3.2. Synthesis
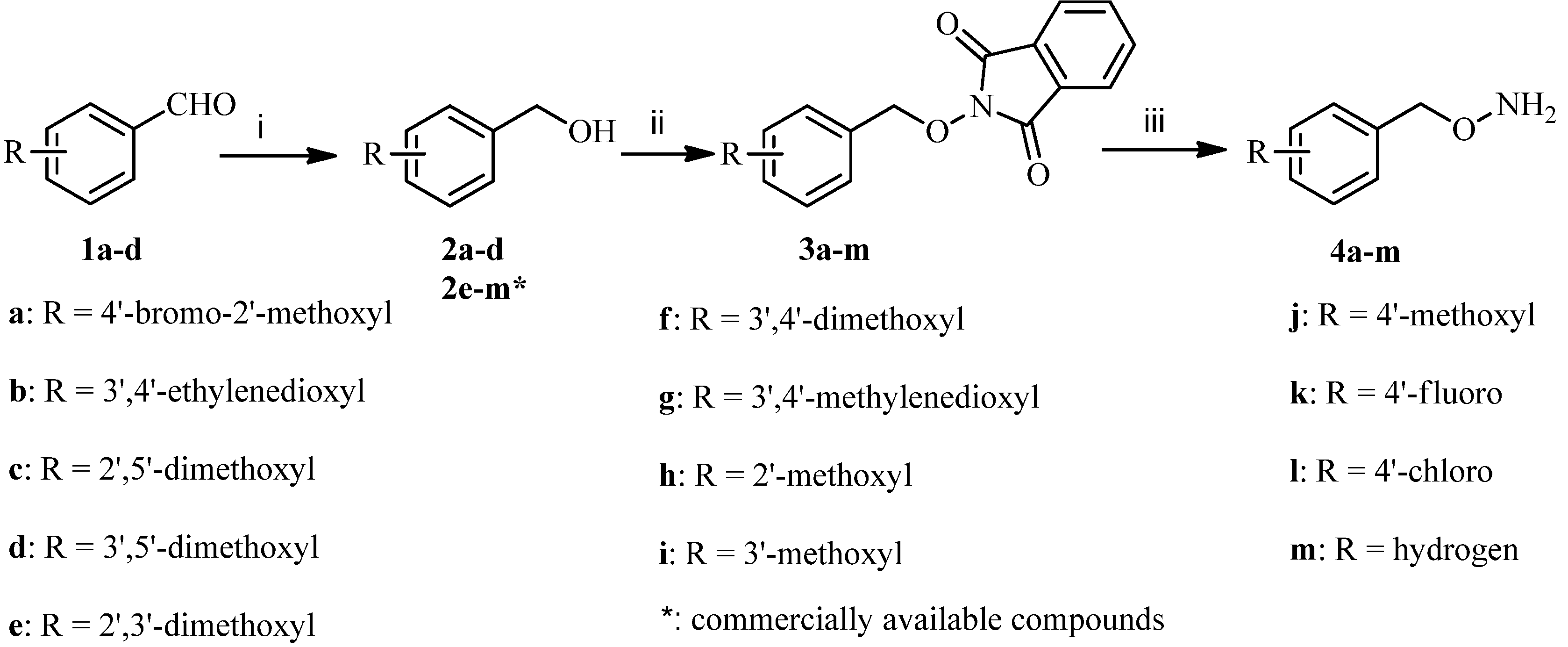
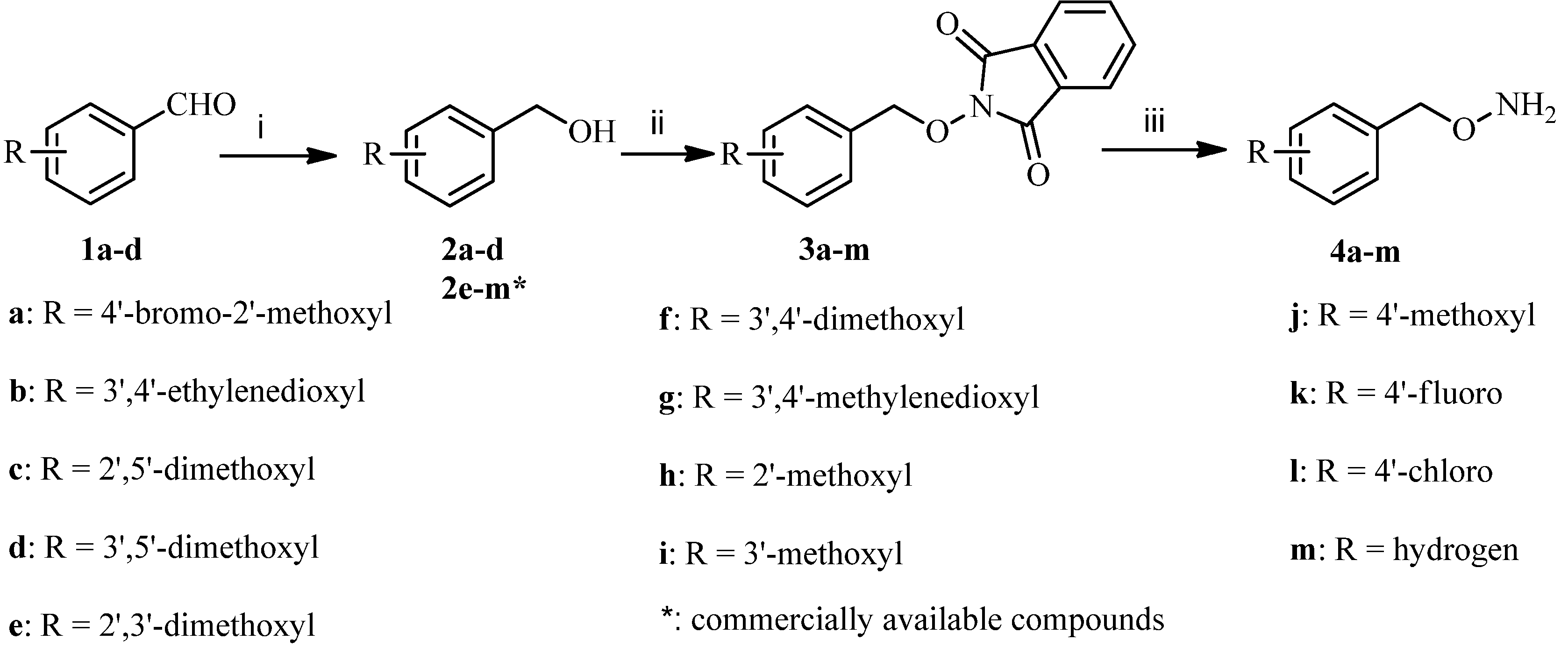
3.2.1. General Procedure for the Synthesis of Substituted O-Benzylhydroxylamines 4a–m
To a solution of benzaldehydes 1a-d (100 mmol) dissolved in methanol (500 mL) was added sodium borohydride (200 mmol) at room temperature, and the mixture was stirred at the same temperature for 2 h and concentrated under reduced pressure. The residue was diluted with dichloromethane (500 mL) and washed with water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give the corresponding phenylmethanols 2a–d as colorless oils.
To a solution of the above obtained phanylmethanols 2a–d or commercially available 2e–m (60 mmol), 2-hydroxyisoindoline-1,3-dione (60 mmol) and triphenylphosphine (75 mmol) in tetrahydrofuran (300 mL) was added dropwise a solution of diethyl azodicarboxylate (75 mmol) in tetrahydrofuran (15 mL) at 0 °C over 0.5 h. The mixture was stirred at the same temperature for 1 h and concentrated under reduced pressure. The residue was dissolved in methanol (300 mL) and stirred at room temperature for 1 h and filtered to give 3a–m as white solids. Next, to a stirred solution of 3a–m (60 mmol) in dichloromethane (200 mL) was added dropwise hydrazine hydrate (120 mmol). The reaction mixture was stirred at room temperature for 3 h, and then filtered. The filtrate was washed successively with 2 mol/L sodium hydroxide solution (200 mL) and brine (200 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to give the corresponding benzyloxyamines 4a–m as colorless oils (56%–70%, from 2a–d; 66%–78%, from 2e–m).
