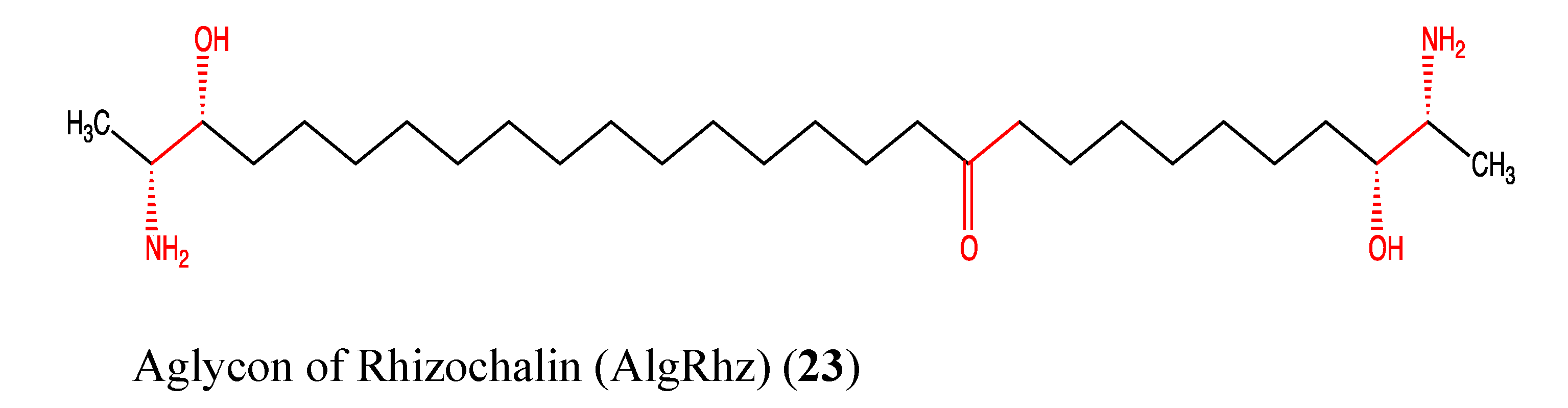
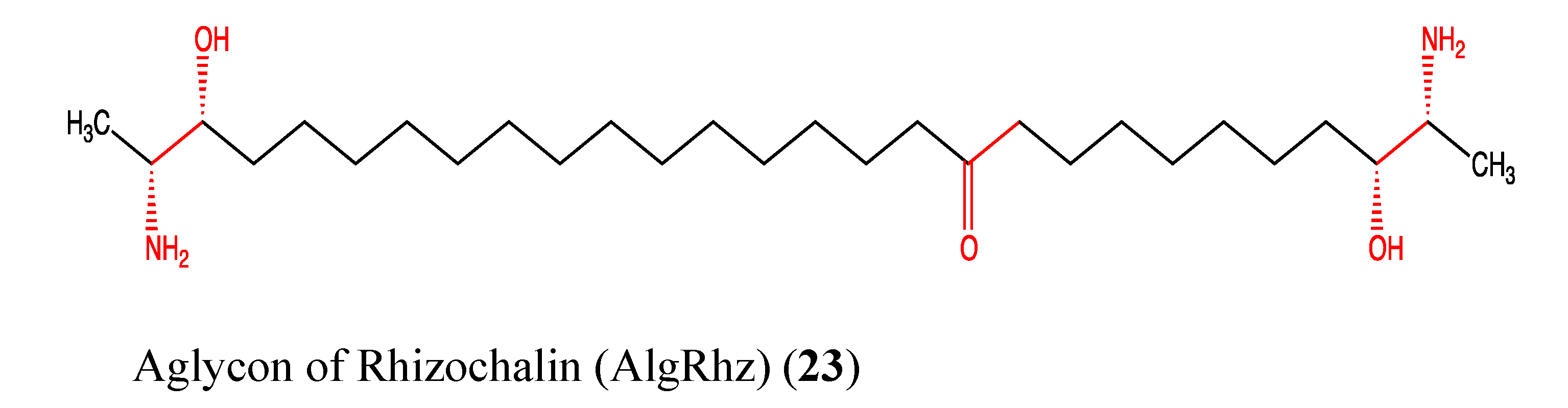
2.6. Sphingolipids
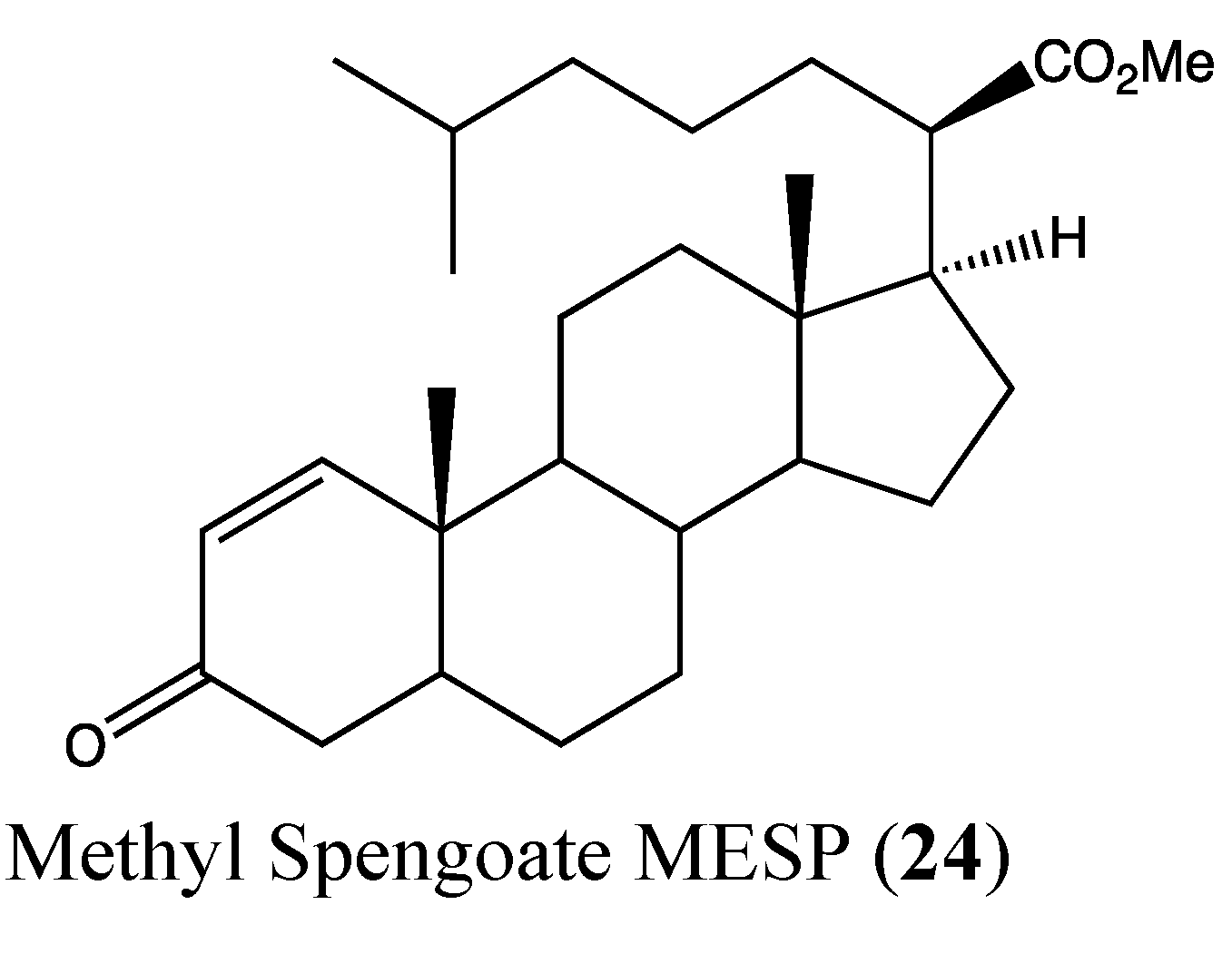
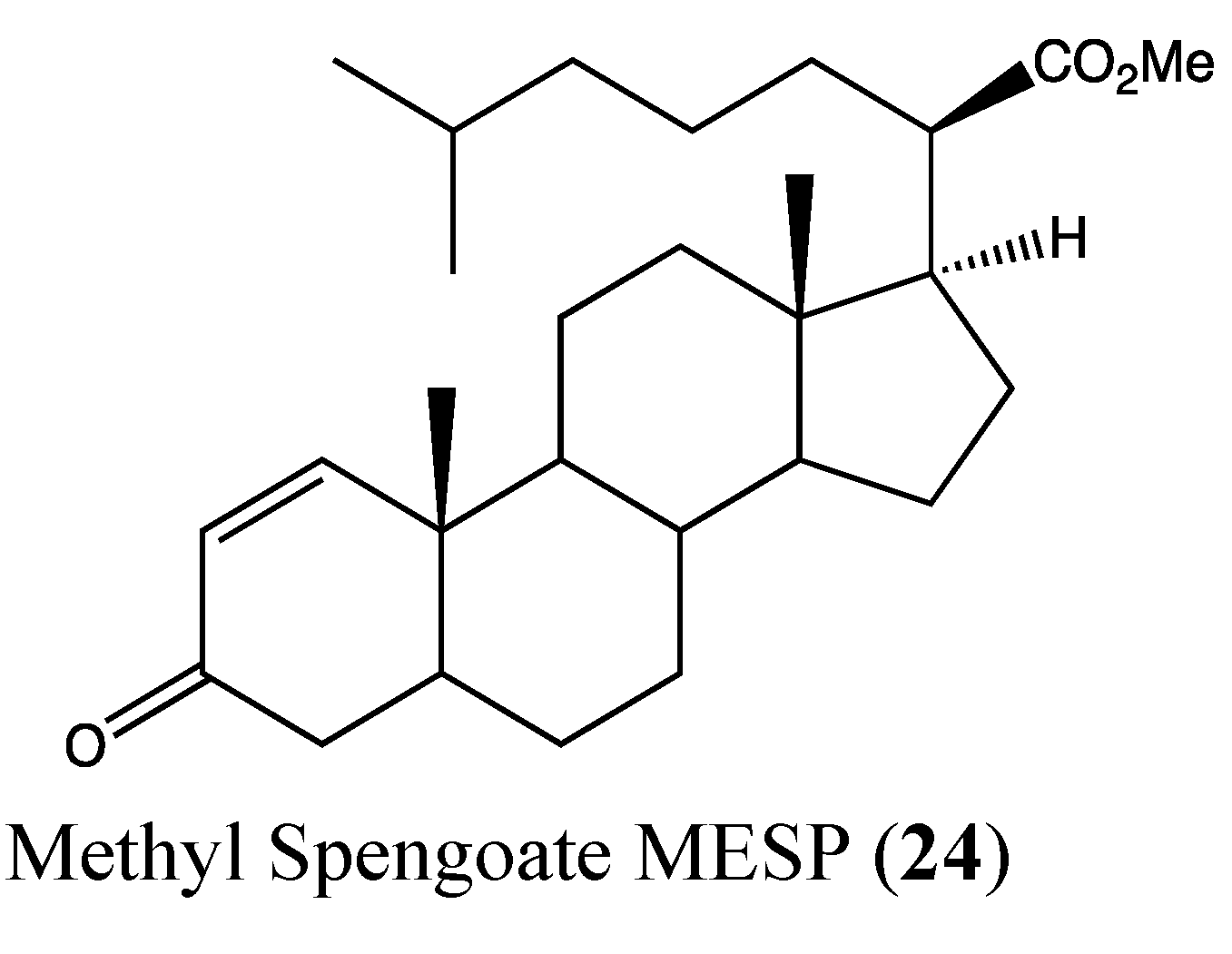
2.7. Steroids
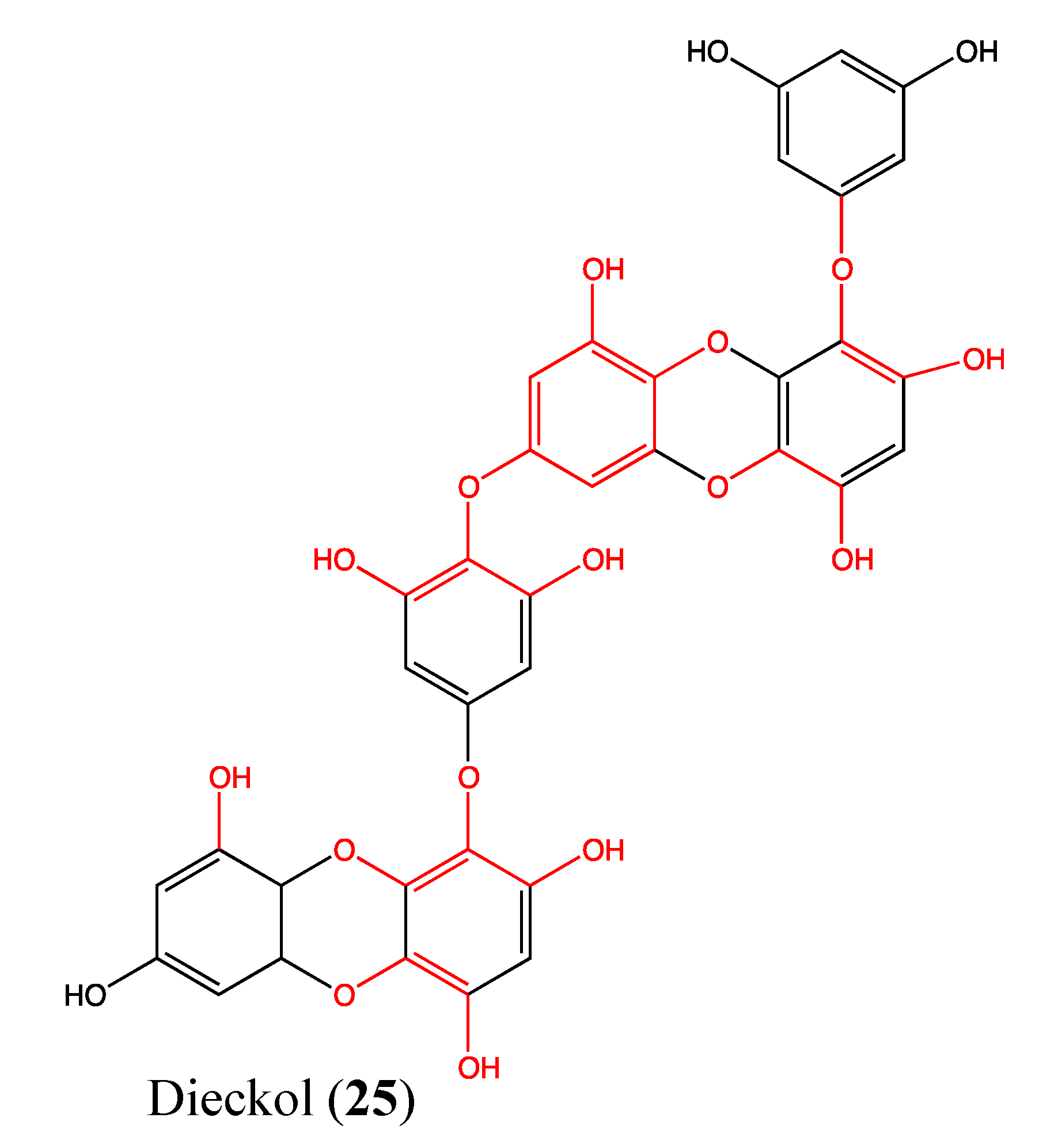
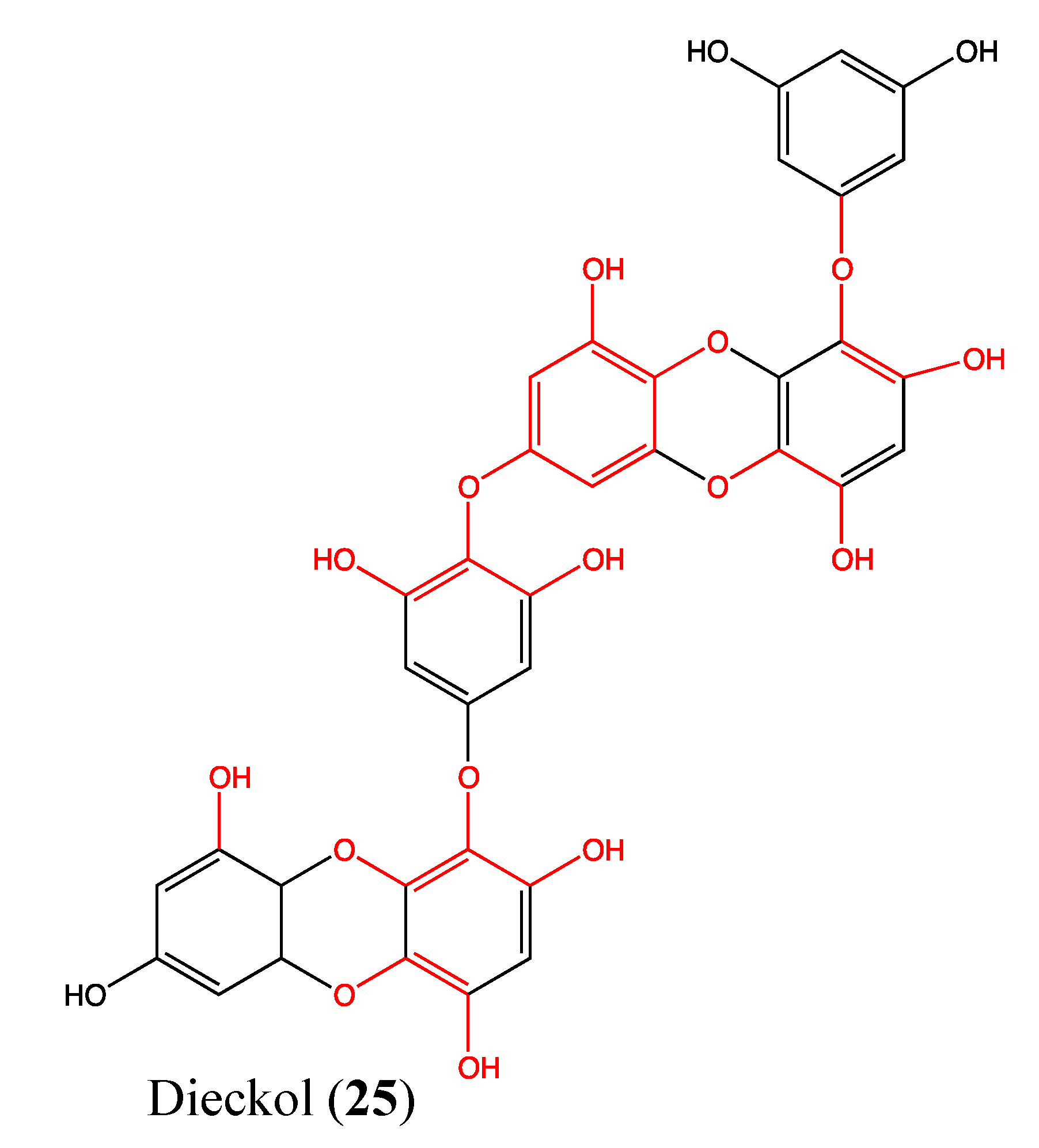
2.8. Tannins
2.9. Terpenes/Terpenoids
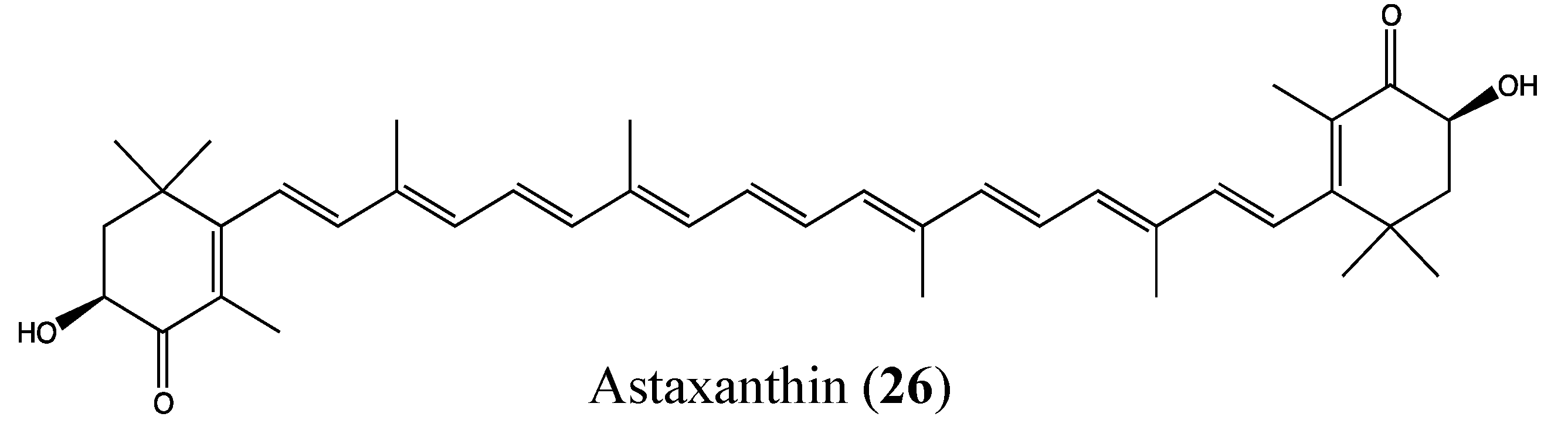
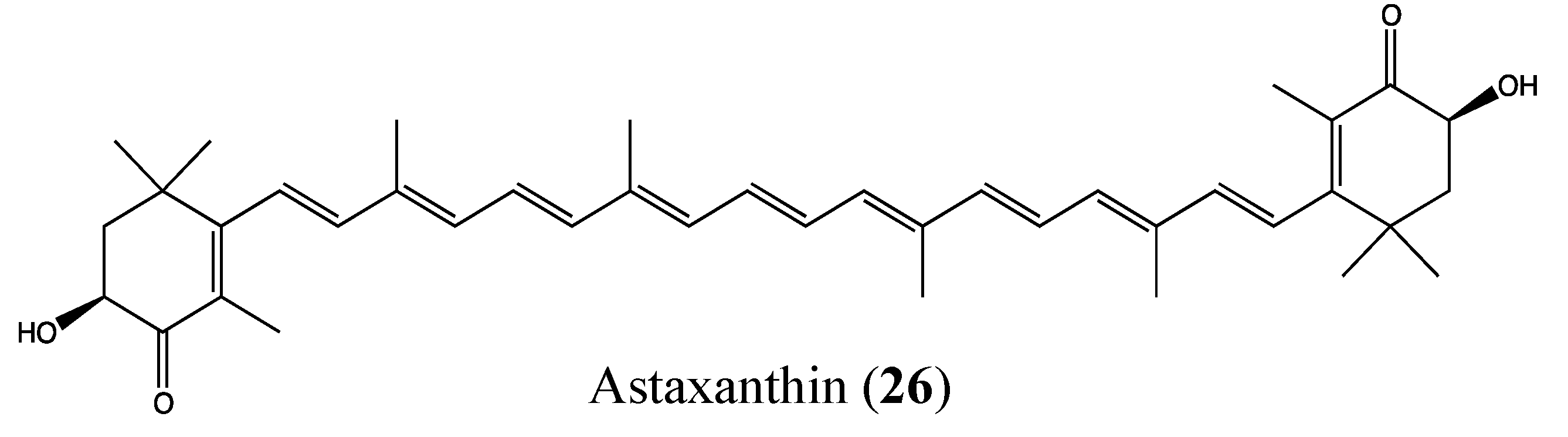
2.9.1. Astaxanthin
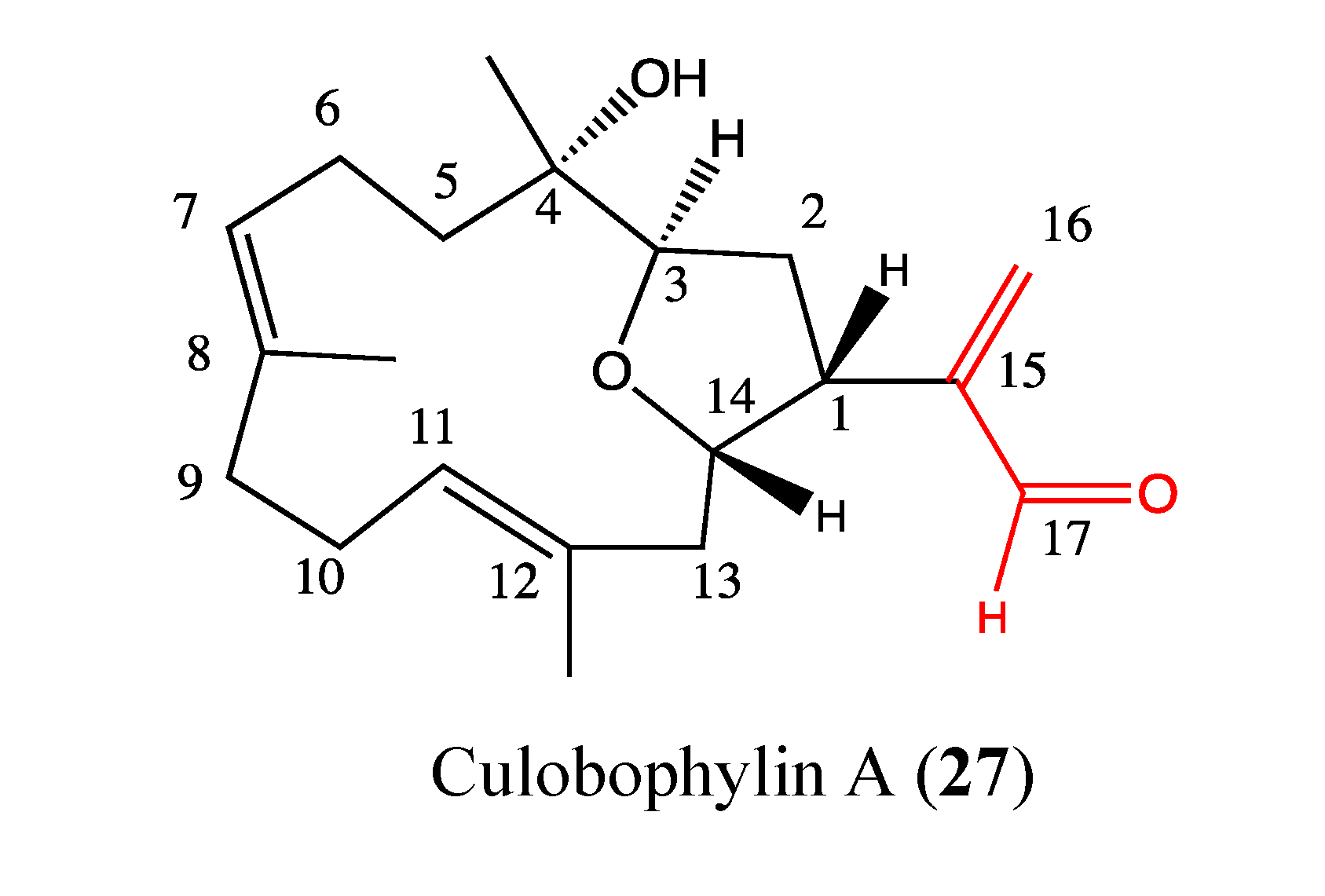
2.9.2. Culobophylin A
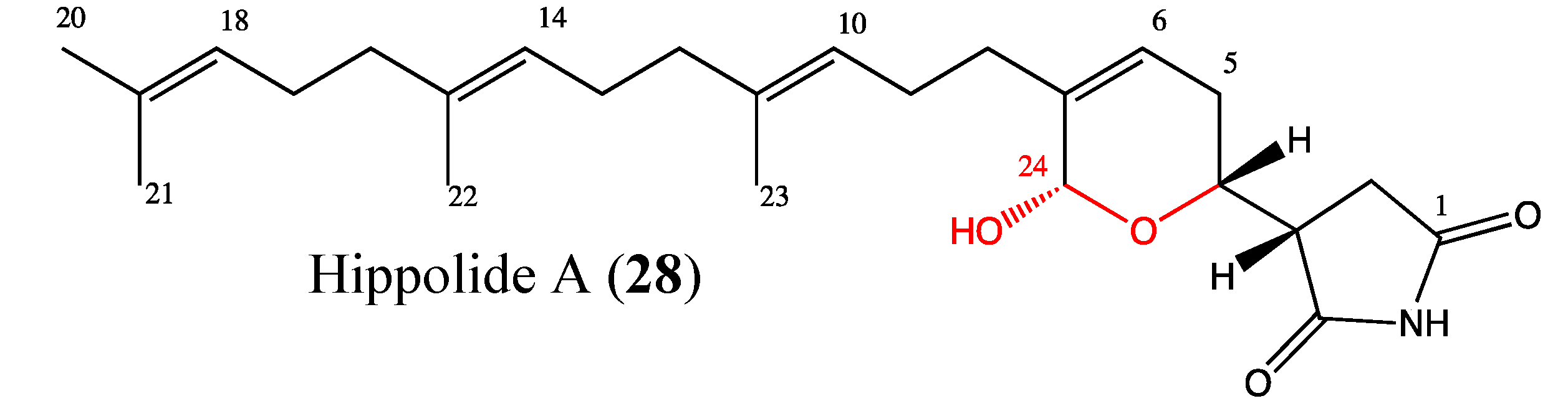
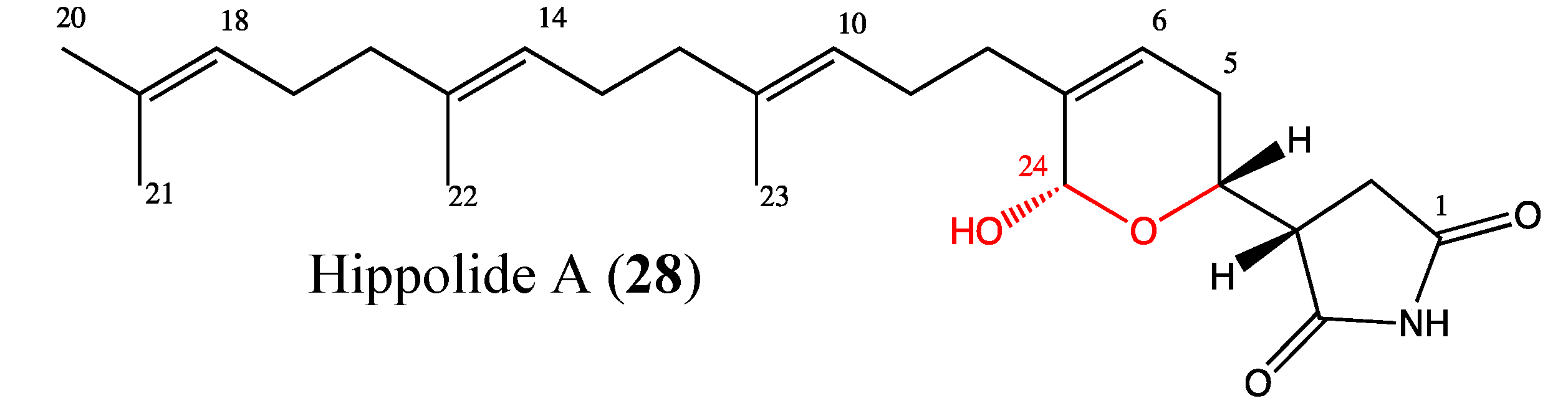
2.9.3. Hippolide A
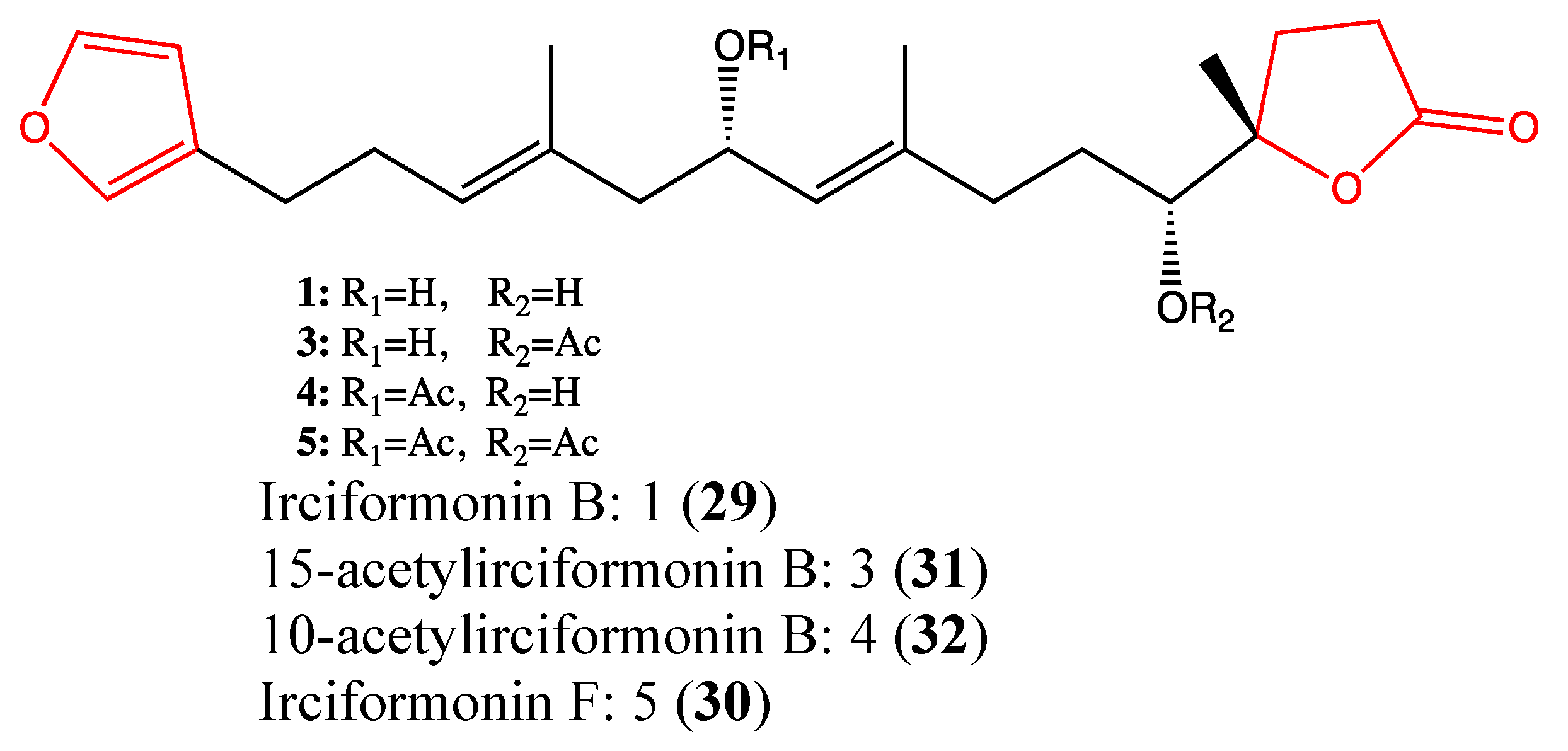
2.9.4. Irciformonin Analogs

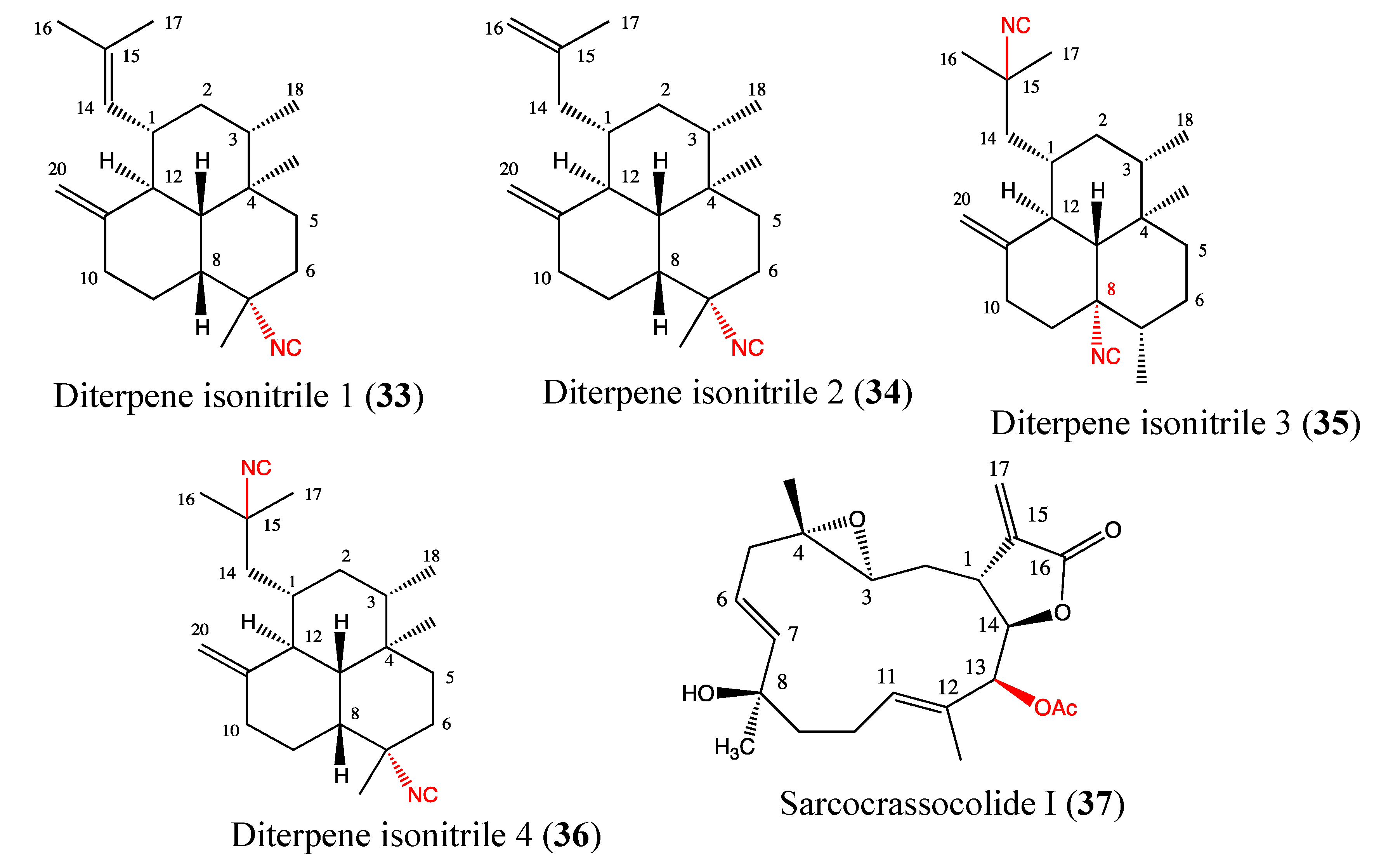
2.9.5. Diterpene Isonitriles 1, 2, 3 and 4
2.9.6. Sarcocrassocolide I
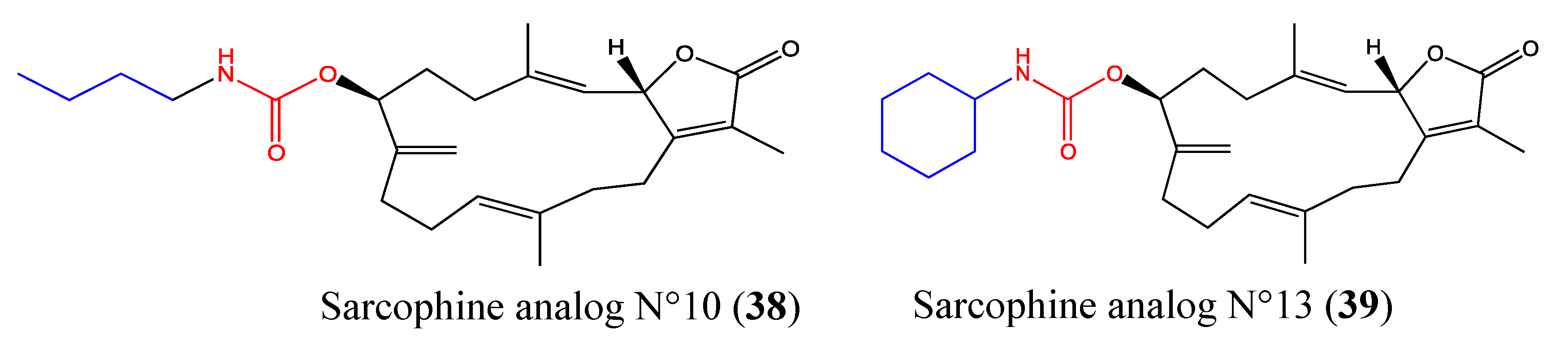
