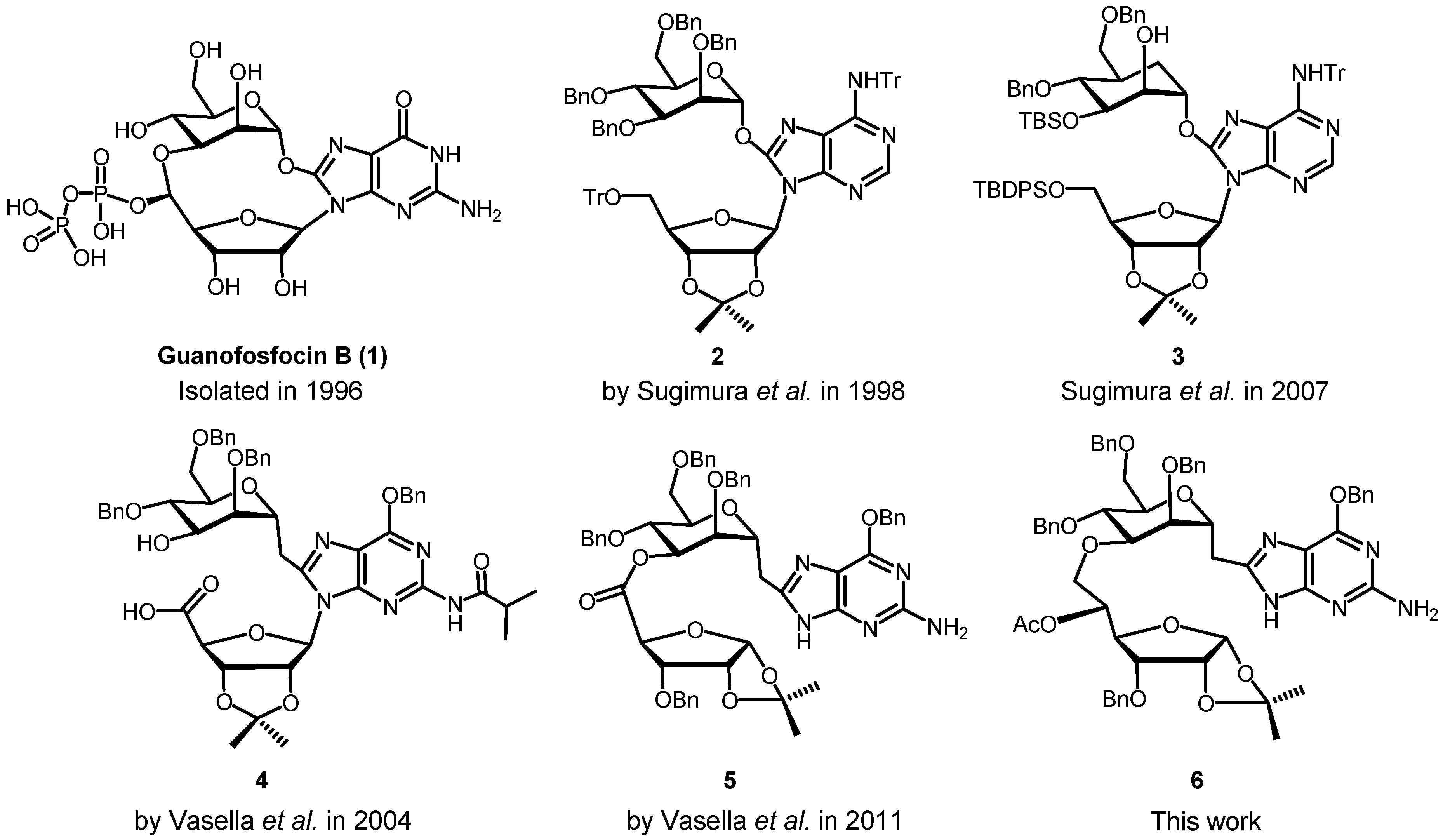
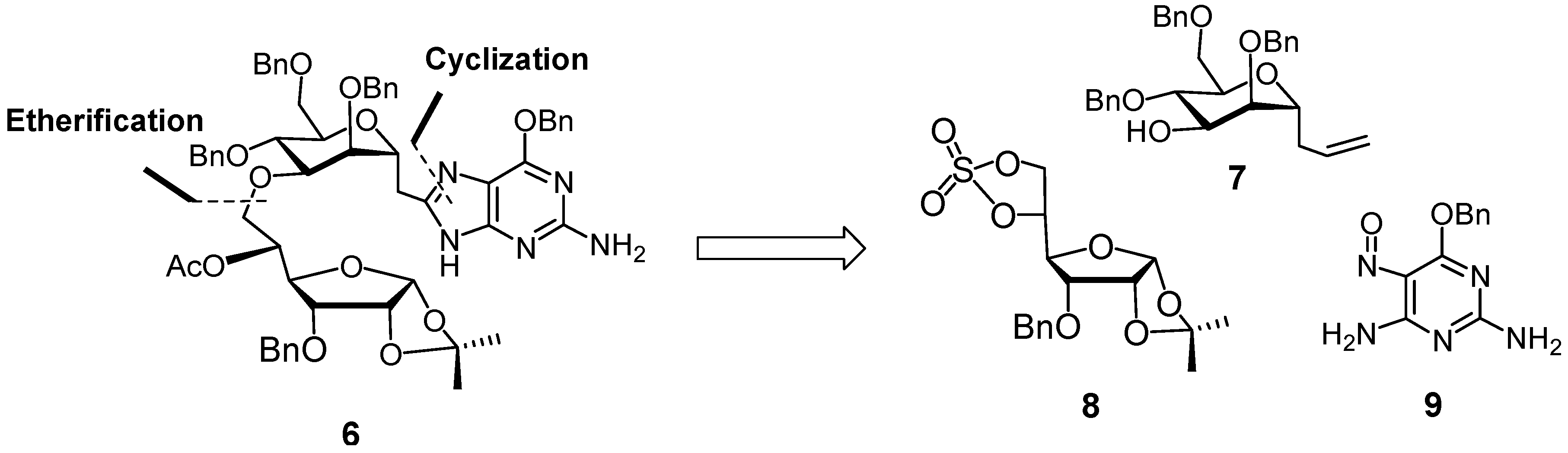
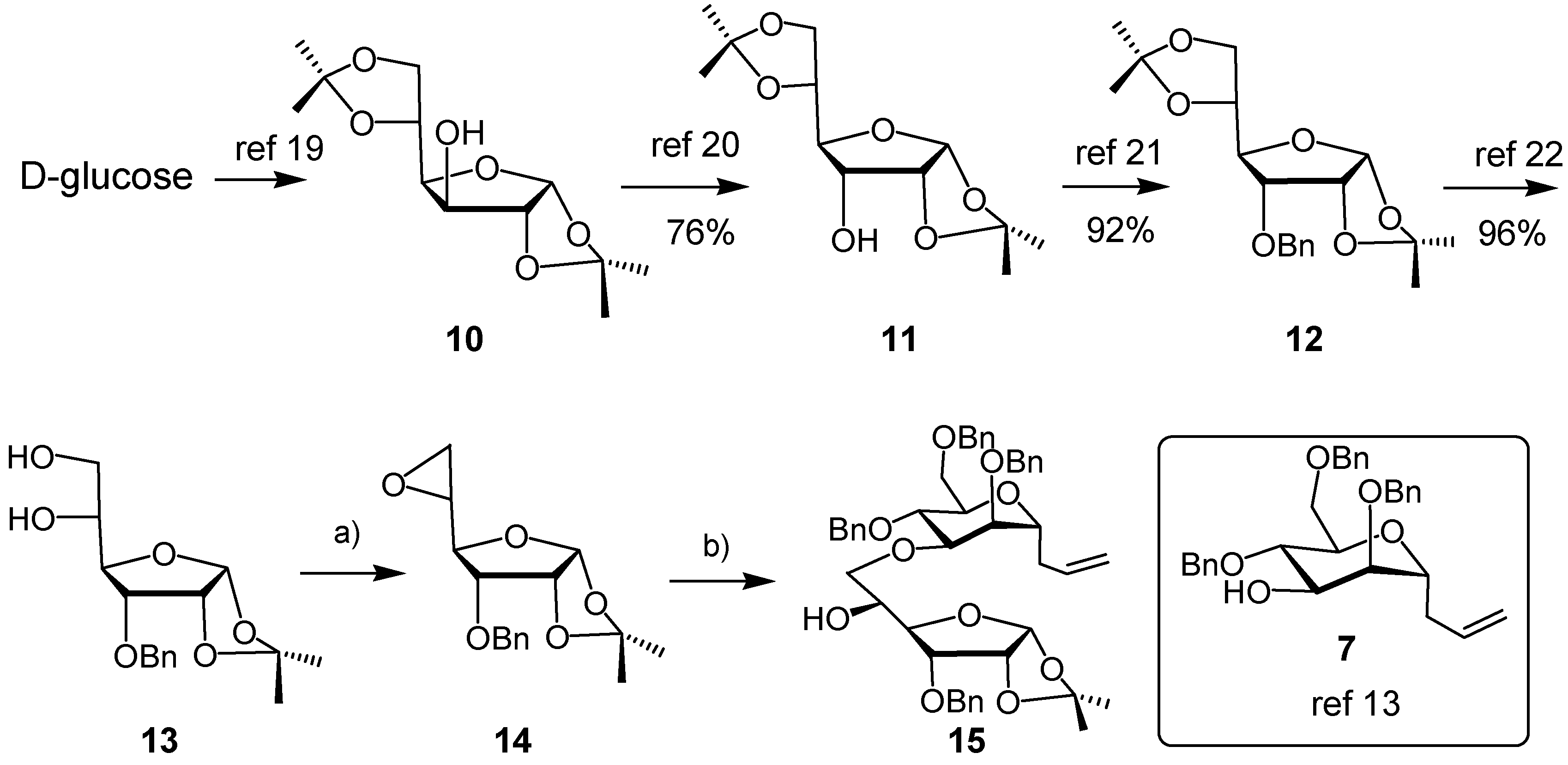
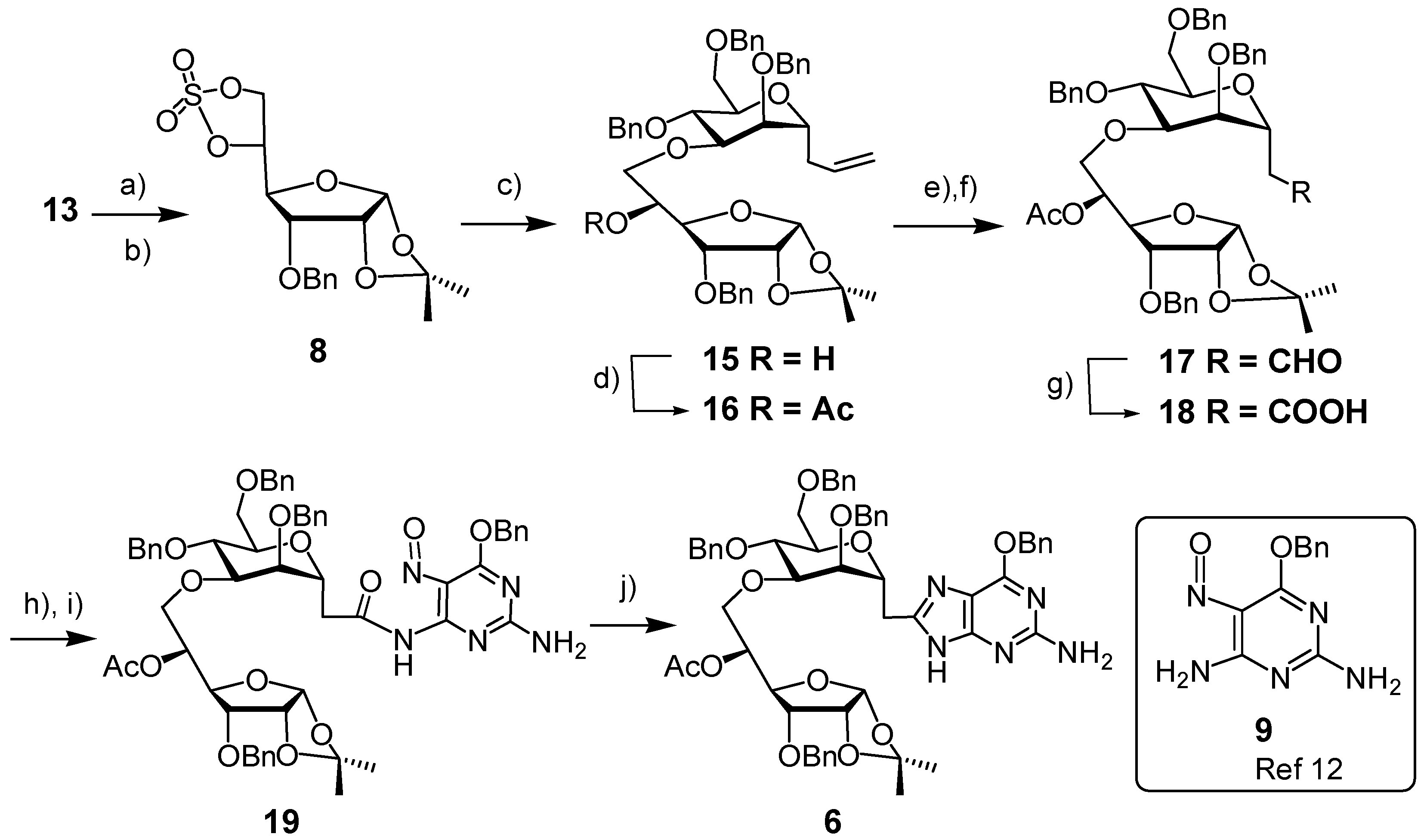
3. Experimental
General
Commercially available reagents were used without further purification. Water-free solvents were dried: THF was distilled from Na/benzophenone; toluene from Na; CH2Cl2, MeCN, MeOH, pyridine, and triethylamine from CaH2; acetone, and chloroform were dried over 4 Å molecular sieves. Technical solvents were distilled: AcOEt, CH2Cl2 from K2CO3; Et2O from FeSO4∙7 H2O; cyclohexane, hexane, MeOH, and