O-(4-Bromo-2-methoxybenzyl) hydroxylamine (4a). The title compound was obtained from 3a as a colorless oil (87%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.53 (1H, d, J = 8.4 Hz, Ar-H), 7.21 (1H, d, J = 2.4 Hz, Ar-H), 6.98 (1H, q, J1 = 8.4 Hz, J2 = 2.4 Hz, Ar-H), 5.21 (2H, s, OCH2Ar), 3.79 (3H, s, O-CH3). MS-ESI (m/z): 233 (M+H)+.
O-((2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)methyl) hydroxylamine (4b). The title compound was obtained from 3b as a colorless oil (85%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 6.81–6.79 (2H, m, Ar-H), 6.77–6.75 (1H, m, Ar-H), 5.95 (2H, s, OCH2Ar), 4.21 (4H, s, 2 × OCH2). MS-ESI (m/z): 182 (M+H)+.
O-(2,5-Dimethoxybenzyl) hydroxylamine (4c). The title compound was obtained from 3c as a colorless oil (90%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 6.89 (2H, m, Ar-H), 6.81 (1H, m, Ar-H), 4.57 (2H, s, OCH2Ar), 3.71 (6H, s, O-CH3). MS-ESI (m/z): 184 (M+H)+.
O-(3,5-Dimethoxybenzyl) hydroxylamine (4d). The title compound was obtained from 3d as a colorless oil (88%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 6.46 (2H, s), 6.39 (1H, s), 4.50 (2H, s), 3.73 (6H, s). MS-ESI (m/z): 184 (M+H)+.
O-(2,3-Dimethoxybenzyl) hydroxylamine (4e). The title compound was obtained from 3e as a colorless oil (91%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.03 (1H, t, J = 7.8 Hz, Ar-H), 6.68 (1H, d, J = 7.8 Hz, Ar-H), 6.90 (1H, d, J = 7.8 Hz, Ar-H), 4.56 (2H, s, OCH2Ar), 3.78 (3H, s, O-CH3), 3.69 (3H, s, O-CH3). MS-ESI (m/z): 184 (M+H)+.
O-(3,4-Dimethoxybenzyl) hydroxylamine (4f). The title compound was obtained from 3f as a colorless oil (89%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.20 (1H, m, Ar-H), 6.93 (2H, m, Ar-H), 5.05 (2H, s, OCH2Ar), 3.78 (6H, s, O-CH3). MS-ESI (m/z): 184 (M+H)+.
O-(Benzo[d][1,3]dioxol-5-ylmethyl) hydroxylamine (4g). The title compound was obtained from 3g as a colorless oil (84%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 6.98 (1H, d, J = 1.2 Hz, Ar-H), 6.94 (1H, d, J = 8 Hz, Ar-H), 6.91 (1H, q, J1 = 8 Hz, J2 = 1.2 Hz, Ar-H), 6.05 (2H, s, OCH2Ar), 4.91 (2H, s, OCH2O). MS-ESI (m/z): 168 (M+H)+.
O-(2-Methoxybenzyl) hydroxylamine (4h). The title compound was obtained from 3h as a colorless oil (85%), 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 7.26 (2H, m, Ar-H), 6.95 (1H, d, J = 7.8 Hz, Ar-H), 6.91 (1H, J = 7.8 Hz, Ar-H), 4.56 (2H, s, Ar-H), 3.76 (3H, s, O-CH3). MS-ESI (m/z): 154 (M+H)+.
O-(3-Methoxybenzyl) hydroxylamine (4i). The title compound was obtained from 3i as a colorless oil (86%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.33 (1H, m, Ar-H), 6.97 (3H, m, Ar-H) 5.00 (2H, s, OCH2Ar), 3.77 (3H, s, O-CH3). MS-ESI (m/z): 154 (M+H)+.
O-(4-Methoxybenzyl) hydroxylamine (4j). The title compound was obtained from 3j as a colorless oil (88%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.34 (2H, d, J = 8.4 Hz, Ar-H), 6.97 (2H, d, J = 8.4 Hz, Ar-H), 4.90 (2H, s, OCH2Ar), 3.77 (3H, s, O-CH3). MS-ESI (m/z): 154 (M+H)+.
O-(4-Fluorobenzyl) hydroxylamine (4k). The title compound was obtained from 3k as a colorless oil (84%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.35 (2H, m, Ar-H), 7.16 (2H, m, Ar-H), 4.54 (2H, s, OCH2Ar). MS-ESI (m/z): 142 (M+H)+.
O-(4-Chlorobenzyl) hydroxylamine (4l). The title compound was obtained from 3l as a colorless oil (90%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.38 (2H, d, J = 8.4 Hz, Ar-H), 7.34 (2H, d, J = 8.4 Hz, Ar-H), 4.56 (2H, s, OCH2Ar). MS-ESI (m/z): 158 (M+H)+.
O-Benzyl hydroxylamine (4m). The title compound was obtained from 3m as a colorless oil (92%), 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.39 (4H, m, Ar-H), 7.33 (1H, m, Ar-H), 5.12 (2H, s, OCH2Ar). MS-ESI (m/z): 124 (M+H)+.
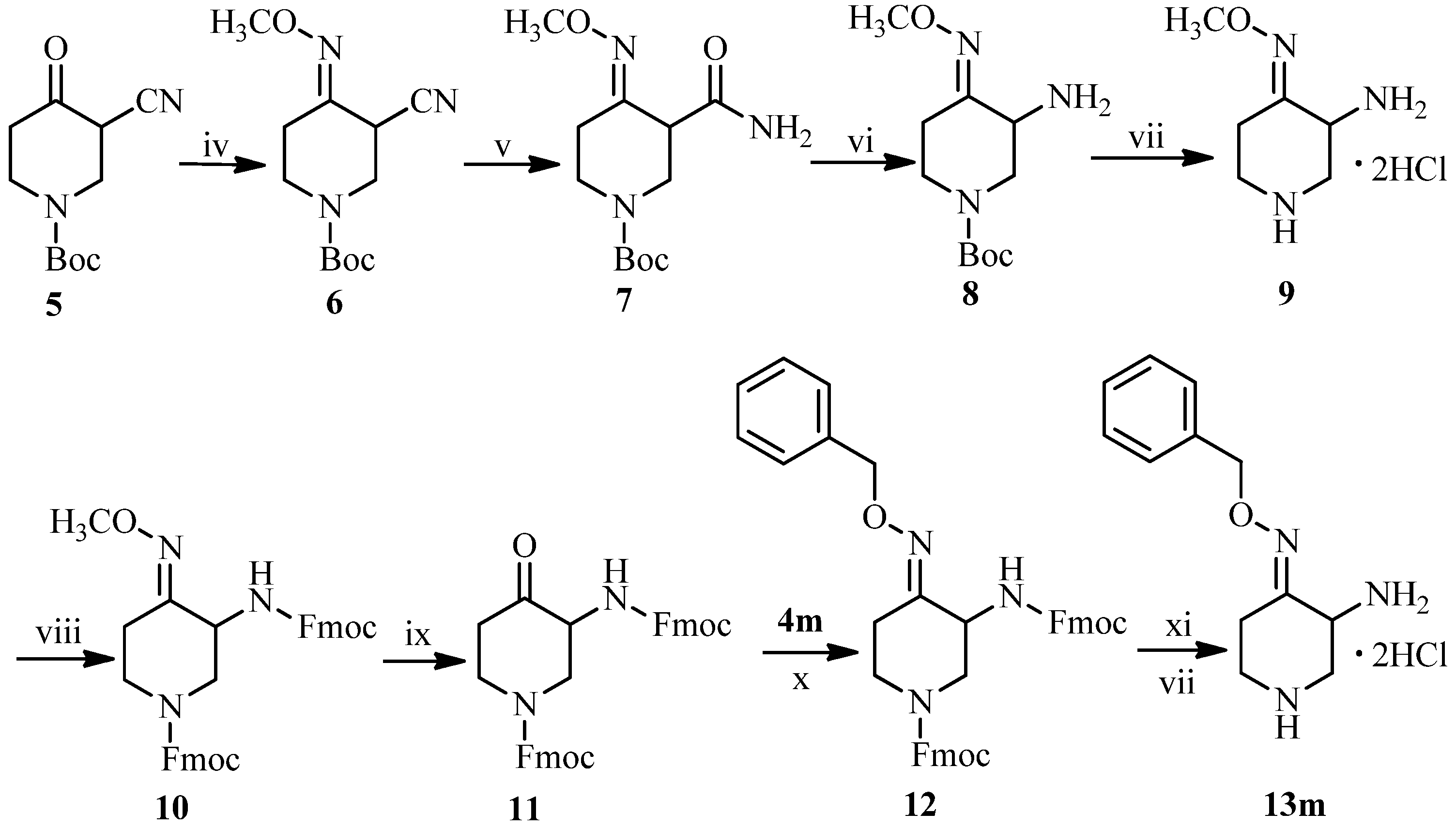
tert-Butyl 3-cyano-4-(methoxyimino)piperidine-1-carboxylate (6). To a solution of hydroxylamine hydrochloride (5.01 g, 60 mmol) in methanol (150 mL) was added sodium hydroxide (2.40 g, 60 mmol), and the mixture stirred at room temperature for 30 minutes. The piperidinone 5 (11.18 g, 50 mmol) was added to the mixture, stirred at 50 °C for 3 h and then filtered. The filtrate was concentrated under reduced pressure. The residue was diluted with ethyl acetate (200 mL), washed with water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford the title compound 6 (14.12 g, 93%) as a yellow oil. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 4.48 (1H, s, piperidine-H), 4.25 (1H, m, piperidine-H), 4.04 (1H, m, piperidine-H), 3.84 (3H, s, NOCH3), 3.14 (1H, brs, piperidine-H), 2.93 (1H, brs, piperidine-H), 2.41 (2H, m, piperidine-H), 1.42 (9H, s, Boc-9H). MS-ESI (m/z): 254 (M+H)+.