2.9.7. Sarcophine Analogs No 10 and No 13
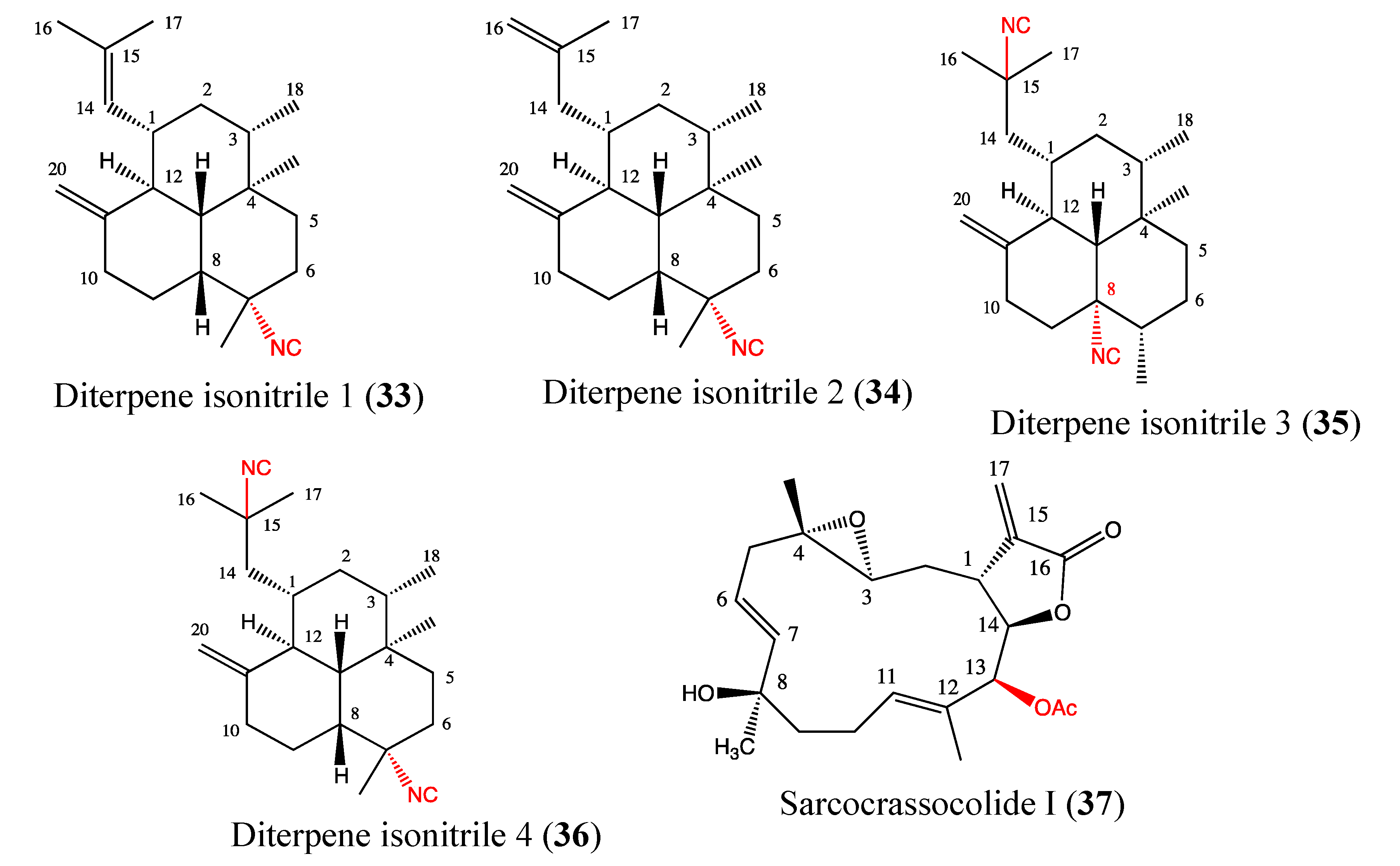

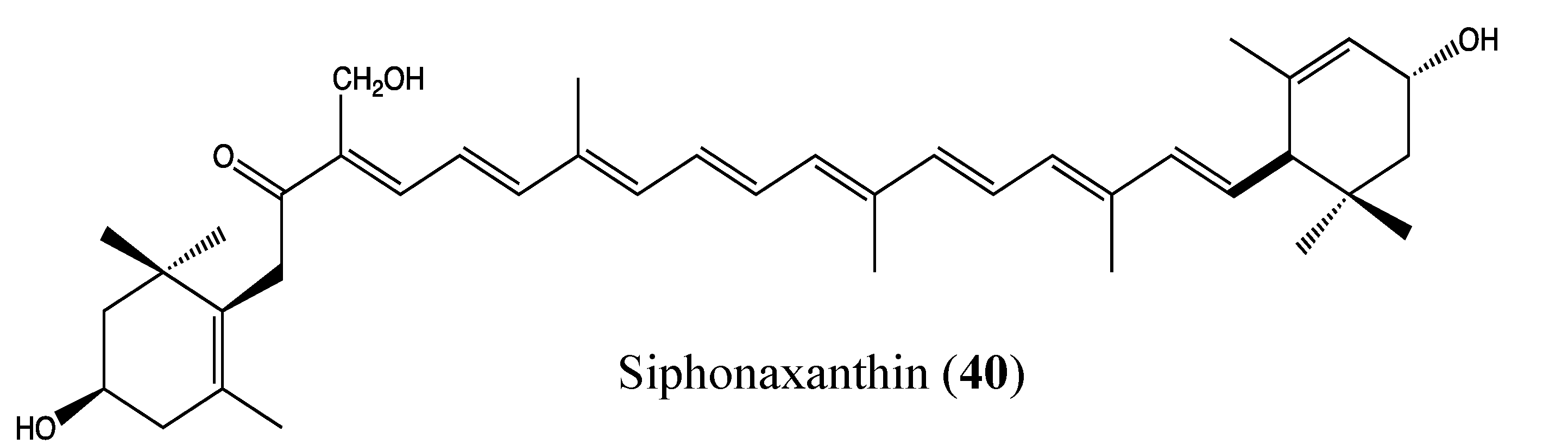
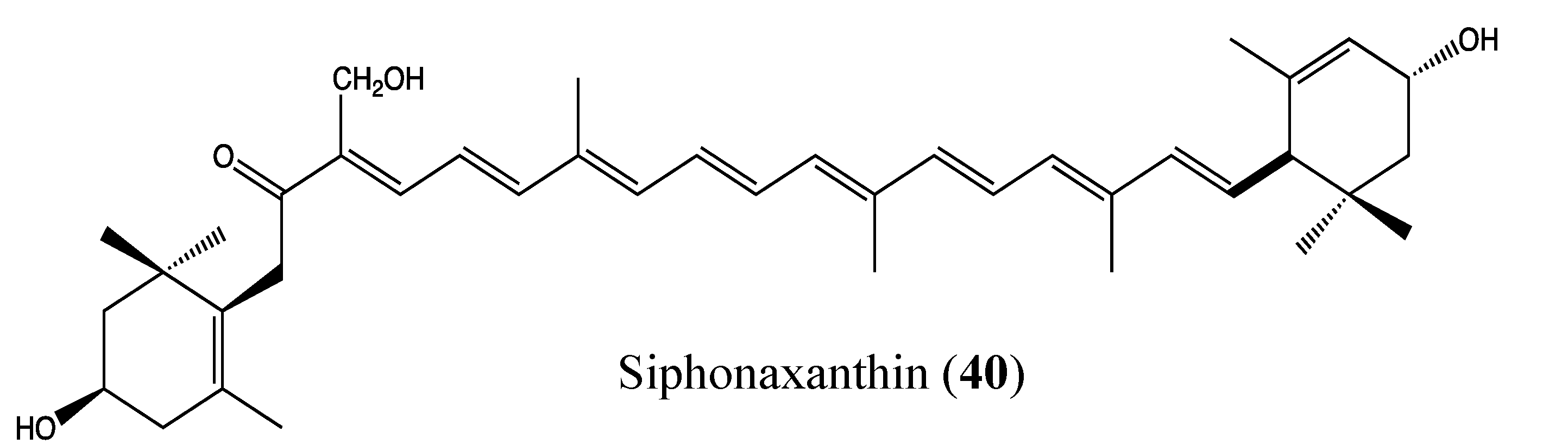
2.9.8. Siphonaxanthin
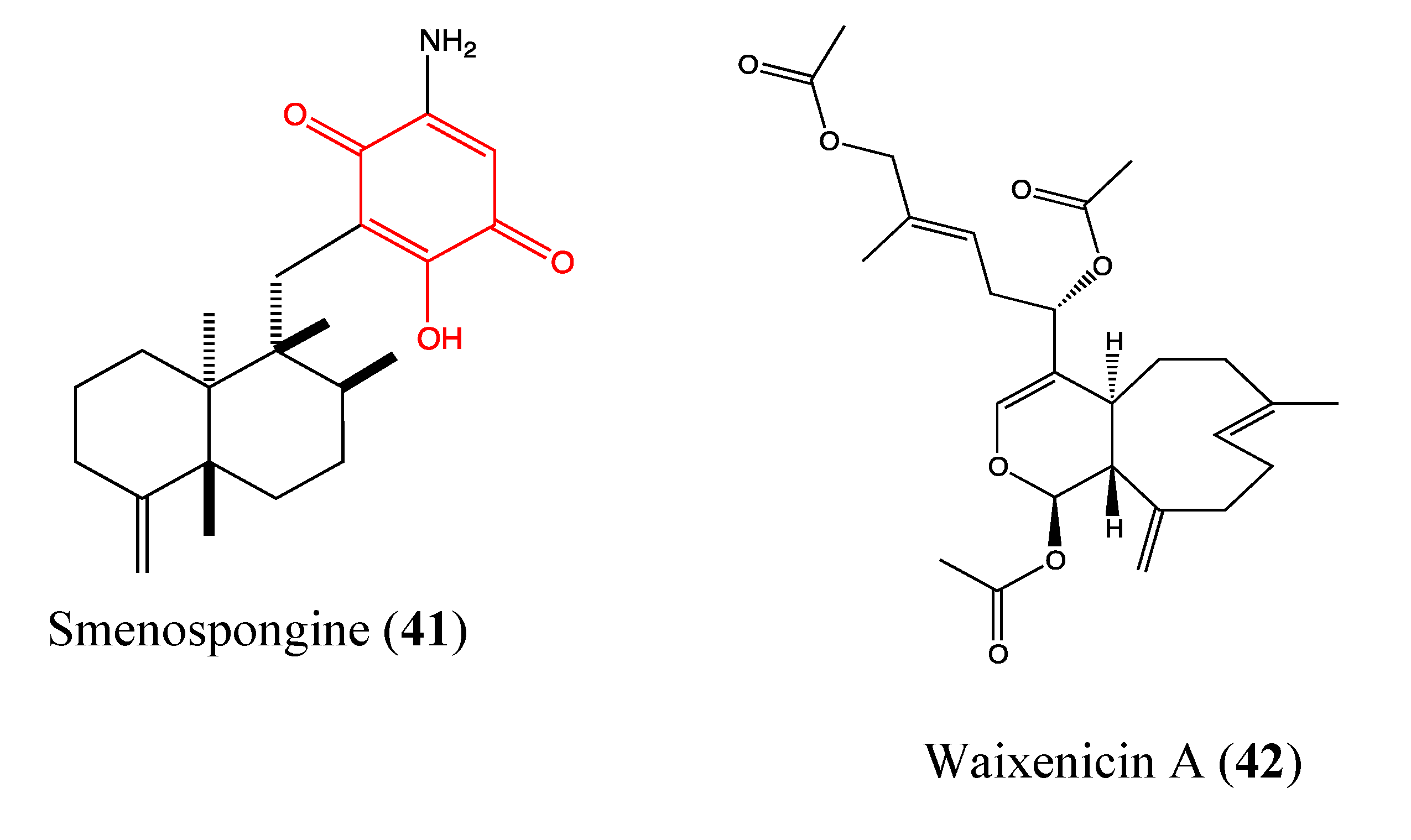
2.9.9. Smenospongine
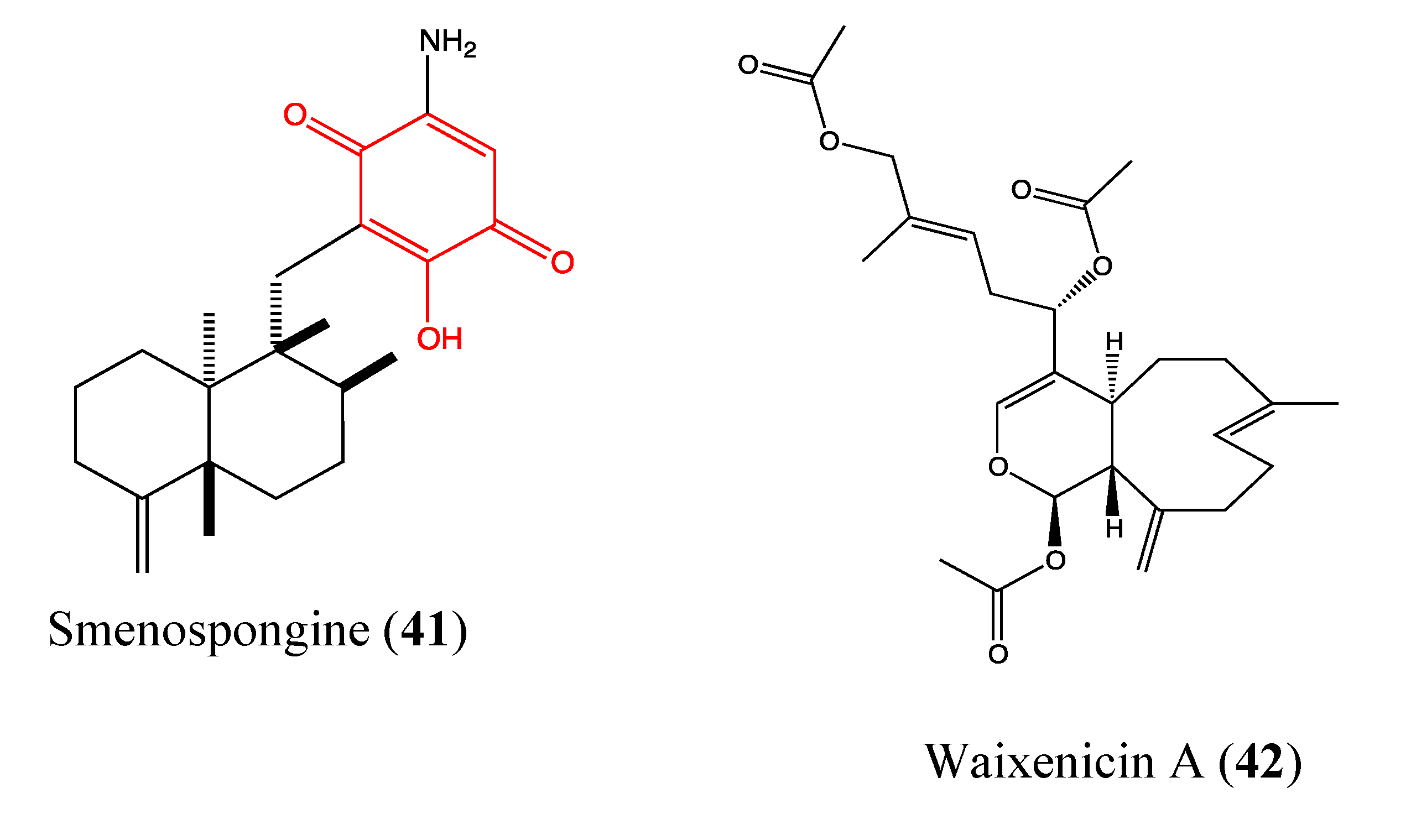
2.9.10. Waixenicin A
3. Discussion
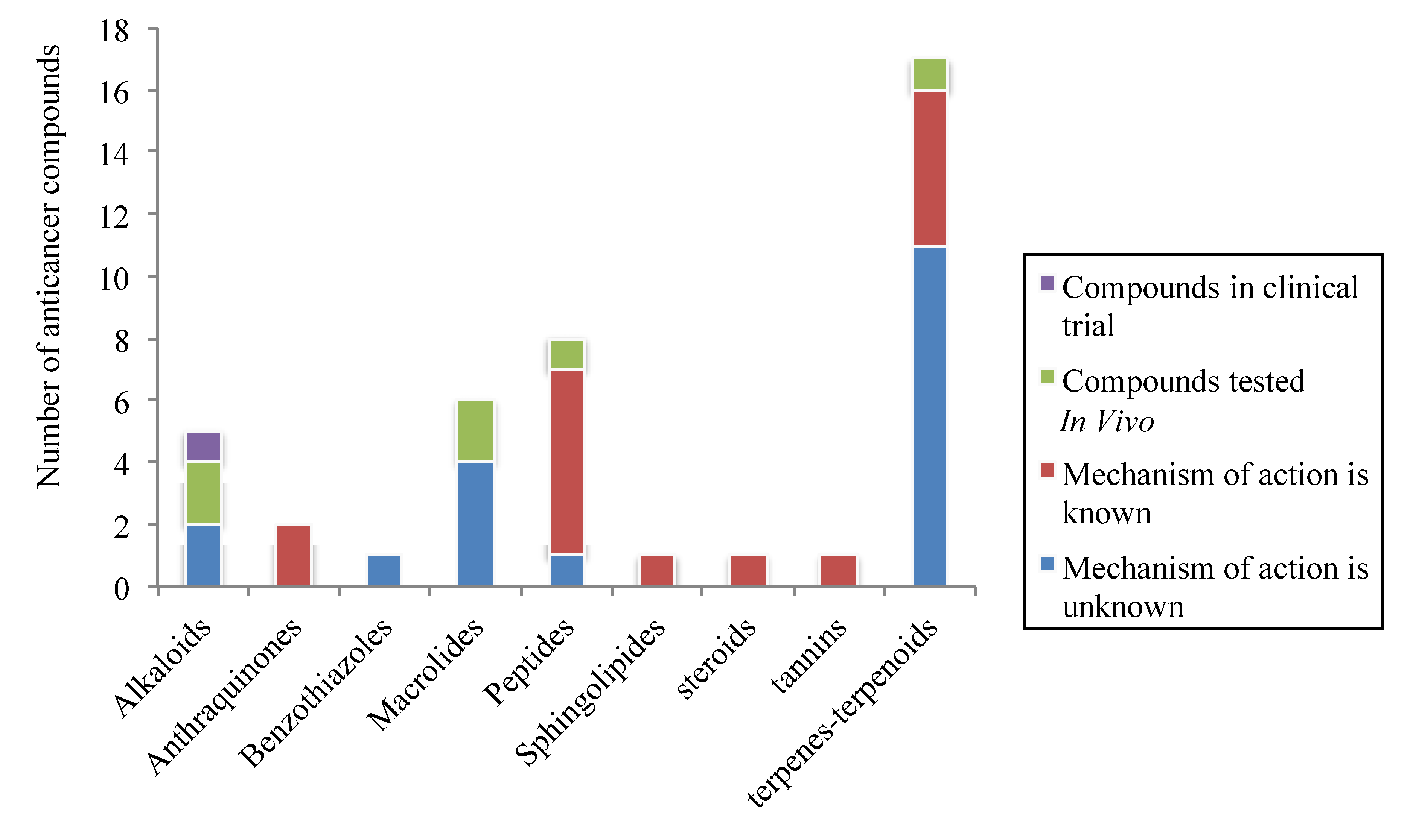
4. Conclusion
Acknowledgments
Abbreviations
Cell Line Abbreviation
| A549 | Lung carcinoma cell line |
| A2780 and OVCAR-3 | Ovarian cancer cells |
| ACHN | Human kidney adenocarcinoma cell |
| AGS | Human stomach adenocarcinoma cell line |
| CEM | leukemia cell |
| DLD1 | Human colorectal carcinoma |
| Daoy | medulloblastoma cells |
| H1395 and H2122 | Lung cancer cell |
| HCC44 and HCC366 | non-small lung cancer cells |
| HCT116 | Human colon carcinoma cell line |