toluene without any other additive. The reactions were carried out in oven-dried glassware, under an N2 or Ar atmosphere, unless stated otherwise. Qualitative TLC: precoated silica-gel plates (Merck silica gel 60 F254); detection by heating with ‘mostain’ (400 mL of 10% H2SO4 soln., 20 g of (NH4)6Mo7O24∙4H2O, 0.4 g of Ce(SO4)2∙4 H2O), or by UV. FCC (flash column chromatography): silica gel Fluka 60 (0.04–0.063 mm) or Merck silica gel 60 (0.063–0.200 mm) under slightly elevated pressure (0.1–0.4 bar). Melting points were measured on a Büchi B-540 melting point apparatus using open glass capillaries and are uncorrected. Optical rotations were measured with a PerkinElmer digital polarimeter: 1-dm cell at 25 °C, 589 nm, concentration (c) in g/100 mL. Infrared spectroscopy (IR) were recorded on a Perkin Elmer Spectrum RX-I FT-IR: ca. 2% soln. in CHCl3; absorptions in cm–1. NMR-spectra were recorded on Bruker magnetic resonance spectrometer (1H at 300 MHz, 13C at 75 MHz): chemical shifts δ in ppm relative to a residual undeuterated solvent peak. MS spectra were recorded on an IONSPEC Ultima ESI-FT-ICR spectrometer at 4.7 T.