tert-Butyl 3-carbamoyl-4-(methoxyimino)piperidine-1-carboxylate (7). To a solution of 6 (10.12 g, 40 mmol) and potassium carbonate (11.06 g, 80 mmol) in dimethyl sulfoxide (100 mL) was added hydrogen peroxide (36.27 mL, 320 mmol) at 15 °C for 1 h, and stirred at room temperature for 3 h. The mixture was diluted with water (200 mL), and extracted by ethyl acetate (200 mL). The combined extracts were washed with brine, dried over anhydrous sodium sulfate, and then concentrated under reduced pressure to give the title compound 7 (7.71 g, 71%) as a light yellow oil. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.37 (1H, s, CONH2), 7.00 (1H, s, CONH2), 4.19–4.10 (1H, brs, piperidine-H), 4.00–3.92 (1H, m, piperidine-H), 3.84 (1H, s, piperidine-H), 3.72 (3H, s, NOCH3), 3.11 (1H, m, piperidine-H), 2.61–2.53 (1H, m, piperidine-H), 2.24 (1H, brs, piperidine-H), 1.42 (1H, s, piperidine-H), 1.38 (9H, s, Boc-9H). MS-ESI (m/z): 294 (M+Na)+.
tert-Butyl 3-amino-4-(methoxyimino)piperidine-1-carboxylate (8). To a stirred solution of 7 (13.55 g, 50 mmol) in acetonitrile (350 mL) was added dropwise 10% sodium hypobromite solution (106.20 mL, 90 mmol) at 5 °C for 1 h. The reaction mixture was stirred at room temperature for 10 h, and adjusted to pH 6.5–7 with 20% acetic acid, and then concentrated under reduced pressure. The residue was dissolved in water (200 mL), adjusted to pH 4–3 with 10% hydrochloride and washed with ethyl acetate. The water layer was adjusted to pH 9 with 15% sodium hydroxide and extracted with ethyl acetate. The combined extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure to yield the title compound 8 (7.65 g, 63%) as a yellow oil. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 4.09–4.02 (1H, m, piperidine-H), 3.74 (3H, s, NOCH3), 3.57 (1H, m, piperidine-H), 3.31–3.25 (1H, m, piperidine-H), 3.11 (1H, s, piperidine-H), 2.99–2.92 (1H, brs, piperidine-H), 2.71–2.66 (1H, m, piperidine-H), 1.98 (1H, brs, piperidine-H), 1.38 (9H, s, Boc-9H). MS-ESI (m/z): 244 (M+H)+.
3-Amino-4-methoxyiminopiperidine dihydrocholoride (9). To a solution of 8 (7.29 g, 30 mmol) in dichloromethane was pumped dried hydrochloride gas at room temperature for 30 minutes. The mixture was stirred for another 1 h at room temperature, and concentrated under reduced pressure. The residue was treated with ethyl acetate. The precipitate was collected by suction, and dried under vacuum to afford the title compound 9 (2.15 g, 52%) as a white solid, m.p.: 224–226 °C. 1H-NMR (300 MHz, D2O) δ (ppm): 4.45–4.50 (1H, m, piperidine-H), 4.01–3.97 (4H, m, piperidine-H, NOCH3), 3.70–3.65 (1H, m, piperidine-H), 3.59–3.53 (1H, m, piperidine-H), 3.34–3.28 (1H, m, piperidine-H), 3.23–3.15 (1H, m, piperidine-H), 2.46–2.37 (1H, m, piperidine-H). M.p.: 178–181 °C. MS-FAB (m/z): 144 (M+H)+.
(9H-Fluoren-9-yl)methyl 3-[(9H-fluoren-9-yl)methoxycarbonylamino]-4-(methoxyimino)piperidine-1-carboxylate (10). To a stirring solution of 9 (3.24g, 15 mmol) and triethylamine (6.24 mL,45 mmol) in dichloromethane (300 mL) was added in portions 9-fluorenylmethyl chloroformate (7.76 g, 30 mmol) at 0 °C, and the mixture stirred at room temperature for 5 h, then washed with water, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude product was purified by column chromatography (silica gel), eluting with petroleum ether and ethyl acetate (v:v = 3:1) to produce the title compound 10 (5.02 g, 57%) as an off-white solid, m.p.: 152–154 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.76 (4H, m, Fmoc-ArH), 7.61 (4H, m, Fmoc-ArH), 7.39 (4H, m, Fmoc-ArH), 7.32 (4H, m, Fmoc-ArH), 4.44 (4H, d, J = 6.8 Hz, 2 × Fmoc-CH2), 4.27 (2H, m, 2 × Fmoc-CH), 4.12 (1H, m, piperidine-H), 3.92 (3H, s, NOCH3), 3.20 (1H, brs, piperidine-H), 3.04 (1H, t, J = 8.8 Hz, piperidine-H), 2.89 (1H, t, J = 11 Hz, piperidine-H), 2.10 (2H, brs, piperidine-H), 1.25 (1H, m, piperidine-H). MS-ESI (m/z): 610 (M+Na)+.
(9H-Fluoren-9-yl)methyl 3-[(9H-fluoren-9-yl)methoxycarbonylamino]-4-oxopiperidine-1-carboxylate (11). To a solution of 10 (11.75 g, 20 mmol) and ethyl acetoacetate (26.02 g, 200 mmol) in methanol (200 mL) was added concentrated hydrochloride (0.4 mmol, 33.33 mL) and stirred at 60 °C for 6 h. The mixture was concentrated under reduced pressure. The residue was dissolved in water (200 mL), adjusted to pH 6.5–7 with saturated sodium bicarbonate solution, and extracted with dichloromethane. The combined extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by column chromatography (silica gel), eluting with petroleum ether and ethyl acetate (v:v = 2:1) to afford the title compound 11 (5.81 g, 52%) as a white solid, m.p.: 141–142 °C. 1H-NMR (600 MHz, CDCl3) δ (ppm): 7.77 (4H, m, Fmoc-ArH), 7.61 (4H, m, Fmoc-ArH), 7.41 (4H, m, Fmoc-ArH), 7.31 (4H, m, Fmoc-ArH), 4.63 (4H, s, 2 × Fmoc-CH2), 4.28 (2H, m, 2 × Fmoc-CH), 4.12 (1H, m, piperidine-H), 3.72 (1H, brs, piperidine-H), 3.08 (1H, brs, piperidine-H), 2.84 (1H, brs, piperidine-H), 2.66–2.59 (2H, m, piperidine-H), 1.26 (1H, m, piperidine-H). MS-ESI (m/z): 581 (M+Na)+.