| HEp-2 | Human epithelial cells |
| Hep3B | Human hepatoma cells |
| HepG2 | Human hepatocellular liver carcinoma cell line |
| Hel | Human erythroleukemia cells |
| HL-60 | Human promyelocytic leukemia cell line |
| HT29 | Human colon adenocarcinoma grade II cell line |
| HT1080 | Fibrosarcoma cell line |
| IOSE-144 | Non-tumorigenic human Ovarian surface epithelial cells |
| JB6 Cl 41 | Mouse epidermal cell line |
| Jurkat | Human T-cell leukemia cell line |
| K562 | Human chronic myeloid leukemia cell line |
| KB | human nasopharynx carcinoma cell line |
| MDR-KBv200 | Multidrug resistant form of KB |
| LoVo | Colon cancer cell line |
| MCF-7 | Breast adenocarcinoma cell line |
| MDA-MB231 | Mammary carcinoma cell line |
| MiaPaCa2 | Human pancreatic carcinoma cell line |
| PC-3 | Prostate cancer cell line |
| RAW264.7 | Mouse macrophage cell line |
| RPMI8226 | Human multiple myeloma cell line |
| S180 | Murine sarcoma cell line |
| U2OS | Human osteosarcoma cell line |
| U937-GTB | Histiocytic lymphoma cell line |
| WiDr | Colorectal adenocarcinoma cell line |
Others
| 1403P-3 | Adriamycin analogue |
| AglRhz | Aglycon of Rhizochalin |
| AMPK | 5' adenosine monophosphate-activated protein kinase |
| AP-1 | Activator protein 1 |
| Bcl-2 | B-cell lymphoma 2 |
| Bax | Bcl-2-associated X protein |
| BF65 and BF78 | Hemiasterlin derivatives |
| BRCA1 | Breast cancer 1 |
| CDK4 | Cyclin-dependent kinase 4 |
| FBA-TPQ | Makaluvamine analog |
| GADD45α | Growth arrest and DNA damage gene |
| IGF | Insulin-like growth factor |
| iNOS | Inducible nitric oxide synthase |
| MMP-9 | Matrix metalloproteinase |
| MDR | Multidrug resistance |
| MESP | Methyl spongoate |
| NF-κB | Nuclear factor-kappa B |
| PARP | Poly (ADP-ribose) polymerase |
| P-gp | P-glycoprotein |
| PI3 kinase/Akt | Phosphoinositol-3-kinase |
| RAD51 and RAD54 | DNA repair protein |
| ROS | Reactive oxygen species |
| S4-1e | Apratoxin analog |
| STAT3 | Signal transducer and activator of transcription 3 |
| TNFα | Tumor necrosis factor α |
| TRPM7 | Transient receptor potential melastatin 7 channels |
| VEGF | Vascular endothelial growth factor |
| XIAP | X-linked inhibitor of apoptosis protein |
Conflicts of Interest
References
- IARC, Cancer Incidence and Mortality Worlwide; International Agency for Research on Cancer: Lyon, France, 2011.
- WHO, Global Status Report on Noncommunicable Diseases 2010; WT 500; WHO: Geneva, Switzerland, 2011.