3-O-Benzyl-1,2-O-isopropylidene-α-D-allofuranose, 5,6-cyclic sulfate (8). A solution of thionyl chloride (0.11 mL, 1.5 mmol) in CH2Cl2 (0.85 mL) was added dropwise to an ice-cooled solution of diol 13 (230 mg, 0.74 mmol) in CH2Cl2 (5 mL) and pyridine (0.24 mL, 3 mmol). The mixture was stirred for 5 min, when TLC revealed the disappearance of starting material. The mixture was diluted with CH2Cl2 and washed with water. The combined aqueous layers were extracted with CH2Cl2. The combined organic layers were dried (Na2SO4) and concentrated. The residue was dissolved in CH2Cl2/MeCN/H2O (2/2/3), to which was added NaIO4 (320 mg, 1.5 mmol) followed by Ru(II)Cl2∙xH2O (10 mg). After 10 min, the mixture was diluted with CH2Cl2, the organic layer was separated, the water layer was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The residue was purified by Flash Column Chromatography (FCC, EtOAc/cyclohexane, 1/3→1/1) to afford the cyclic sulfate 8 as a white solid (191 mg, 69%). m.p. 120–122 °C (dec.) (EtOAc/hexane). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 7.40–7.37 (m, 5 H), 5.74 (d, 1 H, J = 3.3 Hz), 5.17 (dt, 1 H, J = 2.4, 7.2 Hz), 4.79 (dd, 1 H, J = 7.8, 9.0 Hz), 4.75 (d, 1 H, J = 12.0 Hz), 4.62 (d, 1 H, J = 12.0 Hz), 4.60 (d, 1 H, J = 6.9 Hz), 4.57 (dd, 1 H, J = 1.2, 5.4 Hz), 4.23 (dd, 1 H, J = 2.4, 9.0 Hz), 3.99 (dd, 1 H, J = 4.5, 9.0 Hz), 1.59 (s, 3 H), 1.36 (s, 3 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 136.51, 128.53, 128.36, 113.53, 103.95, 79.99, 77.13, 77.05, 76.41, 72.59, 67.86, 26.90, 26.46. HR-MS(ESI), m/z 395.07695 [M+Na]+ (C16H20NaO8S+, required 395.07711).
5,6-Anhydro-3-O-benzyl1,2-O-isopropylidene-α-D-allofuranose (14). To a solution of diol 13 (305 mg, 0.98 mmol) and triphenylphosphine (310 mg, 1.18 mmol) in dry toluene (6 mL) was added diisopropyl azodicarboxylate (0.25 mL, 94% pure, 1.18 mmol) dropwise at room temperature. The mixture was stirred under reflux overnight. The solvent was removed in vacuo, the residue was purified by FCC (EtOAc/cyclohexane, 1/3) to give epoxide 14 as a colorless oil (189 mg, 66%). IR (Film, cm−1): 3021 (s), 2934 (w), 2870 (w), 1751 (w), 1455 (w), 1384 (w), 1375 (w), 1315 (w), 1254 (w), 1163 (w), 1131 (m), 1102 (s), 1022 (s). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 7.40–7.29 (m, 5 H), 5.75 (d, 1 H, J = 3.9 Hz), 4.75 (d, 1 H, J = 11.4 Hz), 4.59–4.55 (m, 2 H), 4.21 (dd, 1 H, J = 3.0, 9.0 Hz), 3.66 (dd, 1 H, J = 4.2, 8.7 Hz), 3.19 (dd, 1 H, J = 3.0, 7.2 Hz), 2.81–2.73 (m, 2 H), 1.59 (s, 3 H), 1.36 (s, 3 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 137.26, 128.57, 128.20, 128.16, 113.12, 104.21, 77.73, 77.57, 77.54, 72.02, 50.70, 44.45, 26.87, 26.60. [α]25 D +85.92° (c 1.05 in CHCl3).