(9H-Fluoren-9-yl)methyl 3-[(9H-fluoren-9-yl)methoxycarbonylamino]-4-benzyloxyiminopiperidine-1-carboxylate (12). To a solution of 11 (5.58 g, 10 mmol) in ethanol (100 mL) was added 4m (2.46 g, 20 mmol) and the mixture stirred at room temperature for 10 h and concentrated under reduced pressure. The residue was dissolved in ethyl acetate (100 mL) and washed with brine (200 mL). The combined extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by column chromatography (silica gel), eluting with petroleum ether acetate (v:v = 2:1) to yield the title compound 12 (4.95 g, 75%) as a white solid, m.p.: 169–171 °C. 1H-NMR (600 MHz, CDCl3) δ (ppm): 7.75 (4H, m, Fmoc-ArH), 7.61 (4H, m, Fmoc-ArH), 7.42–7.37 (9H, m, Fmoc-ArH, Ar-H), 7.31 (4H, m, Fmoc-ArH), 5.15 (2H, s, OCH2Ar), 4.43 (4H, s, 2 × Fmoc-CH2), 4.26 (2H, m, 2 × Fmoc-CH), 4.18 (1H, m, piperidine-H), 3.28 (1H, brs, piperidine-H), 3.00 (1H, m, piperidine-H), 2.85 (1H, m, piperidine-H), 2.15 (1H, brs, piperidine-H), 1.53 (2H, m, piperidine-H). MS-ESI (m/z): 664 (M+H)+, 686 (M+Na)+, 702 (M+K)+.
3-Amino-4-benzyloxyiminopiperidine dihydrocholoride (13m). To a solution of 12 (2 g, 3 mmol) in tetrahydrofuran (50 mL) was added 10% sodium hydroxide (6 mL, 15 mmol) and stirred at 60 °C for 20 h. The mixture was concentrated under reduced pressure. The residue was treated with dichloromethane (100 mL) and washed with water. The organic layer was dried over anhydrous sodium sulfate and filtered. To the filtrate was pumped dried hydrochloride gas at room temperature for 30 minutes and concentrated under reduced pressure to afford the title compound 13m (0.3 g, 45%) as a white solid, m.p.: 153–155 °C. 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 7.39 (4H, m), 7.33 (1H, m, Ar-H), 5.12 (2H, s, OCH2Ar), 3.95 (1H, m, piperidine-H), 3.64 (1H, m, piperidine-H), 3.07 (1H, m, piperidine-H), 2.97 (1H, m, piperidine-H), 2.88 (1H, m, piperidine-H), 2.78 (1H, m, piperidine-H), 2.38 (1H, m, piperidine-H). MS-ESI (m/z): 220 (M+H)+, 242 (M+Na)+. M.p.: 153–155 °C.
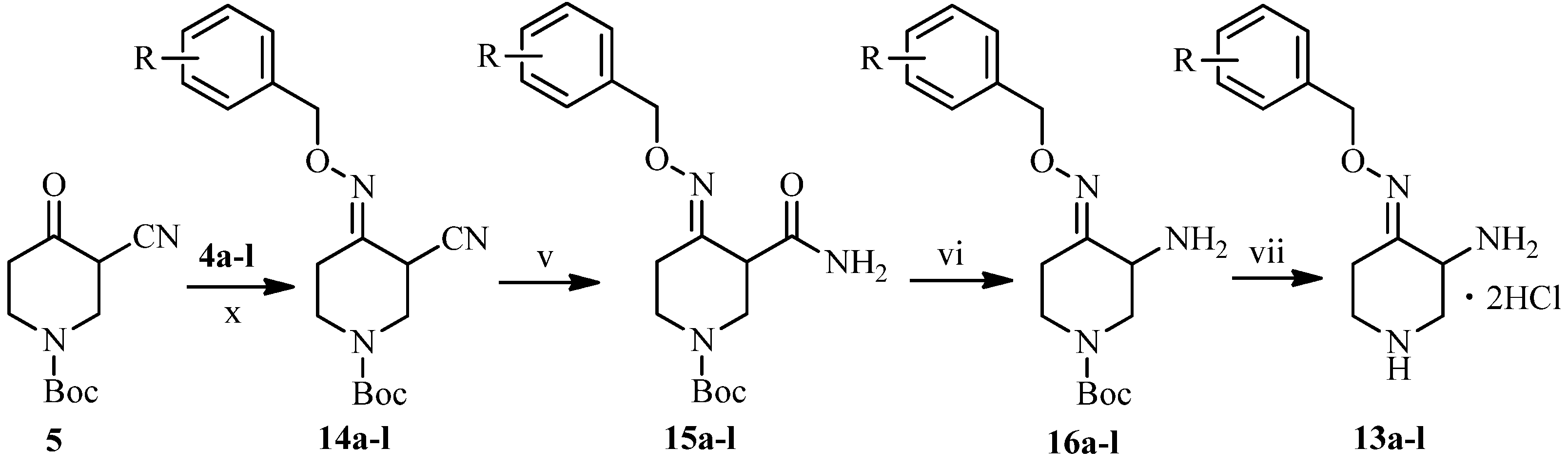
3.2.2. General Procedure for the Synthesis of 3-Amino-4-(substituted benzyloxyimino)piperidine Dihydrocholorides 13a–l
To a solution of 5 (70 mmol) in ethanol (100 mL) was added 4a–l (140 mmol) and stirred at room temperature for 10 h and concentrated under reduced pressure. The residue was dissolved in ethyl acetate (200 mL) and washed with brine (200 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford compounds 14a–l as yellow oils.
To a solution of 14a–l (50 mmol) and potassium carbonate (100 mmol) in dimethyl sulfoxide (150 mL) was added hydrogen peroxide (400 mmol) at 15 °C for 1 h, and stirred at room temperature for 3 h. The mixture was diluted with water (300 mL) and extracted by ethyl acetate (400 mL). The combined extracts were washed with brine (400 mL), dried over anhydrous sodium sulfate, and then concentrated under reduced pressure to give compounds 15a–l as colorless or light yellow oils.
To a stirred solution of 15a–l (50 mmol) in acetonitrile (350 mL) was added dropwise 10% sodium hypobromite solution (90 mmol) at 5 °C for 1 h. The reaction mixture was stirred at room temperature for 10 h, adjusted to pH 6.5–7 by 20% acetic acid and then concentrated under reduced pressure. The residue was dissolved in water (200 mL), adjusted to pH 4 with 10% hydrochloride and washed by ethyl acetate. The water layer was adjusted to pH 9 with 10% sodium hydroxide and extracted by ethyl acetate. The combined extracts were dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford compounds 16a–l as yellow oils.
To a solution of 16a–l (7.29 g, 30 mmol) in dichloromethane was pumped dried hydrochloride gas at room temperature for 30 minutes. The mixture was stirred for another 1 h at room temperature, and concentrated under reduced pressure. The residue was treated with ethyl acetate. The precipitate was collected by suction, and dried under vacuum to afford compounds 13a–l (19%–26%, from 5) as white or yellow solids.
3-Amino-4-(4′-bromo-2′-methoxybenzyloxyimino)piperidine dihydrocholoride (13a). The title compound was obtained from 16a as an off-white solid easily absorbing moisture (50%).
3-Amino-4-(3′,4′-ethylenedioxylbenzyloxyimino)piperidine dihydrocholoride (13b). The title compound was obtained from 16b as a white solid (51%), m.p.: 190–192 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 6.87 (3H, m, Ar-H), 5.02 (2H, s, OCH2Ar), 4.41 (1H, m, piperidine-H), 4.23 (4H, s, 2 × OCH2), 3.71 (1H, m, piperidine-H), 3.53 (1H, m, piperidine-H), 3.37 (1H, m, piperidine-H), 3.23–3.11 (2H, m, piperidine-H), 2.98 (1H, m, piperidine-H). MS-ESI (m/z): 278 (M+H)+.
3-Amino-4-(2′,5′-dimethoxylbenzyloxyimino)piperidine dihydrocholoride (13c). The title compound was obtained from 16c as a light yellow solid (53%), m.p.: 189–192 °C. 1H-NMR (400 MHz, D2O) δ (ppm): 7.07 (1H, m, Ar-H), 7.02 (2H, m, Ar-H), 5.20 (2H, s, OCH2Ar), 4.50 (1H, J1 = 12.4 Hz, J2 = 5.6 Hz, piperidine-H), 4.00 (1H, m, piperidine-H), 3.84 (3H, s, OCH3), 3.82 (3H, s, OCH3), 3.67 (1H, m, piperidine-H), 3.56 (1H, m, piperidine-H), 3.31 (1H, t, J = 12.4 Hz, piperidine-H), 3.18 (1H, m, piperidine-H), 2.44 (1H, m, piperidine-H). MS-ESI (m/z): 280 (M+H)+.