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Sarfaraj, H.M.; Sheeba, F.; Saba, A.; Mohd, S.K. Marine natural products: A lead for anti-cancer. Indian J. Mar. Sci. 2012, 41, 27–39. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef]
- Benkendorff, K. Molluscan biological and chemical diversity: Secondary metabolites and medicinal resources produced by marine molluscs. Biol. Rev. 2010, 85, 757–775. [Google Scholar]
- Nastrucci, C.; Cesario, A.; Russo, P. Anticancer drug discovery from the marine environment. Recent Pat. Anticancer Drug Discov. 2012, 7, 218–232. [Google Scholar] [CrossRef]
- Schumacher, M.; Kelkel, M.; Dicato, M.; Diederich, M. A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2010. Molecules 2011, 16, 5629–5646. [Google Scholar] [CrossRef]
- Nakamura, H.; Wu, H.; Ohizumi, Y.; Hirata, Y. Agelasine-A, -B, -C and -D, novel bicyclic diterpenoids with a 9-methyladeninium unit possessing inhibitory effects on na,K-atpase from the okinawa sea sponge Agelas sp.1). Tetrahedron Lett. 1984, 25, 2989–2992. [Google Scholar] [CrossRef]
- Roggen, H.; Charnock, C.; Burman, R.; Felth, J.; Larsson, R.; Bohlin, L.; Gundersen, L.L. Antimicrobial and antineoplastic activities of agelasine analogs modified in the purine 2-position. Arch. Pharm. (Weinheim) 2011, 344, 50–55. [Google Scholar] [CrossRef]
- Yan, X.; Chen, H.; Lu, X.; Wang, F.; Xu, W.; Jin, H.; Zhu, P. Fascaplysin exert anti-tumor effects through apoptotic and anti-angiogenesis pathways in sarcoma mice model. Eur. J. Pharm. Sci. 2011, 43, 251–259. [Google Scholar]
- Segraves, N.L.; Lopez, S.; Johnson, T.A.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Structures and cytotoxicities of fascaplysin and related alkaloids from two marine phyla—Fascaplysinopsis sponges and Didemnum tunicates. Tetrahedron Lett. 2003, 44, 3471–3475. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Comparison of fascaplysin and related alkaloids: A study of structures, cytotoxicities, and sources. J. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef]
- Lin, J.; Yan, X.J.; Chen, H.M. Fascaplysin, a selective CDK4 inhibitor, exhibit anti-angiogenic activity in vitro and in vivo. Cancer Chemother. Pharmacol. 2007, 59, 439–445. [Google Scholar] [CrossRef]
- Zheng, Y.L.; Lu, X.L.; Lin, J.; Chen, H.M.; Yan, X.J.; Wang, F.; Xu, W.F. Direct effects of fascaplysin on human umbilical vein endothelial cells attributing the anti-angiogenesis activity. Biomed. Pharmacother. 2010, 64, 527–533. [Google Scholar] [CrossRef]
- Hormann, A.; Chaudhuri, B.; Fretz, H. DNA binding properties of the marine sponge pigment fascaplysin. Bioorg. Med. Chem. 2001, 9, 917–921. [Google Scholar] [CrossRef]
- Lu, X.L.; Zheng, Y.L.; Chen, H.M.; Yan, X.J.; Wang, F.; Xu, W.F. Anti-proliferation of human cervical cancer HeLa cell line by fascaplysin through apoptosis induction. Yao Xue Xue Bao 2009, 44, 980–986. [Google Scholar]
- Shafiq, M.I.; Steinbrecher, T.; Schmid, R. Fascaplysin as a specific inhibitor for CDK4: Insights from molecular modelling. PLoS One 2012, 7, e42612. [Google Scholar]
- Wang, W.; Rayburn, E.R.; Velu, S.E.; Nadkarni, D.H.; Murugesan, S.; Zhang, R. In vitro and in vivo anticancer activity of novel synthetic makaluvamine analogues. Clin. Cancer Res. 2009, 15, 3511–3518. [Google Scholar] [CrossRef]
- Shinkre, B.A.; Raisch, K.P.; Fan, L.; Velu, S.E. Synthesis and antiproliferative activity of benzyl and phenethyl analogs of makaluvamines. Bioorg. Med. Chem. 2008, 16, 2541–2549. [Google Scholar] [CrossRef]
- Chen, T.; Xu, Y.; Guo, H.; Liu, Y.; Hu, P.; Yang, X.; Li, X.; Ge, S.; Velu, S.E.; Nadkarni, D.H.; et al. Experimental therapy of ovarian cancer with synthetic makaluvamine analog: In vitro and in vivo anticancer activity and molecular mechanisms of action. PLoS One 2011, 6, e20729. [Google Scholar]
- Ocio, E.M.; Maiso, P.; Chen, X.; Garayoa, M.; Alvarez-Fernandez, S.; San-Segundo, L.; Vilanova, D.; Lopez-Corral, L.; Montero, J.C.; Hernandez-Iglesias, T.; et al. Zalypsis: A novel marine-derived compound with potent antimyeloma activity that reveals high sensitivity of malignant plasma cells to DNA double-strand breaks. Blood 2009, 113, 3781–3791. [Google Scholar] [CrossRef]
- Leal, J.F.; Garcia-Hernandez, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, A.; Gago, F.; Guillen-Navarro, M.J.; Aviles, P.; Cuevas, C.; et al. Molecular pharmacology and antitumor activity of Zalypsis in several human cancer cell lines. Biochem. Pharmacol. 2009, 78, 162–170. [Google Scholar]
- Colado, E.; Paino, T.; Maiso, P.; Ocio, E.M.; Chen, X.; Alvarez-Fernandez, S.; Gutierrez, N.C.; Martin-Sanchez, J.; Flores-Montero, J.; San Segundo, L.; et al. Zalypsis has in vitro activity in acute myeloid blasts and leukemic progenitor cells through the induction of a DNA damage response. Haematologica 2011, 96, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Massard, C.; Margetts, J.; Amellal, N.; Drew, Y.; Bahleda, R.; Stevens, P.; Armand, J.P.; Calvert, H.; Soria, J.C.; Coronado, C.; et al. Phase I study of PM00104 (Zalypsis ®) administered as a 1-hour weekly infusion resting every fourth week in patients with advanced solid tumors. Invest. New Drugs 2012. [Google Scholar] [CrossRef]
- Yap, T.A.; Cortes-Funes, H.; Shaw, H.; Rodriguez, R.; Olmos, D.; Lal, R.; Fong, P.C.; Tan, D.S.; Harris, D.; Capdevila, J.; et al. First-in-man phase I trial of two schedules of the novel synthetic tetrahydroisoquinoline alkaloid PM00104 (Zalypsis) in patients with advanced solid tumours. Br. J. Cancer 2012, 106, 1379–1385. [Google Scholar] [CrossRef]
- Perez-Ruixo, C.; Valenzuela, B.; Fernandez Teruel, C.; Gonzalez-Sales, M.; Miguel-Lillo, B.; Soto-Matos, A.; Perez-Ruixo, J.J. Population pharmacokinetics of PM00104 (Zalypsis((R))) in cancer patients. Cancer Chemother. Pharmacol. 2012, 69, 15–24. [Google Scholar] [CrossRef]
- Jiang, G.C.; Lin, Y.C.; Zhou, S.N.; Vrumoed, I.I.P.; Jones, E.B.G. Study on the secondary metabolites of mangrove fungus N° 1403 from the South China Sea. Acta Sci. Natur. Univ. Sunyatseni 2000, 39, 68–72. [Google Scholar]
- Zhang, J.Y.; Tao, L.Y.; Liang, Y.J.; Chen, L.M.; Mi, Y.J.; Zheng, L.S.; Wang, F.; She, Z.G.; Lin, Y.C.; To, K.K.; et al. Anthracenedione derivatives as anticancer agents isolated from secondary metabolites of the mangrove endophytic fungi. Mar. Drugs 2010, 8, 1469–1481. [Google Scholar] [CrossRef]
- Yuan, J.; He, Z.; Wu, J.; Lin, Y.; Zhu, X. A novel adriamycin analogue derived from marine microbes induces apoptosis by blocking Akt activation in human breast cancer cells. Mol. Med. Report. 2011, 4, 261–265. [Google Scholar]
- Chin, P.C.; D'Mello, S.R. Survival of cultured cerebellar granule neurons can be maintained by Akt-dependent and Akt-independent signaling pathways. Mol. Brain Res. 2004, 127, 140–145. [Google Scholar] [CrossRef]
- Lee, S.; Choi, E.J.; Jin, C.; Kim, D.H. Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol. Oncol. 2005, 97, 26–34. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Kawada, K.; Takano, D.; Tanimura, S.; Ozaki, K.-I.; Kohno, M. Inhibition of the PI3 kinase/Akt pathway enhances doxorubicin-induced apoptotic cell death in tumor cells in a p53-dependent manner. Biochem. Biophys. Res. Commun. 2006, 340, 560–566. [Google Scholar] [CrossRef]
- West, K.A.; Sianna Castillo, S.; Dennis, P.A. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist. Updat. 2002, 5, 234–248. [Google Scholar] [CrossRef]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updat. 2008, 11, 32–50. [Google Scholar]
- Chen, W.S.; Hou, J.N.; Guo, Y.B.; Yang, H.L.; Xie, C.M.; Lin, Y.C.; She, Z.G. DOI: PubMed: Bostrycin inhibits proliferation of human lung carcinoma A549 cells via downregulation of the PI3K/Akt pathway. J. Exp. Clin. Cancer Res. 2011, 30. [Google Scholar] [CrossRef]
- Hu, Y.; MacMillan, J.B. Erythrazoles A-B, cytotoxic benzothiazoles from a marine-derived Erythrobacter sp. Org. Lett. 2011, 13, 6580–6583. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Liu, Y.; Luesch, H. Systematic chemical mutagenesis identifies a potent novel apratoxin A/E hybrid with improved in vivo antitumor activity. ACS Med. Chem. Lett. 2011, 2, 861–865. [Google Scholar] [CrossRef]
- Baselga, J. Targeting tyrosine kinases in cancer: The second wave. Science 2006, 312, 1175–1178. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis: Past, present and the near future. Carcinogenesis 2000, 21, 505–515. [Google Scholar] [CrossRef]
- Matthew, S.; Schupp, P.J.; Luesch, H. Apratoxin E, a cytotoxic peptolide from a guamanian collection of the marine cyanobacterium Lyngbya bouillonii. J. Nat. Prod. 2008, 71, 1113–1116. [Google Scholar] [CrossRef]
- Ma, D.; Zou, B.; Cai, G.; Hu, X.; Liu, J.O. Total synthesis of the cyclodepsipeptide apratoxin A and its analogues and assessment of their biological activities. Chemistry 2006, 12, 7615–7626. [Google Scholar] [CrossRef]
- Doi, T.; Numajiri, Y.; Takahashi, T.; Takagi, M.; Shin-ya, K. Solid-phase total synthesis of (−)-apratoxin A and its analogues and their biological evaluation. Chem. Asian J. 2011, 6, 180–188. [Google Scholar] [CrossRef]
- Kito, K.; Ookura, R.; Yoshida, S.; Namikoshi, M.; Ooi, T.; Kusumi, T. New cytotoxic 14-membered macrolides from marine-derived fungus Aspergillus ostianus. Org. Lett. 2008, 10, 225–228. [Google Scholar] [CrossRef]
- Diaz-Oltra, S.; Angulo-Pachon, C.A.; Murga, J.; Falomir, E.; Carda, M.; Marco, J.A. Synthesis and biological properties of the cytotoxic 14-membered macrolides aspergillide A and B. Chemistry 2011, 17, 675–688. [Google Scholar] [CrossRef]
- Hu, Y.; Espindola, A.P.; Stewart, N.A.; Wei, S.; Posner, B.A.; MacMillan, J.B. Chromomycin SA analogs from a marine-derived Streptomyces sp. Bioorg. Med. Chem. 2011, 19, 5183–5189. [Google Scholar] [CrossRef]
- Menendez, N.; Nur-e-Alam, M.; Brana, A.F.; Rohr, J.; Salas, J.A.; Mendez, C. Biosynthesis of the antitumor chromomycin A3 in Streptomyces griseus: analysis of the gene cluster and rational design of novel chromomycin analogs. Chem. Biol. 2004, 11, 21–32. [Google Scholar]
- Wei, R.B.; Xi, T.; Li, J.; Wang, P.; Li, F.C.; Lin, Y.C.; Qin, S. Lobophorin C and D, new kijanimicin derivatives from a marine sponge-associated actinomycetal strain AZS17. Mar. Drugs 2011, 9, 359–368. [Google Scholar] [CrossRef]
- Paterson, I.; Coster, M.J. Chapter 8 Total synthesis of spongistatin 1 (altohyrtin a): A tale of ten aldols. In Strategies and Tactics in Organic Synthesis; Michael, H., Ed.; Academic Press: Oxford, UK, 2004; Volume 4, pp. 211–245. [Google Scholar]