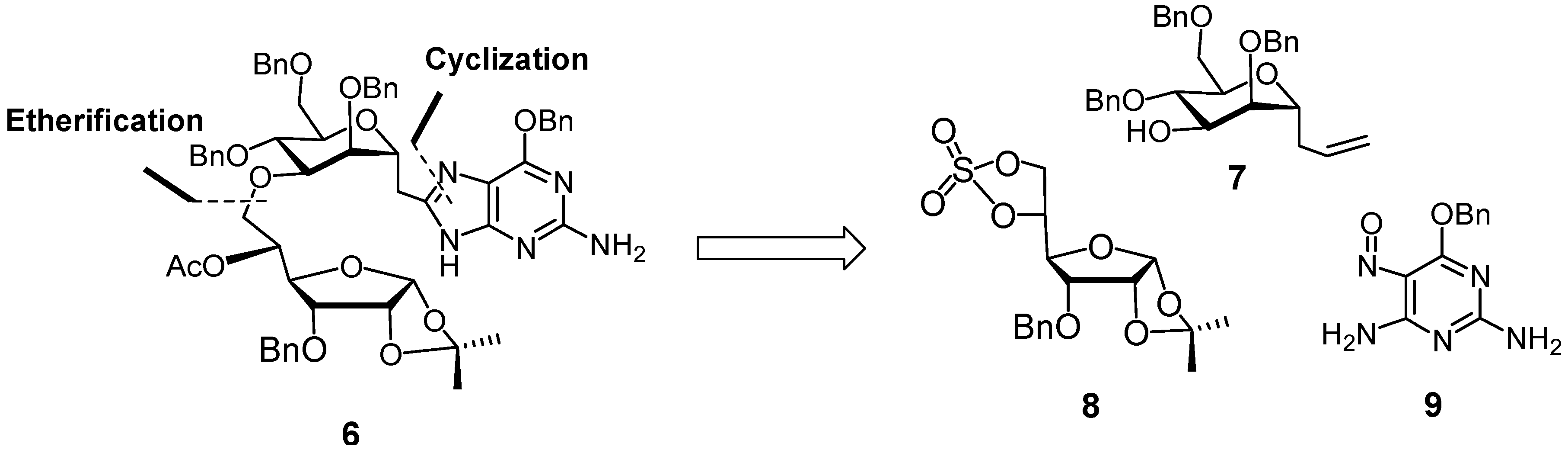
2,6-Anhydro-1,3,5-tri-O-benzyl-4-O-(3-O-benzyl-1,2-O-isopropylidene-6-deoxy-α-d-allofuranos-6-yl)-7,8,9-trideoxy-d-glycero-d-mannonon-8-enitol (15). To a mixture of NaH (60% in oil, 93 mg, 2.32 mmol) in HMPA (3 mL) and THF (1 mL) was added alcohol 7 (1.10 g, 2.32 mmol) in THF (15 mL). The resulting slightly yellow solution was stirred at room temperature for 20 min, and then treated with cyclic sulfate 8 (720 mg, 1.93 mmol) in THF (5 mL). The resulting mixture was stirred at room temperature overnight when TLC revealed the disappearance of the cyclic sulfate. The reaction mixture was treated with H2SO4/THF/H2O (1/100/0.3, 3 mL), stirred for 1 h, treated with saturated aqueous NaHCO3 solution, and extracted with EtOAc. The combined organic layers were dried (MgSO4), concentrated, and the residue purified by FCC (EtOAc/cyclohexane, 1/2→1/1) to afford 15 as a colorless oil (1.28 g, 86%). IR (Film, cm−1): 3475 (w), 3067 (w), 3032 (w), 3009 (m), 2928 (m), 2872 (w), 1732 (w), 1672 (w), 1496 (w), 1454 (w), 1374 (w), 1313 (w), 1224 (m), 1094 (s), 1027 (s). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 7.34–7.23 (m, 20 H), 5.84–5.70 (m, 1 H), 5.72 (d, 1 H, J = 3.3 Hz), 5.08–5.01 (m, 2 H), 5.72–4.48 (m, 9 H), 4.05–3.91 (m, 4 H), 3.86–3.48 (m, 8 H), 2.88 (d, 1 H, J = 2.4 Hz), 2.38–2.25 (m, 2 H), 1.57 (s, 3 H), 1.35 (s, 3 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 138.22, 137.94, 137.90, 137.40, 134.16, 128.29, 128.19, 127.94, 127.87, 127.64, 127.39, 117.17, 112.86, 104.03, 78.66, 78.13, 77.71, 77.60, 75.14, 74.94, 73.86, 73.55, 73.29, 72.28, 72.13, 71.81, 71.10, 70.29, 68.96, 34.61, 26.93, 26.67. ([α]25 D +45.45° (c 1.00 in CHCl3). HR-MS (ESI), m/z 789.36095 [M+Na]+ (C46H54NaO10+, required 789.36092).
2,6-Anhydro-1,3,5-tri-O-benzyl-4-O-(5-O-acetyl-3-O-benzyl-1,2-O-isopropylidene-6-deoxy-α-d-allofuranos-6-yl)-7,8,9-trideoxy-d-glycero-d-mannonon-8-enitol (16). Triethylamine (0.32 mL, 2.29 mmol) and DMAP (61 mg, 0.50 mmol) in dry CH2Cl2 (2 mL), were added dropwise at 0 °C to a solution of alcohol 15 (0.88 g, 1.15 mmol) and acetic anhydride (0.21 mL, 2.29 mmol) in dry CH2Cl2 (3 mL). The resulting mixture was stirred at room temperature overnight. The reaction was quenched by adding water (1 mL), and the solvent was removed in vacuo. A solution of the residue in EtOAc (20 mL) was washed with water and 0.1 N HCl. The aqueous layer was extracted with EtOAc, the combined organic layers were dried (MgSO4) and concentrated to give the acetate 16 as a slightly yellow oil (0.94 g, 100%). IR (cm−1, CHCl3): 3067 (w), 3032 (w), 3010 (m), 2928 (w), 1738 (s), 1642 (w), 1496 (w), 1374 (m), 1374 (m), 1307 (w), 1240 (s), 1093 (s), 1027 (s). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 7.34–7.26 (m, 20 H), 5.85–5.73 (m, 1 H), 5.64 (d, 1 H, J = 3.7 Hz), 5.31 (ddd, 1 H, J = 4.8, 3.0, 6.6 Hz), 5.11–5.04 (m, 2 H), 4.73 (d, 1 H, J = 11.3 Hz), 4.67 (d, 1 H, J = 11.2 Hz), 4.61–4.40 (m, 7 H), 4.12 (dd, 1 H, J = 5.3, 8.6 Hz), 4.05–3.98 (m, 1 H), 3.90 (dd, 1 H, J = 4.3, 8.9 Hz), 3.83–3.62 (m, 8 H), 2.41–2.05 (m, 2 H), 1.85 (s, 3 H), 1.53 (s, 3 H), 1.31 (s, 3 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 170.27 (CO), 138.57, 138.45, 138.37, 137.50, 128.40, 128.38, 128.36, 128.29, 128.14, 127.99, 127.83, 127.74, 127.63, 127.58, 127.43, 117.27, 113.08, 104.17, 79.81, 78.99, 77.20, 76.46, 75.53, 75.22, 73.93, 73.57, 73.97, 73.57, 73.28, 72.84, 72.21, 71.92, 71.75, 69.28, 68.92, 34.43, 26.80, 26.60, 21.00. [α]25 D +43.73° (c 1.65 in CHCl3). HR-MS (ESI), m/z, 831.37633 [M+Na]+ (C48H56NaO11+, requires 831.37148).