3-Amino-4-(3′,5′-dimethoxylbenzyloxyimino)piperidine dihydrocholoride (13d). The title compound was obtained from 16d as a white solid (50%), m.p.: 212–213 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 6.55 (2H, m, Ar-H), 6.45 (1H, m, Ar-H), 5.09 (2H, s, OCH2Ar), 4.45 (1H, m, piperidine-H), 3.75 (6H, s, OCH3), 3.72 (1H, m, piperidine-H), 3.38 (1H, m, piperidine-H), 3.26 (1H, m, piperidine-H), 3.17 (1H, m, piperidine-H), 2.98 (1H, m, piperidine-H), 2.58 (1H, m, piperidine-H). MS-ESI (m/z): 280 (M+H)+, 302 (M+Na)+.
3-Amino-4-(2′,3′-dimethoxylbenzyloxyimino)piperidine dihydrocholoride (13e). The title compound was obtained from 16e as a light yellow solid (54%), m.p.: 194–196 °C. 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 7.07 (2H, m, Ar-H), 6.98 (1H, m, Ar-H), 5.15 (2H, s, OCH2Ar), 4.44 (1H, m, piperidine-H), 3.81 (3H, s, OCH3), 3.74 (3H, s, OCH3), 3.70 (1H, m, piperidine-H), 3.36 (1H, m, piperidine-H), 3.23 (1H, m, piperidine-H), 3.16 (1H, m, piperidine-H), 2.98 (1H, m, piperidine-H), 2.58 (1H, m, piperidine-H). MS-ESI (m/z): 280 (M+H)+.
3-Amino-4-(3′,4′-dimethoxylbenzyloxyimino)piperidine dihydrocholoride (13f). The title compound was obtained from 16f as a light yellow solid (52%), m.p.: 183–184 °C. 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 6.95 (3H, m, Ar-H), 5.05 (2H, s, OCH2Ar), 4.45 (1H, m, piperidine-H), 3.75 (6H, s, OCH3), 3.37 (1H, m, piperidine-H), 3.28 (1H, m, piperidine-H), 3.13 (1H, m, piperidine-H), 2.96 (1H, m, piperidine-H), 2.55 (1H, m, piperidine-H), 1.21 (1H, m, piperidine-H). MS-ESI (m/z): 280 (M+H)+.
3-Amino-4-(3′,4′-methylenedioxybenzyloxyimino)piperidine dihydrocholoride (13g). The title compound was obtained from 16e as a white solid (50%), m.p.: 199–201 °C. 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 7.01 (1H, s, Ar-H), 6.89 (2H, m, Ar-H), 6.02 (2H, s, OCH2Ar), 5.04 (2H, s, 2 × OCH2), 4.36 (1H, m, piperidine-H), 3.68 (1H, brs, piperidine-H), 3.35 (1H, m, piperidine-H), 3.22 (1H, m, piperidine-H), 3.12 (1H, m, piperidine-H), 2.98 (1H, m, piperidine-H), 2.48 (1H, m, piperidine-H). MS-ESI (m/z): 264 (M+H)+, 527 (2M+H)+.
3-Amino-4-(2′-methoxylbenzyloxyimino)piperidine dihydrocholoride (13h). The title compound was obtained from 16e as a light yellow solid (55%), m.p.: 199–200 °C. 1H-NMR (400 MHz, D2O) δ (ppm): 7.44 (2H, m, Ar-H), 7.13 (1H, d, J = 8.4 Hz, Ar-H), 7.07 (1H, t, J = 7.6 Hz, Ar-H), 5.24 (2H, s, OCH2Ar), 4.44 (1H, q, J1 = 12 Hz, J2 = 5.2 Hz, piperidine-H), 3.96 (1H, q, J1 = 12 Hz, J2 = 5.2 Hz, piperidine-H), 3.88 (3H, s, OCH3), 3.63 (1H, q, J1 = 12.8 Hz, J2 = 5.6 Hz, piperidine-H), 3.53 (1H, m, piperidine-H), 3.25 (1H, t, J = 12 Hz, piperidine-H), 3.13 (1H, m, piperidine-H), 2.41 (1H, m, piperidine-H). MS-ESI (m/z): 250 (M+H)+.
3-Amino-4-(3′-methoxylbenzyloxyimino)piperidine dihydrocholoride (13i). The title compound was obtained from 16e as a light yellow solid (51%), m.p.: 215–217 °C. 1H-NMR (400 MHz, D2O) δ (ppm): 7.43 (1H, m, Ar-H), 7.04 (3H, m, Ar-H), 5.15 (2H, s, OCH2Ar), 4.38 (1H, m, piperidine-H), 3.91 (1H, m, piperidine-H), 3.85 (3H, s, OCH3), 3.62–3.52 (2H, m, piperidine-H), 3.24–3.17 (1H, m, piperidine-H), 3.14–3.07 (1H, m, piperidine-H), 2.42–2.34 (1H, m, piperidine-H).MS-ESI (m/z): 250 (M+H)+.
3-Amino-4-(4′-methoxylbenzyloxyimino)piperidine dihydrocholoride (13j). The title compound was obtained from 16e as an off-white solid (53%), m.p.: 221–222 °C. 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 7.35 (2H, d, J = 9 Hz, Ar-H), 6.93 (2H, J = 9 Hz, Ar-H), 5.07 (2H, s, OCH2Ar), 4.41 (1H, m, piperidine-H), 3.76 (3H, s, OCH3), 3.71 (1H, m, piperidine-H), 3.38 (1H, m, piperidine-H), 3.26 (1H, m, piperidine-H), 3.20 (1H, m, piperidine-H), 3.12 (1H, m, piperidine-H), 2.95 (1H, m, piperidine-H). MS-ESI (m/z): 250 (M+H)+.
3-Amino-4-(4′-flourobenzyloxyimino)piperidine dihydrocholoride (13k). The title compound was obtained from 16e as a yellow solid as yellow solid easily absorbing moisture (50%).
3-Amino-4-(4′-chlorobenzyloxyimino)piperidine dihydrocholoride (13l). The title compound was obtained from 16e as a white solid (56%), m.p.: 200–202 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.44 (4H, m, Ar-H), 5.14 (2H, s, OCH2Ar), 4.43 (1H, m, piperidine-H), 3.72 (1H, m, piperidine-H), 3.38 (1H, m, piperidine-H), 3.24 (1H, m, piperidine-H), 3.16 (1H, m, piperidine-H), 2.98 (1H, m, piperidine-H), 2.57 (1H, m, piperidine-H). MS-ESI (m/z): 254 (M+H)+.
