Deuterium Isotope Effects on 13C-NMR Chemical Shifts of 10-Hydroxybenzo[h]quinolines


Abstract
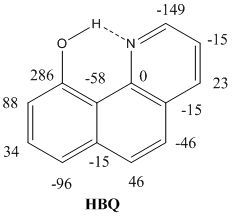
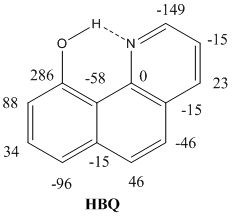
:
1. Introduction
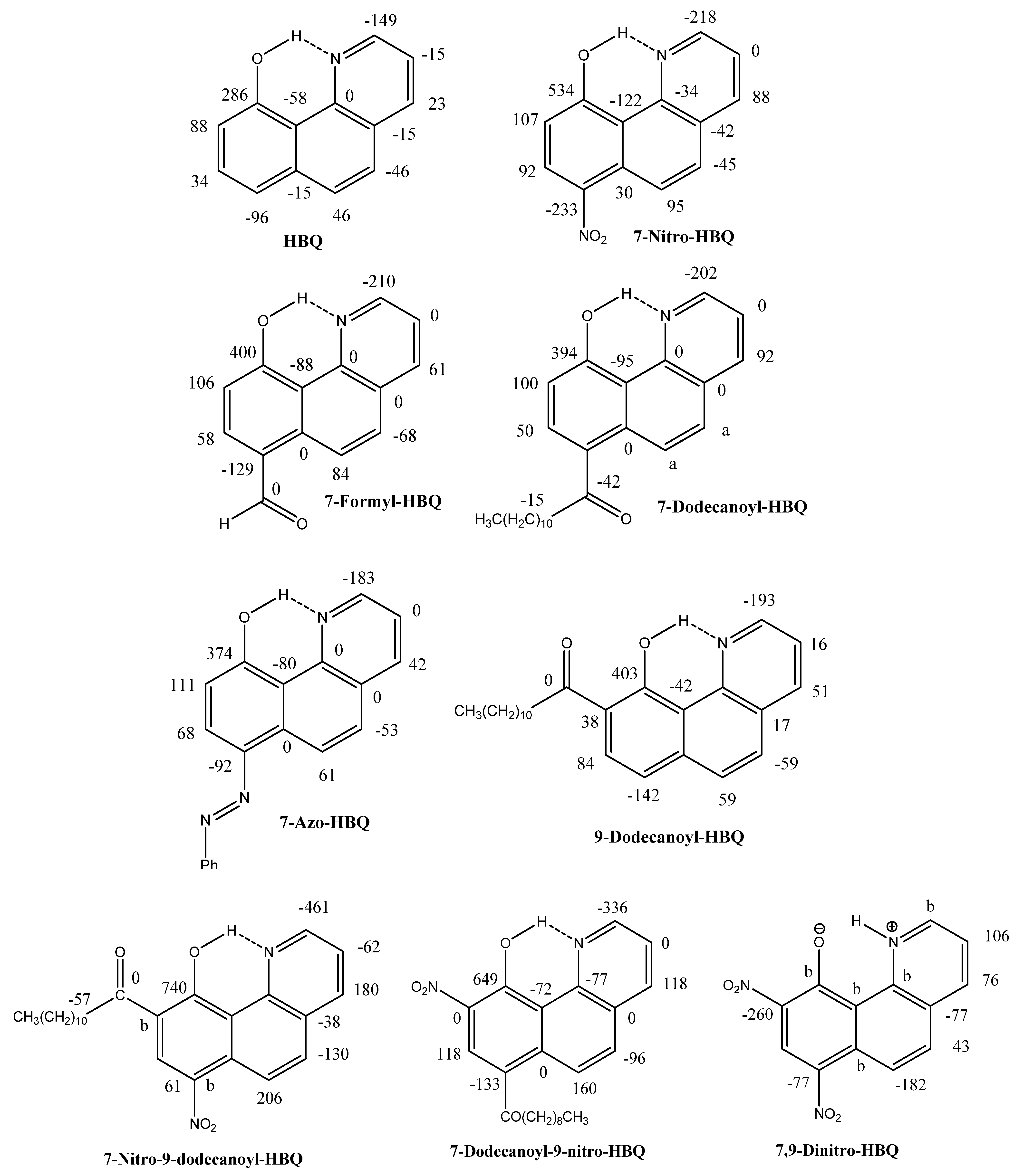
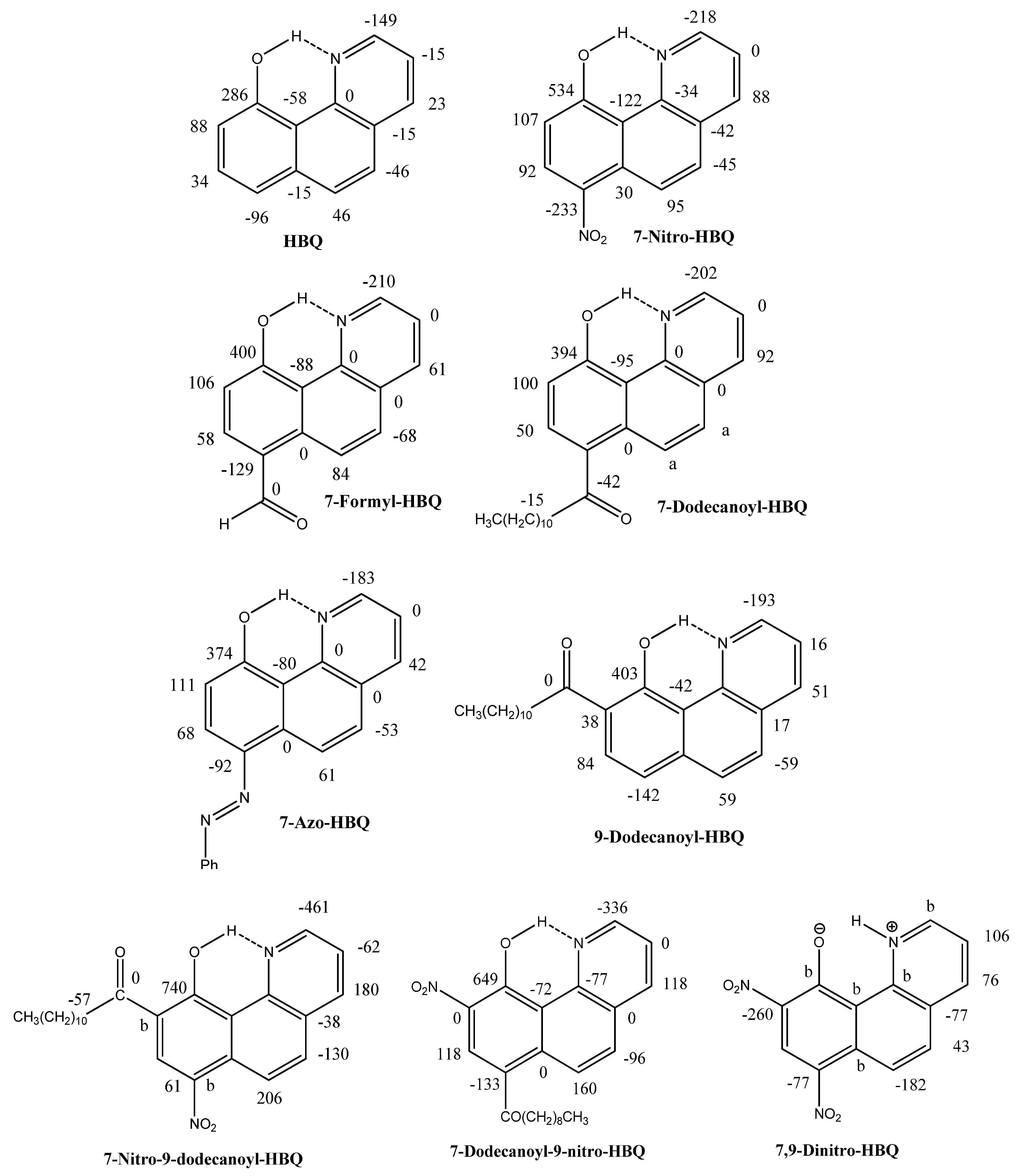
2.1.Assignments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | HBQ | 