- Smith Iii, A.B.; Sfouggatakis, C.; Risatti, C.A.; Sperry, J.B.; Zhu, W.; Doughty, V.A.; Tomioka, T.; Gotchev, D.B.; Bennett, C.S.; Sakamoto, S.; et al. Spongipyran synthetic studies. Evolution of a scalable total synthesis of (+)-spongistatin 1. Tetrahedron 2009, 65, 6489–6509. [Google Scholar] [CrossRef]
- Bai, R.; Cichacz, Z.A.; Herald, C.L.; Pettit, G.R.; Hamel, E. Spongistatin 1, a highly cytotoxic, sponge-derived, marine natural product that inhibits mitosis, microtubule assembly, and the binding of vinblastine to tubulin. Mol. Pharmacol. 1993, 44, 757–766. [Google Scholar]
- Paterson, I.; Chen, D.Y.; Coster, M.J.; Acena, J.L.; Bach, J.; Wallace, D.J. The stereocontrolled total synthesis of altohyrtin A/spongistatin 1: Fragment couplings, completion of the synthesis, analogue generation and biological evaluation. Org. Biomol. Chem. 2005, 3, 2431–2440. [Google Scholar] [CrossRef]
- Rothmeier, A.S.; Schneiders, U.M.; Wiedmann, R.M.; Ischenko, I.; Bruns, C.J.; Rudy, A.; Zahler, S.; Vollmar, A.M. The marine compound spongistatin 1 targets pancreatic tumor progression and metastasis. Int. J. Cancer 2010, 127, 1096–1105. [Google Scholar] [CrossRef]
- Schneiders, U.M.; Schyschka, L.; Rudy, A.; Vollmar, A.M. BH3-only proteins Mcl-1 and Bim as well as endonuclease G are targeted in spongistatin 1-induced apoptosis in breast cancer cells. Mol. Cancer Ther. 2009, 8, 2914–2925. [Google Scholar] [CrossRef]
- Schyschka, L.; Rudy, A.; Jeremias, I.; Barth, N.; Pettit, G.R.; Vollmar, A.M. Spongistatin 1: A new chemosensitizing marine compound that degrades XIAP. Leukemia 2008, 22, 1737–1745. [Google Scholar] [CrossRef]
- Xu, Q.; Huang, K.C.; Tendyke, K.; Marsh, J.; Liu, J.; Qiu, D.; Littlefield, B.A.; Nomoto, K.; Atasoylu, O.; Risatti, C.A.; et al. In vitro and in vivo anticancer activity of (+)-spongistatin 1. Anticancer Res. 2011, 31, 2773–2779. [Google Scholar]
- Choi, E.J.; Park, J.S.; Kim, Y.J.; Jung, J.H.; Lee, J.K.; Kwon, H.C.; Yang, H.O. Apoptosis-inducing effect of diketopiperazine disulfides produced by Aspergillus sp. KMD 901 isolated from marine sediment on HCT116 colon cancer cell lines. J. Appl. Microbiol. 2011, 110, 304–313. [Google Scholar] [CrossRef]
- Gardiner, D.M.; Waring, P.; Howlett, B.J. The epipolythiodioxopiperazine (ETP) class of fungal toxins: Distribution, mode of action, functions and biosynthesis. Microbiology 2005, 151, 1021–1032. [Google Scholar] [CrossRef]
- Hadaschik, B.A.; Adomat, H.; Fazli, L.; Fradet, Y.; Andersen, R.J.; Gleave, M.E.; So, A.I. Intravesical chemotherapy of high-grade bladder cancer with HTI-286, a synthetic analogue of the marine sponge product hemiasterlin. Clin. Cancer Res. 2008, 14, 1510–1518. [Google Scholar] [CrossRef]
- Loganzo, F.; Discafani, C.M.; Annable, T.; Beyer, C.; Musto, S.; Hari, M.; Tan, X.; Hardy, C.; Hernandez, R.; Baxter, M.; et al. HTI-286, a synthetic analogue of the tripeptide hemiasterlin, is a potent antimicrotubule agent that circumvents P-glycoprotein-mediated resistance in vitro and in vivo. Cancer Res. 2003, 63, 1838–1845. [Google Scholar]
- Kuznetsov, G.; TenDyke, K.; Towle, M.J.; Cheng, H.; Liu, J.; Marsh, J.P.; Schiller, S.E.; Spyvee, M.R.; Yang, H.; Seletsky, B.M.; et al. Tubulin-based antimitotic mechanism of E7974, a novel analogue of the marine sponge natural product hemiasterlin. Mol. Cancer Ther. 2009, 8, 2852–2860. [Google Scholar] [CrossRef]
- Hsu, L.C.; Durrant, D.E.; Huang, C.C.; Chi, N.W.; Baruchello, R.; Rondanin, R.; Rullo, C.; Marchetti, P.; Grisolia, G.; Simoni, D.; et al. Development of hemiasterlin derivatives as potential anticancer agents that inhibit tubulin polymerization and synergize with a stilbene tubulin inhibitor. Invest. New Drugs 2012, 30, 1379–1388. [Google Scholar] [CrossRef]
- Watts, K.R.; Morinaka, B.I.; Amagata, T.; Robinson, S.J.; Tenney, K.; Bray, W.M.; Gassner, N.C.; Lokey, R.S.; Media, J.; Valeriote, F.A.; et al. Biostructural features of additional jasplakinolide (jaspamide) analogues. J. Nat. Prod. 2011, 74, 341–351. [Google Scholar] [CrossRef]
- Tannert, R.; Milroy, L.G.; Ellinger, B.; Hu, T.S.; Arndt, H.D.; Waldmann, H. Synthesis and structure-activity correlation of natural-product inspired cyclodepsipeptides stabilizing F-actin. J. Am. Chem. Soc. 2010, 132, 3063–3077. [Google Scholar]
- Tan, L.T.; Chang, Y.Y.; Ashootosh, T. Besarhanamides A and B from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2008, 69, 2067–2069. [Google Scholar] [CrossRef]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Chan, K.P.; Chen, D.Y.; Tan, L.T. Lagunamide C, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2011, 72, 2369–2375. [Google Scholar] [CrossRef]
- Duffy, R.; Wade, C.; Chang, R. Discovery of anticancer drugs from antimalarial natural products: a MEDLINE literature review. Drug Discov. Today 2012, 17, 942–953. [Google Scholar] [CrossRef]
- Hirata, A.; Guan, P.; Fujita, T.; Hirotsu, Y.; Inoue, A.; Yavari, A.R.; Sakurai, T.; Chen, M. Direct observation of local atomic order in a metallic glass. Nat. Mater. 2011, 10, 28–33. [Google Scholar] [CrossRef]
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [Google Scholar]
- Wang, B.; Huang, P.H.; Chen, C.S.; Forsyth, C.J. Total syntheses of the histone deacetylase inhibitors largazole and 2-epi-largazole: application of N-heterocyclic carbene mediated acylations in complex molecule synthesis. J. Org. Chem. 2011, 76, 1140–1150. [Google Scholar] [CrossRef]
- Benelkebir, H.; Marie, S.; Hayden, A.L.; Lyle, J.; Loadman, P.M.; Crabb, S.J.; Packham, G.; Ganesan, A. Total synthesis of largazole and analogues: HDAC inhibition, antiproliferative activity and metabolic stability. Bioorg. Med. Chem. 2011, 19, 3650–3658. [Google Scholar] [CrossRef]
- Bhansali, P.; Hanigan, C.L.; Casero, R.A.; Tillekeratne, L.M. Largazole and analogues with modified metal-binding motifs targeting histone deacetylases: Synthesis and biological evaluation. J. Med. Chem. 2011, 54, 7453–7463. [Google Scholar] [CrossRef]
- Mwakwari, S.C.; Patil, V.; Guerrant, W.; Oyelere, A.K. Macrocyclic histone deacetylase inhibitors. Curr. Top. Med. Chem. 2010, 10, 1423–1440. [Google Scholar] [CrossRef]
- Raju, R.; Piggott, A.M.; Huang, X.C.; Capon, R.J. Nocardioazines: A novel bridged diketopiperazine scaffold from a marine-derived bacterium inhibits P-glycoprotein. Org. Lett. 2011, 13, 2770–2773. [Google Scholar] [CrossRef]
- Huang, T.C.; Lee, J.F.; Chen, J.Y. Pardaxin, an antimicrobial peptide, triggers caspase-dependent and ROS-mediated apoptosis in HT-1080 cells. Mar. Drugs 2011, 9, 1995–2009. [Google Scholar] [CrossRef]
- Hsu, J.C.; Lin, L.C.; Tzen, J.T.; Chen, J.Y. Pardaxin-induced apoptosis enhances antitumor activity in HeLa cells. Peptides 2011, 32, 1110–1116. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Santalova, E.A.; Pokanevich, E.V.; Molinski, T.F.; Stonik, V.A. Rhizochalin A, a novel two-headed sphingolipid from the sponge Rhizochalina incrustata. J. Nat. Prod. 2005, 68, 255–257. [Google Scholar] [CrossRef]
- Khanal, P.; Kang, B.S.; Yun, H.J.; Cho, H.G.; Makarieva, T.N.; Choi, H.S. Aglycon of rhizochalin from the Rhizochalina incrustata induces apoptosis via activation of AMP-activated protein kinase in HT-29 colon cancer cells. Biol. Pharm. Bull. 2011, 34, 1553–1558. [Google Scholar] [CrossRef]
- Fedorov, S.N.; Makarieva, T.N.; Guzii, A.G.; Shubina, L.K.; Kwak, J.Y.; Stonik, V.A. Marine two-headed sphingolipid-like compound rhizochalin inhibits EGF-induced transformation of JB6 P+ Cl41 cells. Lipids 2009, 44, 777–785. [Google Scholar] [CrossRef]
- Jin, J.O.; Shastina, V.; Park, J.I.; Han, J.Y.; Makarieva, T.; Fedorov, S.; Rasskazov, V.; Stonik, V.; Kwak, J.Y. Differential induction of apoptosis of leukemic cells by rhizochalin, two headed sphingolipids from sponge and its derivatives. Biol. Pharm. Bull. 2009, 32, 955–962. [Google Scholar] [CrossRef]
- Lerdrup, M.; Holmberg, C.; Dietrich, N.; Shaulian, E.; Herdegen, T.; Jäättelä, M.; Kallunki, T. Depletion of the AP-1 repressor JDP2 induces cell death similar to apoptosis. Biochim. Biophys. Acta 2005, 1745, 29–37. [Google Scholar]
- Shaulian, E. AP-1—The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell. Signal. 2010, 22, 894–899. [Google Scholar] [CrossRef]
- Jiang, Y.; Miao, Z.H.; Xu, L.; Yu, B.; Gong, J.X.; Tong, L.J.; Chen, Y.; Zhou, Z.L.; Liu, H.C.; Wang, Y.; et al. Drug transporter-independent liver cancer cell killing by a marine steroid methyl spongoate via apoptosis induction. J. Biol. Chem. 2011, 286, 26461–26469. [Google Scholar] [CrossRef]
- Papatheodoridis, G.V.; Lampertico, P.; Manolakopoulos, S.; Lok, A. Incidence of hepatocellular carcinoma in chronic hepatitis B patients receiving nucleos(t)ide therapy: A systematic review. J. Hepatol. 2010, 53, 348–356. [Google Scholar] [CrossRef]
- Thomas, M. Molecular targeted therapy for hepatocellular carcinoma. J. Gastroenterol. 2009, 44, 136–141. [Google Scholar] [CrossRef]
- Al Zaid Siddiquee, K.; Turkson, J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008, 18, 254–267. [Google Scholar] [CrossRef]
- Masuda, M.; Wakasaki, T.; Suzui, M.; Toh, S.; Joe, A.K.; Weinstein, I.B. Stat3 orchestrates tumor development and progression: The Achilles’ heel of head and neck cancers? Curr. Cancer Drug Targets 2010, 10, 117–126. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Y.; Qian, Z.J.; Lee, S.H.; Li, Y.X.; Kim, S.K. Dieckol from ecklonia cava regulates invasion of human fibrosarcoma cells and modulates MMP-2 and MMP-9 expression via NF-kappaB pathway. Evid. Based Complement. Alternat. Med. 2011, 2011, 140462. [Google Scholar]
- Das, S.; Banerji, A.; Frei, E.; Chatterjee, A. Rapid expression and activation of MMP-2 and MMP-9 upon exposure of human breast cancer cells (MCF-7) to fibronectin in serum free medium. Life Sci. 2008, 82, 467–476. [Google Scholar] [CrossRef]
- Baker, A.H.; Edwards, D.R.; Murphy, G. Metalloproteinase inhibitors: Biological actions and therapeutic opportunities. J. Cell. Sci. 2002, 115, 3719–3727. [Google Scholar] [CrossRef]
- Tanimura, S.; Kadomoto, R.; Tanaka, T.; Zhang, Y.J.; Kouno, I.; Kohno, M. Suppression of tumor cell invasiveness by hydrolyzable tannins (plant polyphenols) via the inhibition of matrix metalloproteinase-2/-9 activity. Biochem. Biophys. Res. Commun. 2005, 330, 1306–1313. [Google Scholar] [CrossRef]