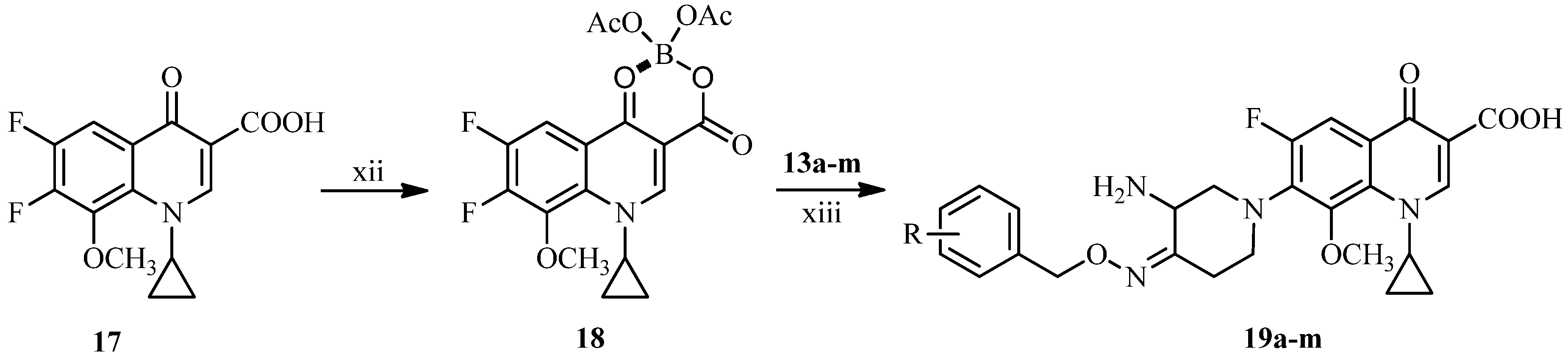
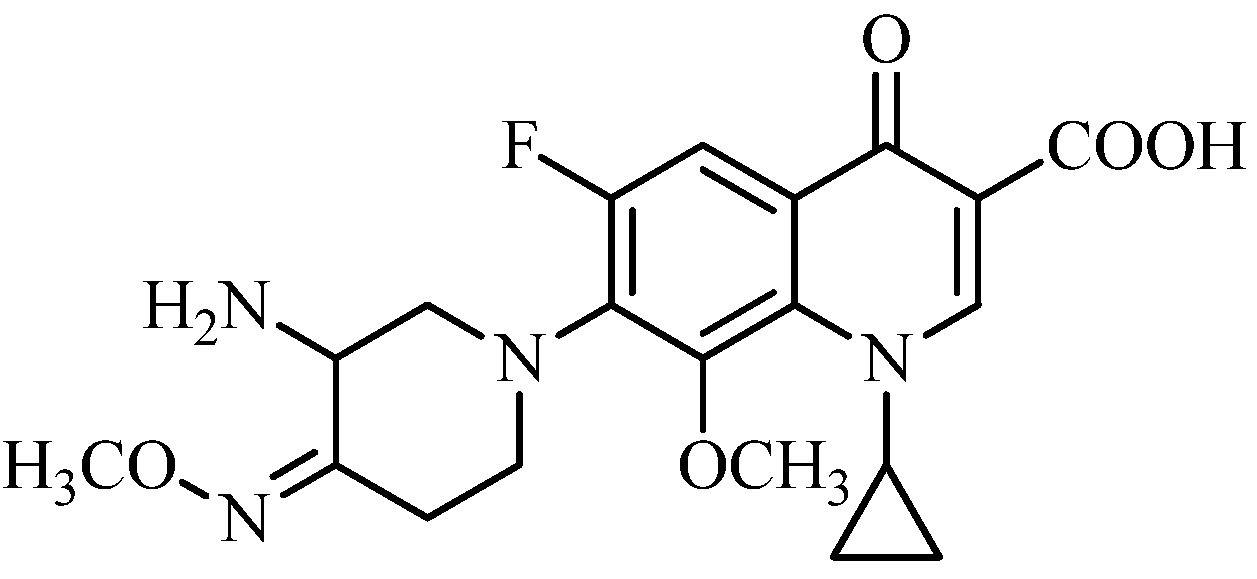
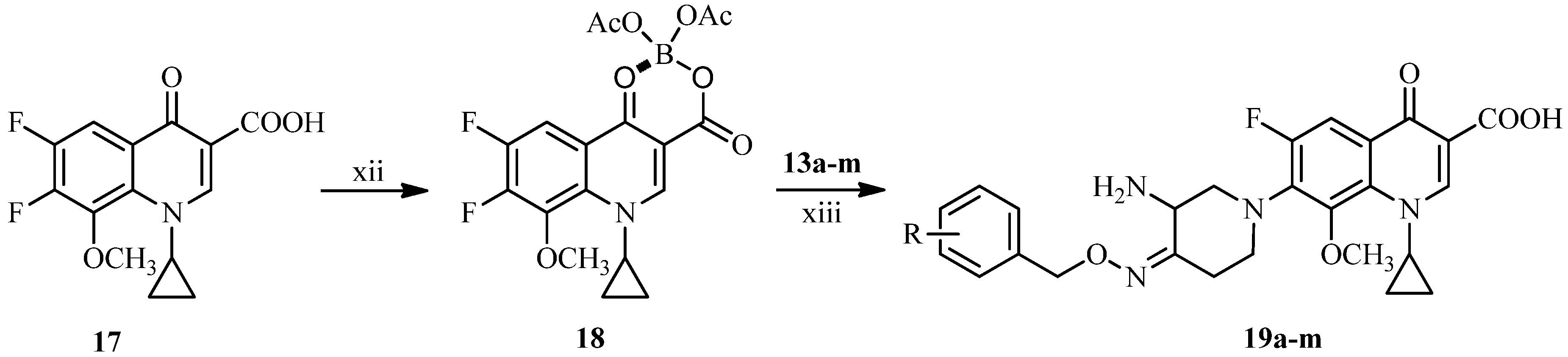
3.2.3. General Procedure for the Synthesis of 7-[3-Amino-4-(substituted benzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic Acids 19a–m
A mixture of boric acid (9.27 g, 150 mmol) and acetic anhydride (54.06 g, 530 mmol) was stirred at 110 °C for 1.5 h, added acetic acid (50 mL) and then stirred for 1h at the same temperature. To the reaction mixture temperature was added 17 (32.3 g, 100 mmol) at 95 °C and stirred at the same temperature for 2 h. After cooling to room temperature, the mixture was poured into ice water (500 mL) slowly and stirred for 0.5 h. The resulting solid was collected by suction and washed successively with water (50 mL), chilled ethanol (50 mL) and ethyl ether (50 mL), and dried under vacuum to afford compound 18 (29.4 g, 69%) as a white solid, m.p.: 195–196 °C.
To a solution of 13a–m (1 mmol) and triethylamine (3 mmol) in acetonitrile (10 mL) was added 18 (0.8 mmol) at room temperature. The reaction mixture was stirred overnight at 50 °C, and concentrated under reduced pressure. The residue was dissolved in a solution of 5% sodium hydroxide solution (8 mL) and stirred for 1 h at 50 °C. After cooling to room temperature, the mixture was adjusted to pH 7.0–7.5 with 5% acetic acid, and extracted with dichloromethane. The combined extracts were concentrated under reduced pressure. The residue was dissolved in 20% acetic acid (10 mL), stirred for 0.5 h at 50 °C, and filtered. The filtrate was adjusted to pH 6.5–7.5 with 15% sodium hydroxide and extracted by dichloromethane. The combined extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by column chromatography (silica gel), eluting with dichloromethane and methanol (v:v = 10:1) to afford the target compounds 19a–m as off-white or yellow solids.
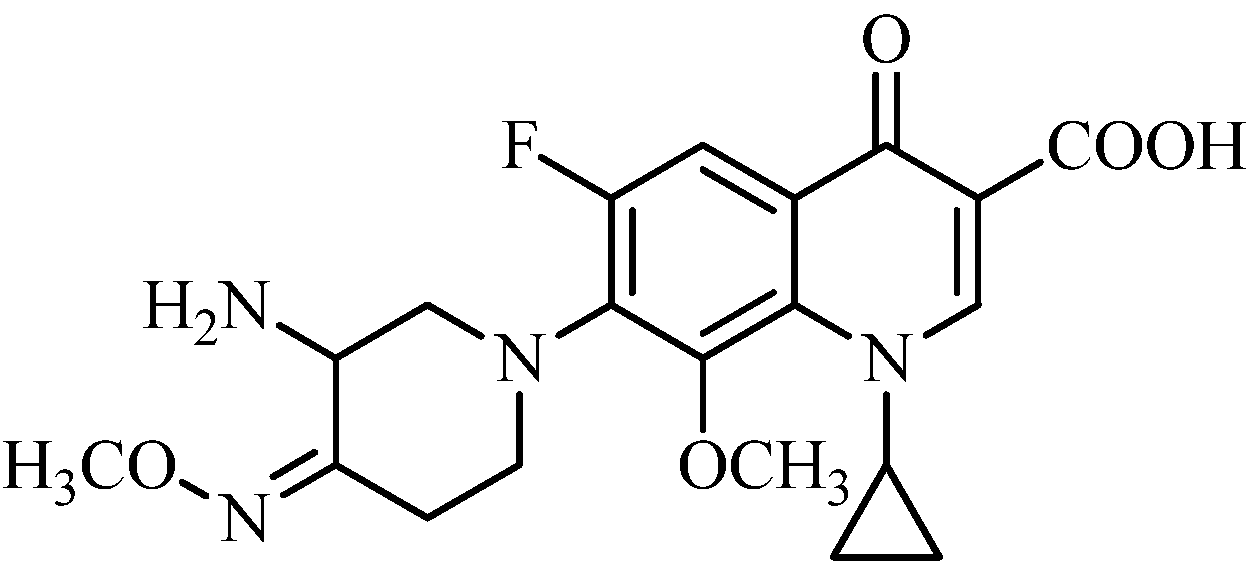
7-[3-Amino-4-(4′-bromo-2′-methoxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19a). Prepared from 13a as an off-white solid (19%), m.p.: >230 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.74 (1H, s, C2-H), 7.65 (1H, d, J = 12 Hz, C5-H), 7.42 (1H, d, J = 8 Hz, Ar-H), 7.22 (1H, d, J = 2 Hz, Ar-H), 6.97 (1H, q, J1 = 8 Hz, J2 = 6 Hz, Ar-H), 5.10 (2H, s, O-CH2Ar), 4.86 (1H, m, cyclopropyl CH), 4.45 (1H, m, piperidine-H), 4.15 (1H, m, piperidine-H), 3.68 (6H, s, O-CH3), 2.96 (1H, m, piperidine-H), 2.85 (1H, m, piperidine-H), 2.77 (1H, m, piperidine-H), 2.68 (1H, m, piperidine-H), 2.40 (1H, m, piperidine-H), 0.93 (4H, m, 2 × cyclopropyl CH2). MS-ESI (m/z): 603 (M+H)+. HRMS-ESI (m/z): Calcd. for C27H29O6N4FBr (M+H)+: 603.1254; Found 603.1276.