2-[3-O-(5-O-Acetyl-3-O-benzyl-1,2-O-isopropylidene-6-deoxy-α-d-allofuranos-6-yl)-2,4,6-tri-O-benzyl-α-D-mannopyranosyl]acetaldehyde (17). To a mixture of alkene 16 (0.850 g, 1.05 mmol) and N-methylmorpholine N-oxide (NMO) (213 mg, 1.58 mmol) in acetone (6 mL) and water (2 mL) was added osmium tetroxide (0.2 w% in water, 2.6 mL, 0.021 mmol, 0.02 eq.) at 0 °C. The resulting mixture was stirred for 45 h when TLC revealed the disappearance of the alkene 16. Upon addition of sodium sulfite (1.12 g) the yellow suspension turned into a slightly yellow two-layered solution. The upper organic layer was separated, the aqueous layer was extracted with EtOAc, the combined organic layers were dried (MgSO4) and concentrated to give the crude diol (802 mg, 91%) as a mixture of two epimers in a ratio of ca. 0.75:1 (based on the integrals in the 1H-NMR spectrum). IR (cm−1, CHCl3): 3473 (w), 3031 (vw), 2931 (w), 2871 (w), 1737 (m), 1496 (w), 1454 (m), 1372 (m), 1236 (s), 1164 (w), 1090 (s), 1070 (s), 1024 (s). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 7.32–7.24 (m, 32.3 H), 5.63 (d, 0.6 H, J = 3.0 Hz), 5.60 (d, 0.8 H, J = 3.3 Hz), 5.29–5.23 (m, 1.5 H), 5.67–4.39 (m, 13.9 H), 4.17–4.06 (m, 4.0 H), 3.95–3.78 (m, 7.2 H), 3.72–3.42 (m, 13.9 H), 4.17–4.06 (m, 4.0 H), 3.95–3.78 (m, 7.2 H), 3.72–3.42 (m, 13.9 H), 2.04 (d, 1.2 H, J = 0.9 Hz), 1.84–1.83 (m, 5.1 H), 1.78–1.51 (m, 2.9 H), 1.52 (s, 5.3 H), 1.31 (br s, 5.2 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 170.29, 138.17, 138.10, 138.07, 138.00, 137.98, 137.49, 137.46, 128.46, 128.42, 128.16, 128.14, 128.02, 127.94, 127.90, 127.83, 127.80, 127.74, 127.70, 127.66, 133.11, 133.09, 104.15, 79.84, 77.20, 76.79, 76.67, 76.40, 76.31, 75.25, 73.61, 73.45, 73.42, 73.18, 71.93, 71.80, 69.47, 69.39, 66.38, 32.81, 32.69, 26.82, 26.60, 21.01. HR-MS(ESI), m/z, 865.37635 [M+Na]+ (C48H58NaO13+, required 865.37696).
To the diol (630 mg, 0.747 mmol) in MeOH (3 mL) and H2O (5 mL) was added sodium periodate (190 mg, 0.897 mmol) in H2O (3 mL) at 0 °C, the mixture was stirred for 60 min, and then extracted with EtOAc. The combined organic layers were dried (Mg2SO4) and concentrated to give the aldehyde 17 as a slightly yellow oil which was pure according to the NMR spectrum. IR (cm−1, CHCl3): 3030 (vw), 2933 (w), 2870 (w), 1739 (m), 1726 (m), 1496 (w), 1454 (w), 1371 (m), 1307 (m), 1234 (s), 1091 (s), 1071 (s), 1025 (s). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 9.68 (t, 1 H, J = 2.3 Hz), 7.36–7.21 (m, 20 H), 5.63 (d, 1 H, J = 3.7 Hz), 5.28 (ddd, 1 H, J = 4.6, 3.4, 6.4 Hz), 4.74–4.42 (m, 10 H), 4.13 (dd, 1 H, J = 5.2, 8.9 Hz), 3.96–3.61 (m, 9 H), 2.62 (dd, 1 H, J = 1.1, 2.0 Hz), 2.60 (t, 1 H, J = 2.3 Hz), 1.87 (s, 3 H), 1.55 (s, 3 H), 1.34 (s, 3 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 200.50, 170.21, 138.40, 138.07, 137.92, 137.79, 137.54, 129.09, 128.50, 128.48, 128.44, 128.38, 128.11, 128.08, 128.03, 127.90, 127.87, 127.82, 127.79, 127.57, 125.36, 113.11, 104.17, 79.80, 77.21, 76.52, 76.43, 76.00, 74.91, 74.43, 73.24, 72.85, 72.18, 71.80, 71.49, 69.65, 68.36, 66.57, 45.32, 26.53, 26.62, 21.52, 21.00. [α]25 D +56.27° (c 2.00 in CHCl3). HR-MS(ESI), m/z, 833.35133 [M+Na]+ (C47H54NaO12+, required 833.35075).