4-mor-pho-lino-HBQ | 4-tosyl-HBQ a | 7-formyl-HBQ | 7-azo-HBQ b | 7-nitro-HBQ c | 7-dode-canoyl-HBQ | 9-dode-canoyl-HBQ | 7-dode-canoyl-9-nitro-HBQ | 7-nitro-9-dode-canoyl-HBQ | 7,9-dinitro-HBQ (NH-form) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C-2 | 144.8 | 145.5 | 144.7 | 145.5 | 145.4 | 147.8 | 145.0 | 145.0 | 145.6 | 145.3 | 131.9 d |
| C-3 | 120.7 | 109.4 | 120.0 | 121.7 | 121.3 | 124.0 | 121.4 | 121.2 | 122.5 | 122.5 | 124.6 |
| C-4 | 136.0 | 156.9 | 149.9 | 136.9 | 136.6 | 138.9 | 132.9 | 136.9 | 138.0 | 138 | 145.7 |
| C-4a | 126.1 | 119.9 | 121.7 | 126.1 | 126.4 | 126.7 | 126.7 | 126.5 | 126.1 | 126.4 | 127.3 d |
| C-5 | 124.6 | 120.3 | 119.2 | 128.1 | 124.4 | 130.3 | 126.0 | 126.8 | 123.9 | 128.7 | 132.5 e |