7-[3-Amino-4-(3′,4′-ethylenedioxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19b). Prepared from 13b as an off-white solid (33%), m.p.: 78–81 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.70 (1H, s, C2-H), 7.77 (1H, d, J = 12 Hz, C5-H), 6.87 (1H, s, Ar-H), 6.83 (2H, m, Ar-H), 4.96 (2H, s, O-CH2Ar), 4.80 (1H, m, cyclopropyl CH), 4.42 (1H, m, piperidine-H), 4.23 (4H, s, 2 × OCH2), 3.74 (3H, s, O-CH3), 3.40 (1H, m, piperidine-H), 3.19 (1H, m, piperidine-H), 3.03 (1H, m, piperidine-H), 2.89 (1H, m, piperidine-H), 2.72 (1H, m, piperidine-H), 2.33 (1H, m, piperidine-H), 1.05 (4H, m, 2 × cyclopropyl CH2). MS-ESI (m/z): 553 (M+H)+, 555 (M+Na)+. HRMS-ESI (m/z): Calcd. for C28H30O7N4F (M+H)+: 553.2093; Found 553.2109. M.p.: 78–81 °C.
7-[3-Amino-4-(2′,5′-dimethoxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19c). Prepared from 13c as an off-white solid (21%), m.p.: 80–81 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.62 (1H, s, C2-H), 7.78 (1H, d, J = 12 Hz, C5-H), 6.94 (1H, d, J = 8 Hz, Ar-H), 6.87 (1H, q, J1 = 8H, J2 = 3 Hz, Ar-H), 6.84 (1H, d, J = 3 Hz, Ar-H), 5.08 (2H, s, O-CH2Ar), 4.93 (1H, m, cyclopropyl CH), 4.42 (1H, m, piperidine-H), 4.17 (1H, m, piperidine-H), 3.69 (9H, s, O-CH3), 3.17 (1H, m, piperidine-H), 3.11 (1H, m, piperidine-H), 2.91–2.86 (1H, m, piperidine-H), 2.75 (1H, m, piperidine-H), 2.39 (1H, m, piperidine-H), 0.98 (4H, m, 2 × cyclopropyl CH2). MS-ESI (m/z): 555 (M+H)+. HRMS-ESI (m/z): Calcd. for C28H32O7N4F (M+H)+: 555.2249; Found 555.2252. M.p.: 80–81 °C.
7-[3-Amino-4-(3′,5′-dimethoxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19d). Prepared from 13d as an off-white solid (23%), m.p.: 68–70 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.70 (1H, s, C2-H), 7.78 (1H, d, J = 12 Hz, C5-H), 6.52 (1H, s, Ar-H), 6.42 (1H, s, Ar-H), 6.25 (1H, s, Ar-H), 5.03 (2H, s, O-CH2Ar), 4.87 (1H, m, cyclopropyl CH), 4.41 (1H, m, piperidine-H), 4.16 (1H, m, piperidine-H), 3.67 (9H, s, O-CH3), 3.43 (1H, m, piperidine-H), 3.18 (1H, m, piperidine-H), 2.88 (1H, m, piperidine-H), 2.75 (1H, m, piperidine-H), 2.41 (1H, m, piperidine-H), 1.03 (4H, m, 2 × cyclopropyl CH2). MS-ESI (m/z): 555 (M+H)+. HRMS-ESI (m/z): Calcd. for C28H32O7N4F (M+H)+: 555.2249; Found 555.2264. M.p.: 68–70 °C.
7-[3-Amino-4-(2′,3′-dimethoxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19e). Prepared from 13e as an off-white solid (18%), m.p.: 73–74 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.70 (1H, s, C2-H), 7.78 (1H, d, J = 12 Hz, C5-H), 6.83 (1H,d, J = 8 Hz, Ar-H), 6.74 (1H, t, J = 8 Hz, Ar-H ), 6.45 (1H, d, J = 8 Hz, Ar-H), 5.11 (2H, s, O-CH2Ar), 4.41 (1H, m, cyclopropyl CH), 4.17–4.13 (2H, m, piperidine-H), 3.80 (3H, s, O-CH3), 3.74 (6H, s, O-CH3), 3.09–3.61 (1H, m, piperidine-H), 2.92–2.81 (1H, m, piperidine-H), 2.75–2.62 (2H, m, piperidine-H), 2.38–2.32 (1H, m, piperidine-H), 1.04–0.96 (4H, m, 2 × cyclopropyl CH2). MS-ESI (m/z): 555 (M+H)+. HRMS-ESI (m/z): Calcd. for C28H32O7N4F (M+H)+: 555.2249; Found 555.2254. M.p.: 73–74 °C.
7-[3-Amino-4-(3′,4′-dimethoxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19f). Prepared from 13f as an off-white solid (20%), m.p.: 88–91 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.69 (1H, s, C2-H), 7.83 (1H, d, J = 11 Hz, C5-H), 6.79 (1H, d, J = 1.6 Hz, Ar-H), 6.68 (1H, d, J = 8, Ar-H), 6.7 (1H, q, J1 = 8 Hz, J2 = 1.6 Hz, Ar-H), 5.08 (2H, s, O-CH2Ar), 4.87 (1H, m, cyclopropyl CH), 4.58 (1H, m, piperidine-H), 4.20–4.16 (1H, m, piperidine-H), 3.77 (9H, O-CH3s), 3.53–3.49 (1H, m, piperidine-H), 3.18–3.15 (1H, m, piperidine-H), 3.02–2.96 (1H, m, piperidine-H), 2.82–2.80 (1H, m, piperidine-H), 2.42–2.35 (1H, m, piperidine-H), 1.12–1.04 (4H, m, 2 × cyclopropyl CH2). MS-ESI (m/z): 555 (M+H)+. HRMS-ESI (m/z): Calcd. for C28H32O7N4F (M+H)+: 555.2249; Found 555.2265.
7-[3-Amino-4-(3′,4′-methylenedioxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19g). Prepared from 13g as an off-white solid (25%), m.p.: 79–80 °C. 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 8.68 (1H, s, C2-H), 7.78 (1H, d, J = 11 Hz, C5-H), 6.96 (1H, s, Ar-H), 6.90 (1H, d, J = 7.8 Hz, Ar-H), 6.87 (1H, d, J = 7.8 Hz, Ar-H), 6.01 (2H, s, OCH2O), 4.99 (2H, s, O-CH2Ar), 4.83 (1H, m, cyclopropyl CH), 4.17 (1H, m, piperidine-H), 3.74 (3H, s, O-CH3), 3.04–3.02 (1H, m, piperidine-H), 2.91–2.88 (1H, m, piperidine-H), 2.82–2.88 (1H, m, piperidine-H), 2.73–2.70 (1H, m, piperidine-H), 2.63–2.60 (1H, m, piperidine-H), 2.38–2.33 (1H, m, piperidine-H), 0.92 (4H, m, 2 × cyclopropyl CH2). 13C-NMR (150 MHz, DMSO-d6) δ (ppm): 176.22, 165.60, 157.19 (d, J = 268 Hz), 150.57, 150.05, 147.19, 146.92, 146.79, 139.05, 134.09, 133.03, 131.73, 131.55, 121.80, 121.16, 108.71, 106.78 (d, J = 24 Hz), 74.75, 63.10, 58.17, 51.31, 49.60, 40.73, 24.39, 9.02. MS-ESI (m/z): 539 (M+H)+, 561 (M+Na)+. HRMS-ESI (m/z): Calcd. for C27H28O7N4F (M+H)+: 539.1941; Found 539.1946.