2-[3-O-(5-O-Acetyl-3-O-benzyl-1,2-O-isopropylidene-6-deoxy-α-d-allofuranos-6-yl)-2,4,6-tri-O-benzyl-α-D-mannopyranosyl]acetic acid (18). The crude aldehyde 17 in acetonitrile (6 mL) was treated with NaH2PO4 (34 mg, 0.25 mmol) in H2O (2 mL) and H2O2 (30%, 0.12 mL, 1.12 mmol), respectively, at 0 °C, followed by addition NaClO2 (0.17 g, 80%, 1.49 mmol) in H2O (2 mL) at 0 °C. The resulting mixture was stirred overnight, brought to pH 2.0 with 1N HCl, and extracted with EtOAc. The combined organic layers were dried (MgSO4) and concentrated to give acid 18 as a colorless gum (0.60 g, 88% from 16). IR(cm−1, CHCl3): 3062 (w), 3030 (w), 2933 (w), 2872 (w), 1739 (s), 1714 (m), 1496 (w), 1454 (w), 1372 (m), 1306 (w), 1236 (s), 1162 (m), 1095 (s), 1026 (s). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 7.33–7.25 (m, 20 H), 5.61 (d, 1 H, J = 3.6 Hz), 5.23 (dd, 1 H, J = 5.3, 10.2 Hz), 4.68–4.36 (m, 10 H), 4.34–4.30 (m, 1 H), 4.10 (dd, 1 H, J = 5.3, 8.9 Hz), 3.97 (br. s, 1 H), 3.89–3.78 (m, 3 H), 3.73 (d, 1 H , J = 2.2 Hz), 3.64–3.56 (m, 3 H), 2.70 (dd, 1 H, J = 4.3, 15.7 Hz), 2.56 (dd, 1 H, J = 8.6, 15.9 Hz), 1.84 (s, 3 H), 1.52 (s, 3 H), 1.31 (s, 3 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 175.43, 170.35, 138.31, 138.06, 137.88, 137.50, 128.47, 128.43, 128.36, 128.17, 128.04, 127.97, 127.89, 127.86, 127.81, 127.56, 133.13, 104.17, 79.80, 77.20, 76.86, 76.45, 75.66, 74.84, 74.47, 73.33, 73.05, 72.24, 71.84, 71.50, 69.45, 68.40, 68.16, 36.25, 26.98, 26.81, 26.61, 20.98. [α]25 D +50.97° (c 4.52 in CHCl3). HR-MS(ESI), m/z, 849.34625 [M+Na]+ (C47H54NaO13+, requires 849.34621), 871.32809 [M−H+Na2]+ (C47H534Na2O13+, requires 871.32816).
2-[3-O-(5-O-Acetyl-3-O-benzyl-1,2-O-isopropylidene-6-deoxy-α-d-allofuranos-6-yl)-2,4,6-tri-O-benzyl-α-D-mannopyranosyl]-N-[2-amino-6-(benzyloxy)-5-nitrosopyrimidin-4-yl]acetamide (19). To a solution of acid 18 (0.470 g, 0.57 mmol) in CH2Cl2 (4 mL) was added oxalyl chloride (0.15 mL, 1.7 mmol) followed by a drop of DMF at 0 °C. The resulting mixture was stirred for 1 h and concentrated in vacuo. The crude acid chloride was pure according to its NMR spectra and used directly for next step. IR (cm−1, CHCl3): 3031 (vw), 2932 (vw), 2868 (vw), 1798 (m), 1740 (s), 1496 (w), 1454 (m), 1371 (m), 1234 (s), 1091 (s), 1069 (s), 1023 (s). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 7.36–7.24 (m, 20 H,), 5.61 (d, 1 H, J = 3.7 Hz), 5.23 (dd, 1 H, J = 6.0, 10.0 Hz), 4.95 (d, 1 H, J = 11.0 Hz), 4.73–4.31 (m, 9 H), 4.10 (dd, 1 H, J = 5.4, 8.9 Hz), 3.97 (t, 1 H, J = 5.8 Hz), 3.86 (dd, 1 H, J = 4.3, 8.8 Hz), 3.81–3.67 (m, 4 H), 3.64–3.57 (m, 3 H), 3.21–3.14 (m, 1 H), 2.99–2.90 (m, 1 H), 1.84 (s, 3 H), 1.52 (s, 3 H), 1.31 (s, 3 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 171.27, 170.22, 138.39, 137.93, 137.54, 137.48, 128.55, 128.48, 128.34, 128.16, 128.02, 127.99, 127.84, 127.77, 127.52, 113.12, 104.13, 79.89, 77.32, 77.23, 76.30, 75.93, 75.25, 74.78, 74.64, 73.34, 72.56, 72.23, 71.74, 71.21, 69.72, 68.25, 67.23, 49.09, 26.82, 26.61, 20.95, 13.97. [α]25 D +43.68° (c 2.35 in CHCl3).