| C-6 | 128.9 | 129.8 | 131.9 | 125.4 | 125.6 | 130.2 | 127.0 | 129.0 | 130.0 | 127.0 | 127.9 e |
| C-6a | 134.9 | 134.8 | 134.4 | 134.4 | 134.1 | 129.2 | 133.9 | 137.8 | 137.3 | 132.7 | 134.3 |
| C-7 | 118.0 | 117.7 | 118.6 | 122.4 | 139.8 | 138.0 | 125.8 | 117.8 | 116.1 | 136.4 | 132.2 f |
| C-8 | 129.8 | 127.6 | 131.0 | 140.4 | 117.3 | 123.5 | 136.4 | 130.5 | 130.0 | 129.8 | 125.2 |
| C-9 | 113.8 | 113.8 | 115.2 | 113.8 | 114.7 | 113.9 | 112.8 | 123.4 | 130.4 | 124.3 | 143.3 |
| C-10 | 159.3 | 159.6 | 159.1 | 165.8 | 165.4 | 166.4 | 163.5 | 161.2 | 166.1 | 160.6 | 167.2 d |
| C-10a | 115.8 | 116.6 | 115.5 | 115.6 | 115.2 | 115.5 | 116.0 | 116.2 | 122.3 | 117.2 | 112 f |