7-[3-Amino-4-(2′-methoxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19h). Prepared from 13h as an off-white solid (19%), m.p.: 208–211 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.71 (1H, s, C2-H), 7.88 (1H, d, J = 12 Hz, C5-H), 7.34–7.29 (2H, m, Ar-H), 7.03–6.89 (2H, m, Ar-H), 5.12 (2H, s, O-CH2Ar), 4.16 (1H, brs, cyclopropyl CH), 3.80–3.68 (5H, m, piperidine-H), 3.50–3.41 (1H, brs, piperidine-H), 3.32 (6H, s, O-CH3), 2.05–1.90 (1H, m, piperidine-H), 1.12–0.84 (4H, m, 2 × cyclopropyl CH2). MS-ESI (m/z): 525 (M+H)+, 547 (M+Na)+. HRMS-ESI (m/z): Calcd. for C27H30O6N4F (M+H)+: 525.2143; Found 525.2147.
7-[3-Amino-4-(3′-methoxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19i). Prepared from 13i as an off-white solid (23%), m.p.: 94–95 °C. 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 8.65 (1H, s, C2-H), 7.75 (1H, d, J = 11 Hz, C5-H), 7.14 (1H, s, Ar-H), 6.86–6.83 (3H, m, Ar-H), 5.03 (2H, s, O-CH2Ar), 4.43–4.41 (1H, brs, cyclopropyl CH), 4.17–4.14 (1H, brs, piperidine-H), 3.75 (6H, s, O-CH3), 3.45–3.43 (1H, m, piperidine-H), 3.23–3.18 (1H, m, piperidine-H), 3.06–3.04 (1H, m, piperidine-H), 2.87–2.84 (1H, m, piperidine-H), 2.70–2.66 (1H, m, piperidine-H), 2.34–2.29 (1H, m, piperidine-H), 1.17–1.12 (4H, m, 2 × cyclopropyl CH2). 13C-NMR (150 MHz, DMSO-d6) δ (ppm): 176.23, 165.83, 165.60, 158.29 (d, J = 235 Hz), 156.98, 154.63, 150.63, 150.12, 129.88, 129.76, 129.24, 113.68, 113.29, 106.92, 106.76, 106.63, 106.25 (d, J = 25 Hz), 74.36, 63.18, 55.06, 54.87, 49.57, 45.27, 40.73, 24.50, 8.59. MS-ESI (m/z): 525 (M+H)+, 547 (M+Na)+. HRMS-ESI (m/z): Calcd. for C27H30O6N4F (M+H)+: 525.2143; Found 525.2149.
7-[3-Amino-4-(4′-methoxybenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19j). Prepared from 13j as an off-white solid (27%), m.p.: 80–82 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.66 (1H, s, C2-H), 7.76–7.68 (1H, m, C5-H), 7.31 (2H, d, J = 8 Hz, Ar-H), 6.91 (2H, d, J = 8 Hz, Ar-H), 4.99 (2H, s, O-CH2Ar), 4.14 (1H, brs, cyclopropyl CH), 3.74 (6H, s, O-CH3), 3.65 (1H, m, piperidine-H), 3.37 (1H, brs, piperidine-H), 3.27 (1H, brs, piperidine-H), 3.16 (1H, brs, piperidine-H), 2.99 (1H, brs, piperidine-H), 2.30 (1H, m, piperidine-H), 1.96 (1H, m, piperidine-H), 1.09–1.02 (4H, m, 2 × cyclopropyl CH2). 13C-NMR (150 MHz, DMSO-d6) δ (ppm): 176.32, 165.71, 158.91, 153.05 (d, J = 250 Hz), 150.58, 134.09, 129.82, 129.65, 129.23, 122.12, 113.65, 107.00, 106.48 (d, J = 22 Hz), 74.63, 63.08, 55.04, 51.44, 49.64, 45.40, 25.09, 8.90. MS-ESI (m/z): 525 (M+H)+. HRMS-ESI (m/z): Calcd. for C27H30O6N4F (M+H)+: 525.2143; Found 525.2146.
7-[3-Amino-4-(4′-flourobenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19k). Prepared from 13k as an off-white solid (30%), m.p.: 74–77 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.63 (1H, s, C2-H), 7.77 (1H, d, J =12 Hz, C5-H), 7.45 (2H, m, Ar-H), 7.11 (2H, m, Ar-H), 5.09 (2H, s, O-CH2Ar), 4.91(1H, m, cyclopropyl CH), 4.11 (1H, m, piperidine-H), 3.73 (3H, s, O-CH3), 3.47 (1H, m, piperidine-H), 3.25 (1H, m, piperidine-H), 3.12 (1H, m, piperidine-H), 2.91 (1H, m, piperidine-H), 2.72 (1H, m, piperidine-H), 2.35 (1H, m, piperidine-H), 0.96 (4H, m, 2 × cyclopropyl CH2). 13C-NMR (150 MHz, DMSO-d6) δ (ppm): 176.32, 165.56, 163.78 (d, J = 243 Hz), 155.42 (d, J = 247 Hz), 150.68, 149.61, 145.21, 133.75, 133.04, 130.24, 129.62, 129.54, 115.22, 114.62, 106.31 (d, J = 24 Hz), 74.43, 63.25, 55.98, 50.23, 49.52, 40.94, 24.71, 8.67. MS-ESI (m/z): 513 (M+H)+, 535 (M+Na)+. HRMS-ESI (m/z): Calcd. for C26H27O5N4F2 (M+H)+: 513.1949; Found 513.1952.
7-[3-Amino-4-(4′-chlorobenzyloxyimino)piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (19l). Prepared from 13l as an off-white solid (34%), m.p.: 67–69 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.67 (1H, s, C2-H), 7.79 (1H, d, J = 12 Hz, C5-H), 7.14 (2H, m, Ar-H), 7.07 (2H, m, Ar-H), 5.11 (2H, s, O-CH2Ar), 4.94 (1H, m, cyclopropyl CH), 4.40 (1H, m, piperidine-H), 4.01 (1H, m, piperidine-H), 3.75 (3H, s, O-CH3), 3.18–3.14 (1H, m, piperidine-H), 3.07–3.05 (1H, m, piperidine-H), 2.92–2.90 (1H, m, piperidine-H), 2.79–2.76 (1H, m, piperidine-H), 2.43–2.38 (1H, m, piperidine-H), 1.07–1.04 (4H, m, 2 × cyclopropyl CH2). 13C-NMR (150 MHz, DMSO-d6) δ (ppm): 176.18, 165.59, 157.75, 155.47 (d, J = 248 Hz), 150.59, 146.11, 137.12, 134.08, 132.25, 129.79, 129.00, 121.19, 117.10, 106.67 (d, J = 23 Hz), 106.61, 73.98, 63.13, 57.79, 51.14, 45.26, 40.72, 24.91, 8.86. MS-ESI (m/z): 529 (M+H)+. HRMS-ESI (m/z): Calcd. for C26H27O5N4FCl (M+H)+: 529.1648; Found 529.1637.
7-(3-Amino-4-benzyloxyiminopiperidin-1-yl)-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (19m). Prepared from 13m as an off-white solid (32%), m.p.: 84–85 °C. 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 8.68 (1H, s, C2-H), 8.74 (1H, d, J = 12 Hz, C5-H), 7.29–7.21 (5H, m, Ar-H), 5.10 (2H, s, O-CH2Ar), 4.97–4.92 (1H, m, cyclopropyl CH), 4.43–4.39 (1H, m, piperidine-H), 4.16–4.13 (1H, m, piperidine-H), 3.72 (3H, s, O-CH3), 3.65–3.63 (1H, m, piperidine-H), 3.2–3.15 (1H, m, piperidine-H), 2.86–2.83 (1H, m, piperidine-H), 2.71–2.67 (1H, m, piperidine-H), 2.37–2.34 (1H, m, piperidine-H), 1.08–0.95 (4H, m, 2 × cyclopropyl CH2). 13C-NMR (150 MHz, DMSO-d6) δ (ppm): 176.26, 165.73, 155.65 (d, J = 255 Hz), 150.60, 150.19, 146.05, 137.89, 133.09, 128.14, 127.98, 127.68, 127.73, 127.29, 106.86 (d, J = 21 Hz), 106.35, 74.62, 61.29, 53.11, 51.35, 45.39, 40.75, 24.78, 8.98. MS-ESI (m/z): 495 (M+H)+, 517 (M+Na)+. HRMS-ESI (m/z): Calcd. for C26H28O5N4F (M+H)+: 495.2038; Found 495.2048. M.p.: 84–85 °C.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}