To a solution of 2,4-diamino-5-nitrosopyrimidine 9 (0.21 g, 0.86 mmol) and pyridine (0.07 mL) in dry THF (12 mL) was added the above acid chloride (0.47 g, 0.57 mmol) in THF (7 mL) at 0 °C dropwise. Once the acid chloride was added, the deep-blue solution turned green. After stirring for 1 h, the purple solid was filtered off, and the filtrate was concentrated in vacuo to afford the amide 19 (600 mg). IR (cm−1, CHCl3): 3328 (w), 3228 (w), 3031 (w), 2933 (w), 2870 (w), 1739 (m), 1631 (m), 1598 (s), 1535 (s), 1497 (m), 1454 (s), 1371 (m), 1347 (s), 1255 (s), 1209 (s), 1091 (s), 1072 (s), 1026 (s). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 12.38 (br. s, 1 H, exchange with D2O), 7.51–7.23 (m, 20 H), 7.17 (br. s, 1 H, exchange with D2O), 6.03 (br. s, 1 H, exchange with D2O), 5.65 (d, 1 H, J = 3.7 Hz), 5.63 (s, 2 H), 5.27 (dd, 1 H, J = 5.8, 10.0 Hz), 4.70 (d, 1 H, J = 11.0 Hz), 4.64 (d, 1 H, J = 11.6 Hz), 4.63 (d, 1 H, J = 11.8 Hz), 4.57–4.43 (m, 6 H), 4.14 (dd, 1 H, J = 5.5, 8.9 Hz), 4.06 (br. s, 1 H), 3.88 (dd, 1 H, J = 4.3, 8.9 Hz), 3.83–3.65 (m, 7 H), 2.97 (dd, 1 H, J = 5.3, 14.7 Hz), 2.89 (dd, 1 H, J = 7.6, 14.9 Hz), 1.83 (s, 3 H), 1.51 (s, 3 H), 1.32 (s, 3 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 171.45, 170.35, 163.78, 138.91, 138.31, 138.04, 137.86, 137.46, 135.49, 128.64, 128.46, 128.44, 128.32, 128.26, 128.20, 128.06, 127.95, 127.80, 127.72, 127.51, 113.12, 104.15, 79.92, 77.33, 77.24, 76.20, 76.05, 74.74, 74.12, 73.13, 72.25, 71.91, 71.62, 69.40, 68.72, 68.62, 41.33, 26.72, 26.61, 20.97. [α]25 D +81.92° (c 0.053 in CHCl3). HR-MS (ESI), m/z, 1076.4270 [M+Na]+ (C58H63N5NaO14+, required 1076.4264).
8-[(3-O-((5-O-Acetyl-3-O-benzyl-1,2-O-isopropylidene-6-deoxy-α-d-allofuranos-6-yl)-2,4,6-tri-O-benzyl-α-D-mannopyranosyl)methyl]-O6-benzylguanine (6). A mixture of the amide 19 (600 mg, 0.57 mmol) and triphenyl phosphine (449 mg, 1.71 mmol) in xylene was stirred at 140 °C overnight and then cooled to room temperature. The mixture was passed through silica gel (EtOAc/cyclohexane 1/1→1/0) to give guanine 6 as a yellowish oil (291 mg, 50% from acid 18). IR (cm−1, CHCl3): 3528 (w), 3420 (w), 3067 (w), 3010 (m), 2928 (m), 1738 (m), 1625 (s), 1591 (s), 1496 (w), 1455 (m), 1374 (m), 1325 (m), 1227 (s), 1146 (m), 1090 (s). 1H-NMR (CDCl3, 300 MHz): δ (ppm) 11.65 (br s, 1 H, NH, exchange with D2O), 7.50–7.20 (m, 25 H), 5.57 (d, 1 H, J = 3.7 Hz), 5.53 (s, 2 H), 5.18 (dd, 1 H, J = 5.0, 10.7 Hz), 4.80 (br. s, 2 H, exchange with D2O), 4.71–4.35 (m, 10 H), 4.21 (t, 1 H, J = 8.1 Hz), 4.08 (dd, 2 H, J = 5.0, 8.9 Hz), 3.86–3.70 (m, 3 H), 3.61 (dd, 1 H, J = 2.6, 7.7 Hz), 3.58–3.53 (m, 2 H), 3.47 (dd, 1 H, J = 4.0, 10.2 Hz), 3.30 (d, 1 H, J = 14.0 Hz), 2.97 (dd, 1 H, J = 10.8, 16.0 Hz), 1.84 (s, 3 H), 1.49 (s, 3 H), 1.30 (s, 3 H). 13C-NMR (CDCl3, 75 MHz): δ (ppm) 170.14, 160.10, 158.72, 155.66, 148.61, 137.74, 137.65, 137.42, 136.79, 132.20, 132.07, 128.61, 128.56, 128.49, 128.46, 128.40, 128.35, 128.32, 128.22, 128.04, 127.98, 127.92, 127.82, 127.69, 114.66, 13.06, 104.16, 79.68, 77.26, 77.11, 76.47, 75.95, 75.27, 74.37 (CH2), 73.14, 72.70, 72.15, 71.79, 69.86, 67.79, 26.95, 26.78, 26.55, 20.98. [α]25 D +31.76° (c 1.41 in CHCl3). HR-MS (ESI), m/z, 1044.4378 [M+Na]+ (C58H63N5NaO12+, required 1044.4365).

{kind=link}
{kind=link}
{kind=link}
{kind=link}