- Park, S.J.; Jeon, Y.J. Dieckol from Ecklonia cava suppresses the migration and invasion of HT1080 cells by inhibiting the focal adhesion kinase pathway downstream of Rac1-ROS signaling. Mol. Cells 2012, 33, 141–149. [Google Scholar] [CrossRef]
- Lee, J.; Jung, E.; Lee, J.; Huh, S.; Hwang, C.H.; Lee, H.Y.; Kim, E.J.; Cheon, J.M.; Hyun, C.G.; Kim, Y.S.; et al. Emodin inhibits TNF alpha-induced MMP-1 expression through suppression of activator protein-1 (AP-1). Life Sci. 2006, 79, 2480–2485. [Google Scholar] [CrossRef]
- Fassett, R.G.; Coombes, J.S. Astaxanthin: A potential therapeutic agent in cardiovascular disease. Mar. Drugs 2011, 9, 447–465. [Google Scholar] [CrossRef]
- Choi, H.D.; Kim, J.H.; Chang, M.J.; Kyu-Youn, Y.; Shin, W.G. Effects of astaxanthin on oxidative stress in overweight and obese adults. Phytother. Res. 2011, 25, 1813–1818. [Google Scholar] [CrossRef]
- Palozza, P.; Torelli, C.; Boninsegna, A.; Simone, R.; Catalano, A.; Mele, M.C.; Picci, N. Growth-inhibitory effects of the astaxanthin-rich alga Haematococcus pluvialis in human colon cancer cells. Cancer Lett. 2009, 283, 108–117. [Google Scholar] [CrossRef]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef]
- Nagendraprabhu, P.; Sudhandiran, G. Astaxanthin inhibits tumor invasion by decreasing extracellular matrix production and induces apoptosis in experimental rat colon carcinogenesis by modulating the expressions of ERK-2, NFkB and COX-2. Invest. New Drugs 2011, 29, 207–224. [Google Scholar] [CrossRef]
- Prabhu, P.N.; Ashokkumar, P.; Sudhandiran, G. Antioxidative and antiproliferative effects of astaxanthin during the initiation stages of 1,2-dimethyl hydrazine-induced experimental colon carcinogenesis. Fundam. Clin. Pharmacol. 2009, 23, 225–234. [Google Scholar] [CrossRef]
- Yasui, Y.; Hosokawa, M.; Mikami, N.; Miyashita, K.; Tanaka, T. Dietary astaxanthin inhibits colitis and colitis-associated colon carcinogenesis in mice via modulation of the inflammatory cytokines. Chem. Biol. Interact. 2011, 193, 79–87. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Lee, N.L.; Su, J.H. Tetrahydrofuran cembranoids from the cultured soft coral Lobophytum crassum. Mar. Drugs 2011, 9, 2526–2536. [Google Scholar] [CrossRef]
- Ebada, S.S.; Lin, W.; Proksch, P. Bioactive sesterterpenes and triterpenes from marine sponges: Occurrence and pharmacological significance. Mar. Drugs 2010, 8, 313–346. [Google Scholar] [CrossRef]
- Piao, S.J.; Zhang, H.J.; Lu, H.Y.; Yang, F.; Jiao, W.H.; Yi, Y.H.; Chen, W.S.; Lin, H.W. Hippolides A-H, acyclic manoalide derivatives from the marine sponge Hippospongia lachne. J. Nat. Prod. 2011, 74, 1248–1254. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Lo, K.-L.; Lin, Y.-C.; Khalil, A.T.; Kuo, Y.-H.; Shih, P.-S. Novel linear C22-sesterterpenoids from sponge Ircinia formosana. Tetrahedron Lett. 2006, 47, 4007–4010. [Google Scholar] [CrossRef]
- Su, J.H.; Tseng, S.W.; Lu, M.C.; Liu, L.L.; Chou, Y.; Sung, P.J. Cytotoxic C21 and C22 terpenoid-derived metabolites from the sponge Ircinia sp. J. Nat. Prod. 2011, 74, 2005–2009. [Google Scholar] [CrossRef]
- Su, J.H.; Chang, W.B.; Chen, H.M.; El-Shazly, M.; Du, Y.C.; Kung, T.H.; Chen, Y.C.; Sung, P.J.; Ho, Y.S.; Kuo, F.W.; et al. 10-acetylirciformonin B, a sponge furanoterpenoid, induces DNA damage and apoptosis in leukemia cells. Molecules 2012, 17, 11839–11848. [Google Scholar] [CrossRef]
- Wright, A.D.; Wang, H.; Gurrath, M.; Konig, G.M.; Kocak, G.; Neumann, G.; Loria, P.; Foley, M.; Tilley, L. Inhibition of heme detoxification processes underlies the antimalarial activity of terpene isonitrile compounds from marine sponges. J. Med. Chem. 2001, 44, 873–885. [Google Scholar] [CrossRef]
- Lamoral-Theys, D.; Fattorusso, E.; Mangoni, A.; Perinu, C.; Kiss, R.; Costantino, V. Evaluation of the antiproliferative activity of diterpene isonitriles from the sponge Pseudoaxinella flava in apoptosis-sensitive and apoptosis-resistant cancer cell lines. J. Nat. Prod. 2011, 74, 2299–2303. [Google Scholar] [CrossRef]
- Lin, W.Y.; Lu, Y.; Su, J.H.; Wen, Z.H.; Dai, C.F.; Kuo, Y.H.; Sheu, J.H. Bioactive cembranoids from the dongsha atoll soft coral Sarcophyton crassocaule. Mar. Drugs 2011, 9, 994–1006. [Google Scholar] [CrossRef]
- Hassan, H.M.; Sallam, A.A.; Mohammed, R.; Hifnawy, M.S.; Youssef, D.T.; El Sayed, K.A. Semisynthetic analogues of the marine cembranoid sarcophine as prostate and breast cancer migration inhibitors. Bioorg. Med. Chem. 2011, 19, 4928–4934. [Google Scholar] [CrossRef]
- Ganesan, P.; Noda, K.; Manabe, Y.; Ohkubo, T.; Tanaka, Y.; Maoka, T.; Sugawara, T.; Hirata, T. Siphonaxanthin, a marine carotenoid from green algae, effectively induces apoptosis in human leukemia (HL-60) cells. Biochim. Biophys. Acta 2011, 1810, 497–503. [Google Scholar]
- Ganesan, P.; Matsubara, K.; Ohkubo, T.; Tanaka, Y.; Noda, K.; Sugawara, T.; Hirata, T. Anti-angiogenic effect of siphonaxanthin from green alga, Codium fragile. Phytomedicine 2010, 17, 1140–1144. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Aoki, S.; Sowa, Y.; Sakai, T.; Kobayashi, M. Smenospongine, a sesquiterpene aminoquinone from a marine sponge, induces G1 arrest or apoptosis in different leukemia cells. Mar. Drugs 2008, 6, 480–488. [Google Scholar]
- Aoki, S.; Kong, D.; Matsui, K.; Kobayashi, M. Smenospongine, a spongean sesquiterpene aminoquinone, induces erythroid differentiation in K562 cells. Anticancer Drug 2004, 15, 363–369. [Google Scholar] [CrossRef]
- Kong, D.; Yamori, T.; Kobayashi, M.; Duan, H. Antiproliferative and antiangiogenic activities of smenospongine, a marine sponge sesquiterpene aminoquinone. Mar. Drugs 2011, 9, 154–161. [Google Scholar] [CrossRef]
- Coval, S.J.; Scheuer, P.J.; Matsumoto, G.K.; Clardy, J. Two new xenicin diterpenoids from the octocoral anthelia edmondsoni. Tetrahedron 1984, 40, 3823–3828. [Google Scholar] [CrossRef]
- Zierler, S.; Yao, G.; Zhang, Z.; Kuo, W.C.; Porzgen, P.; Penner, R.; Horgen, F.D.; Fleig, A. Waixenicin A inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J. Biol. Chem. 2011, 286, 39328–39335. [Google Scholar]
- Guilbert, A.; Gautier, M.; Dhennin-Duthille, I.; Haren, N.; Sevestre, H.; Ouadid-Ahidouch, H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am. J. Physiol. Cell. Physiol. 2009, 297, C493–C502. [Google Scholar] [CrossRef]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell. Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [CrossRef]
- Jiang, J.; Li, M.H.; Inoue, K.; Chu, X.P.; Seeds, J.; Xiong, Z.G. Transient receptor potential melastatin 7-like current in human head and neck carcinoma cells: Role in cell proliferation. Cancer Res. 2007, 67, 10929–10938. [Google Scholar]
- Kim, B.J.; Park, E.J.; Lee, J.H.; Jeon, J.H.; Kim, S.J.; So, I. Suppression of transient receptor potential melastatin 7 channel induces cell death in gastric cancer. Cancer Sci. 2008, 99, 2502–2509. [Google Scholar] [CrossRef]
- Paravicini, T.M.; Chubanov, V.; Gudermann, T. TRPM7: A unique channel involved in magnesium homeostasis. Int. J. Biochem. Cell. Biol. 2012, 44, 1381–1384. [Google Scholar] [CrossRef]
- Li, M.; Du, J.; Jiang, J.; Ratzan, W.; Su, L.T.; Runnels, L.W.; Yue, L. Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J. Biol. Chem. 2007, 282, 25817–25830. [Google Scholar]
- Li, M.; Jiang, J.; Yue, L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef]
- Ryazanova, L.V.; Rondon, L.J.; Zierler, S.; Hu, Z.; Galli, J.; Yamaguchi, T.P.; Mazur, A.; Fleig, A.; Ryazanov, A.G. TRPM7 is essential for Mg2+ homeostasis in mammals. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef]
- Schmitz, C.; Perraud, A.L.; Johnson, C.O.; Inabe, K.; Smith, M.K.; Penner, R.; Kurosaki, T.; Fleig, A.; Scharenberg, A.M. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 2003, 114, 191–200. [Google Scholar] [CrossRef]
- Schmitz, C.; Perraud, A.L.; Fleig, A.; Scharenberg, A.M. Dual-function ion channel/protein kinases: novel components of vertebrate magnesium regulatory mechanisms. Pediatr. Res. 2004, 55, 734–737. [Google Scholar] [CrossRef]
- Cappadone, C.; Merolle, L.; Marraccini, C.; Farruggia, G.; Sargenti, A.; Locatelli, A.; Morigi, R.; Iotti, S. Intracellular magnesium content decreases during mitochondria-mediated apoptosis induced by a new indole-derivative in human colon cancer cells. Magnes. Res. 2012, 25, 104–111. [Google Scholar]
- Wolf, F.I.; Trapani, V. Magnesium and its transporters in cancer: A novel paradigm in tumour development. Clin. Sci. 2012, 123, 417–427. [Google Scholar] [CrossRef]
- Chen, J.P.; Luan, Y.; You, C.X.; Chen, X.H.; Luo, R.C.; Li, R. TRPM7 regulates the migration of human nasopharyngeal carcinoma cell by mediating Ca2+ influx. Cell Calcium 2010, 47, 425–432. [Google Scholar] [CrossRef]
- Carvalho, M.; Ferreira, P.J.; Mendes, V.S.; Silva, R.; Pereira, J.A.; Jerónimo, C.; Silva, B.M. Human cancer cell antiproliferative and antioxidant activities of Juglans regia L. Food Chem. Toxicol. 2010, 48, 441–447. [Google Scholar] [CrossRef]
- Dia, V.P.; Gonzalez de Mejia, E. Lunasin potentiates the effect of oxaliplatin preventing outgrowth of colon cancer metastasis, binds to α5β1 integrin and suppresses FAK/ERK/NF-κB signaling. Cancer Lett. 2011, 313, 167–180. [Google Scholar] [CrossRef]
- Rea, G.; Antonacci, A.; Lambreva, M.; Pastorelli, S.; Tibuzzi, A.; Ferrari, S.; Fischer, D.; Johanningmeier, U.; Oleszek, W.; Doroszewska, T.; et al. Integrated plant biotechnologies applied to safer and healthier food production: The Nutra-Snack manufacturing chain. Trends Food Sci. Technol. 2011, 22, 353–366. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sawadogo, W.R.; Schumacher, M.; Teiten, M.-H.; Cerella, C.; Dicato, M.; Diederich, M. A Survey of Marine Natural Compounds and Their Derivatives with Anti-Cancer Activity Reported in 2011. Molecules 2013, 18, 3641-3673. https://doi.org/10.3390/molecules18043641
Sawadogo WR, Schumacher M, Teiten M-H, Cerella C, Dicato M, Diederich M. A Survey of Marine Natural Compounds and Their Derivatives with Anti-Cancer Activity Reported in 2011. Molecules. 2013; 18(4):3641-3673. https://doi.org/10.3390/molecules18043641
Chicago/Turabian StyleSawadogo, Wamtinga Richard, Marc Schumacher, Marie-Hélène Teiten, Claudia Cerella, Mario Dicato, and Marc Diederich. 2013. "A Survey of Marine Natural Compounds and Their Derivatives with Anti-Cancer Activity Reported in 2011" Molecules 18, no. 4: 3641-3673. https://doi.org/10.3390/molecules18043641