| C-10b | 148.1 | 149.4 | 145.2 | 147.4 | 148.1 | 147.2 | 147.7 | 148.6 | 147.2 | 147.5 | 144.3 d |
| C=O | - | - | - | 192.1 | 203.2 | 202.6 | 200.2 | 201.9 | - |
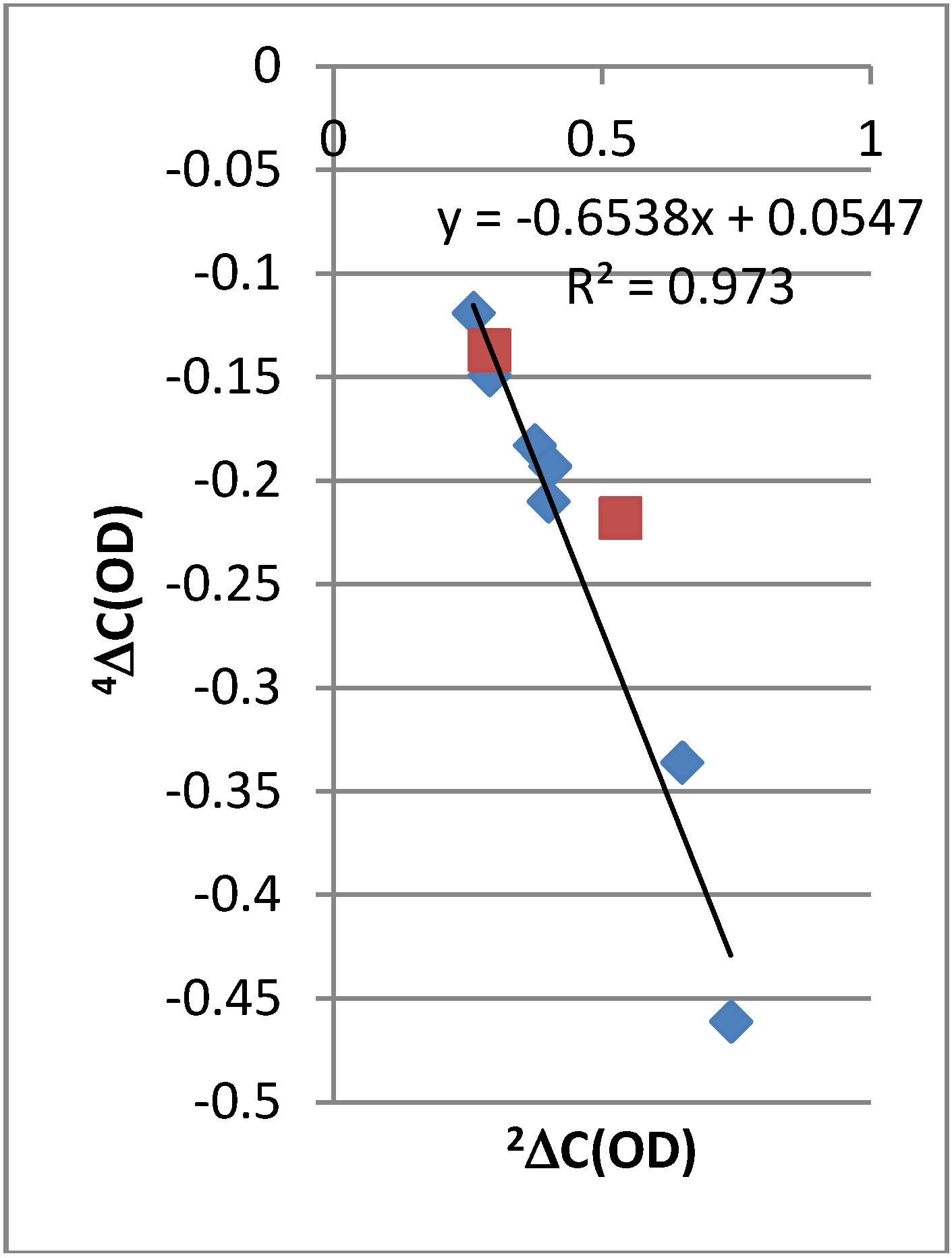
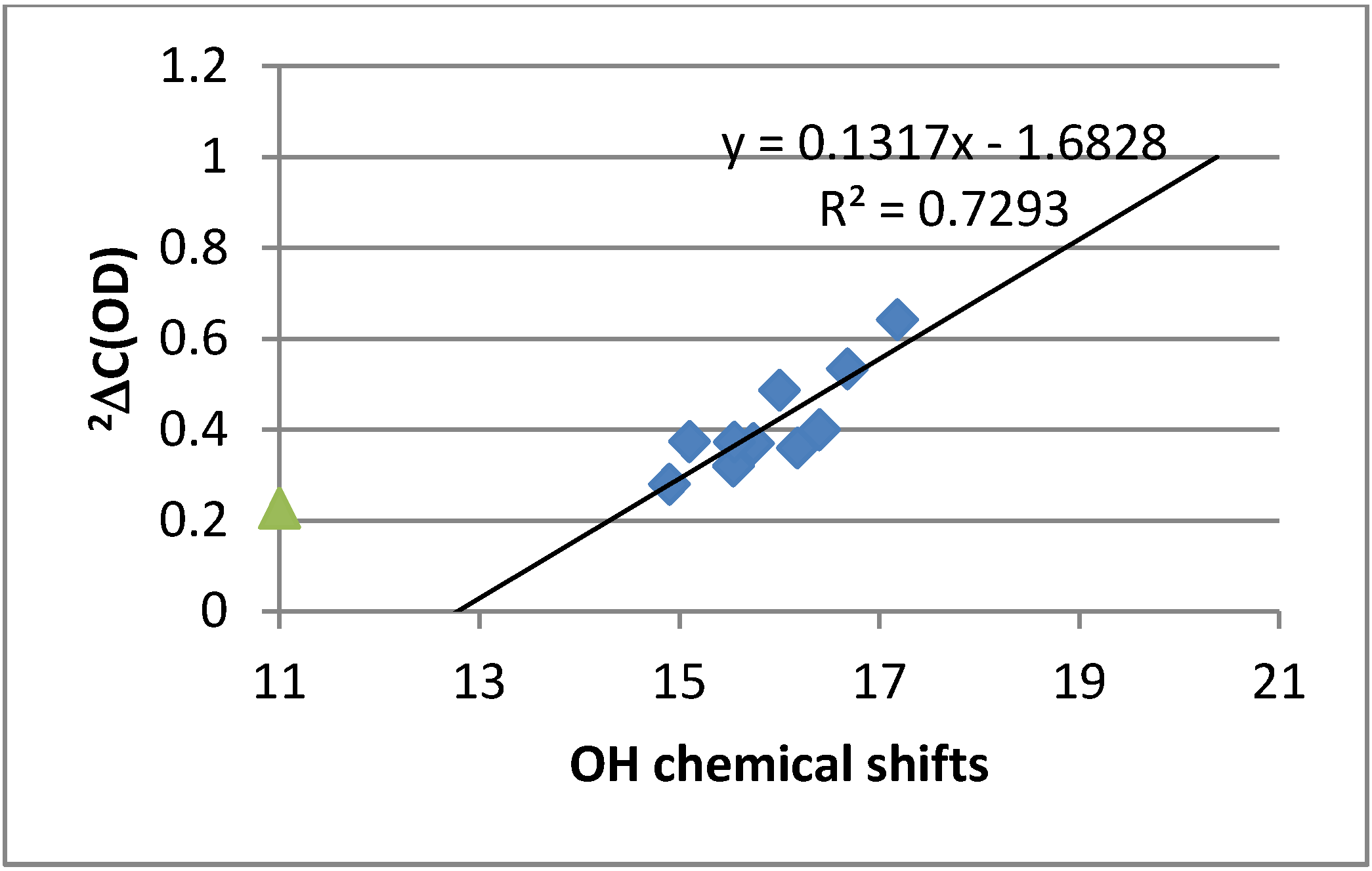
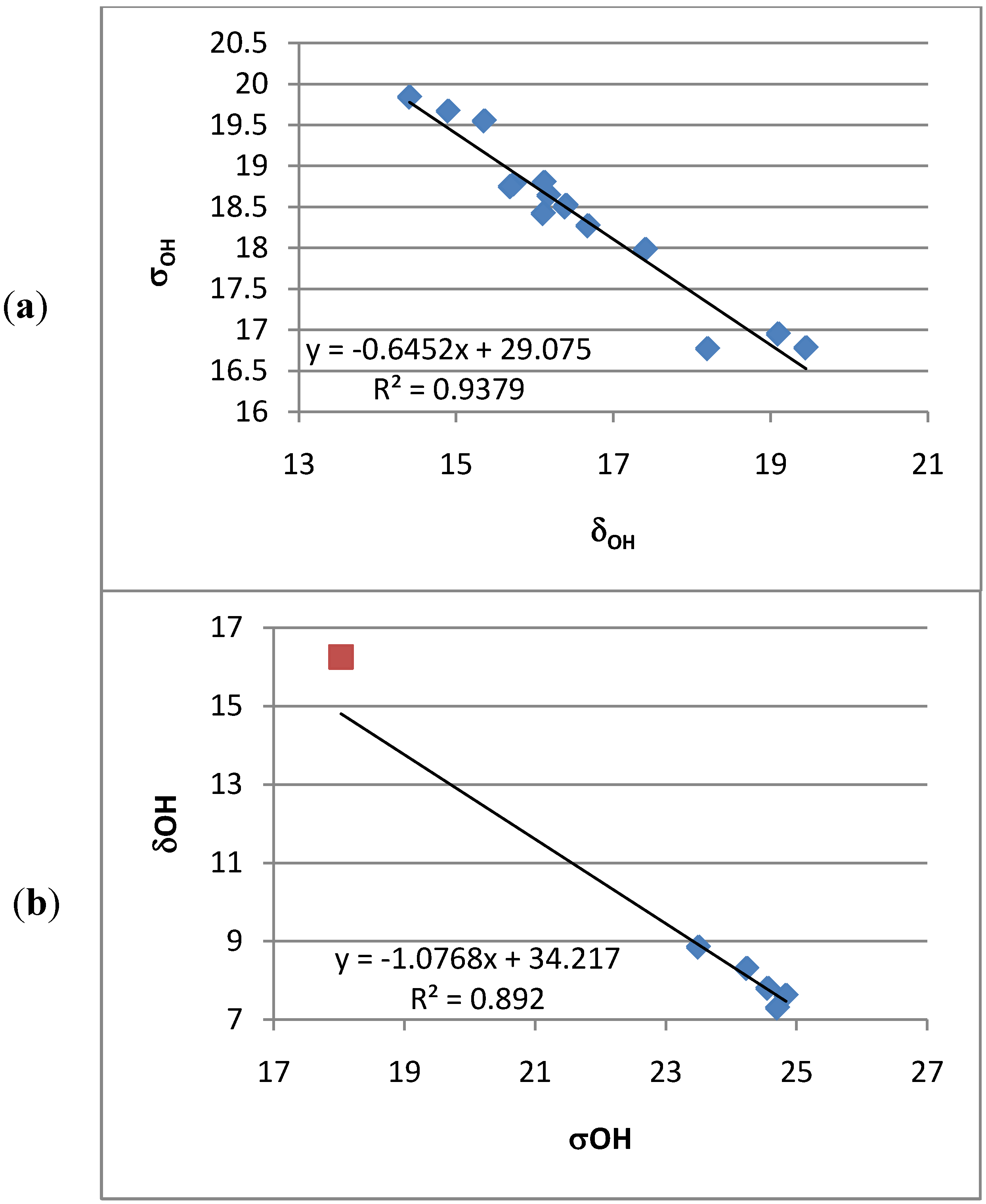
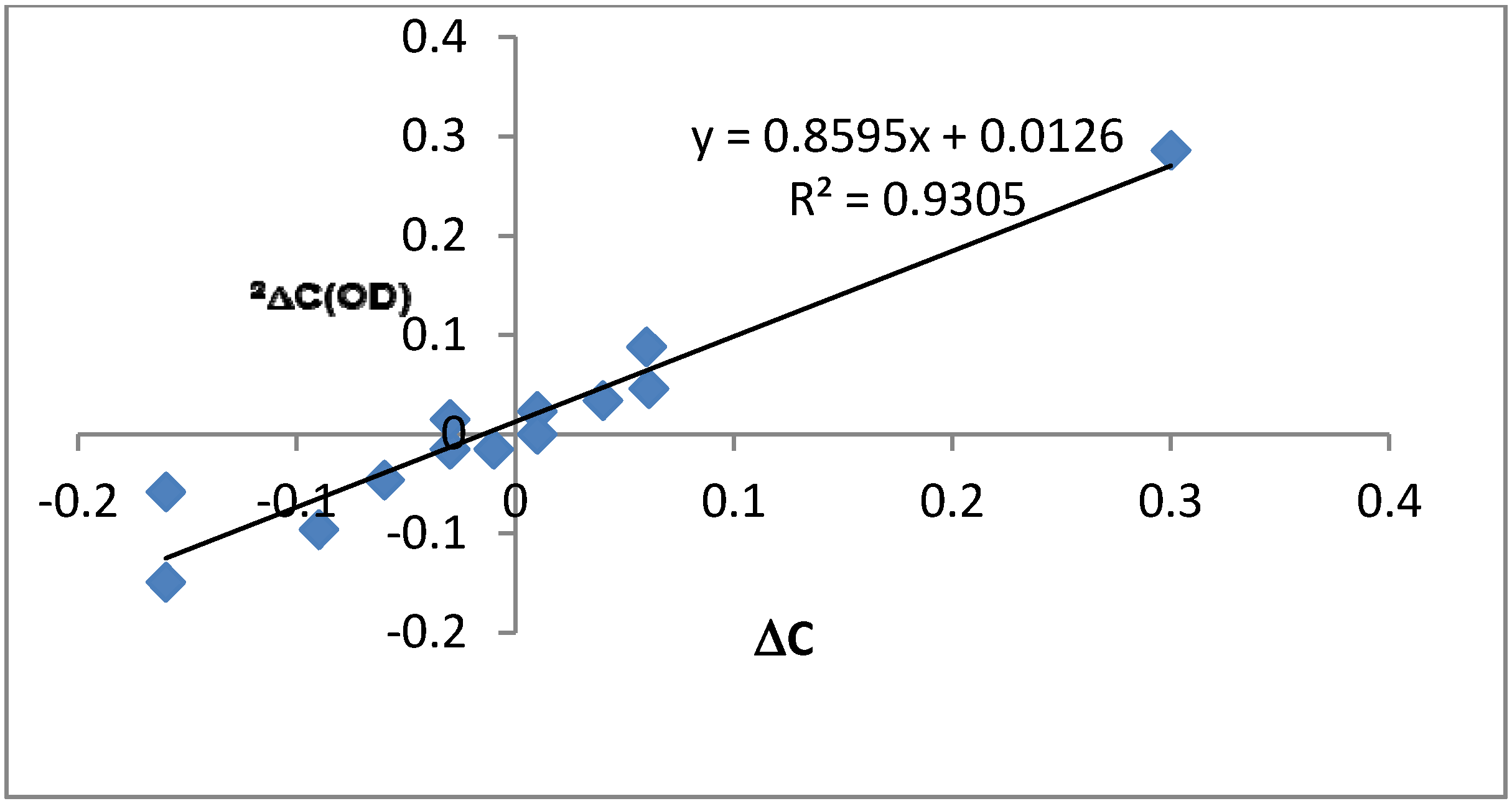
2.2. Deuterium Isotope Effects
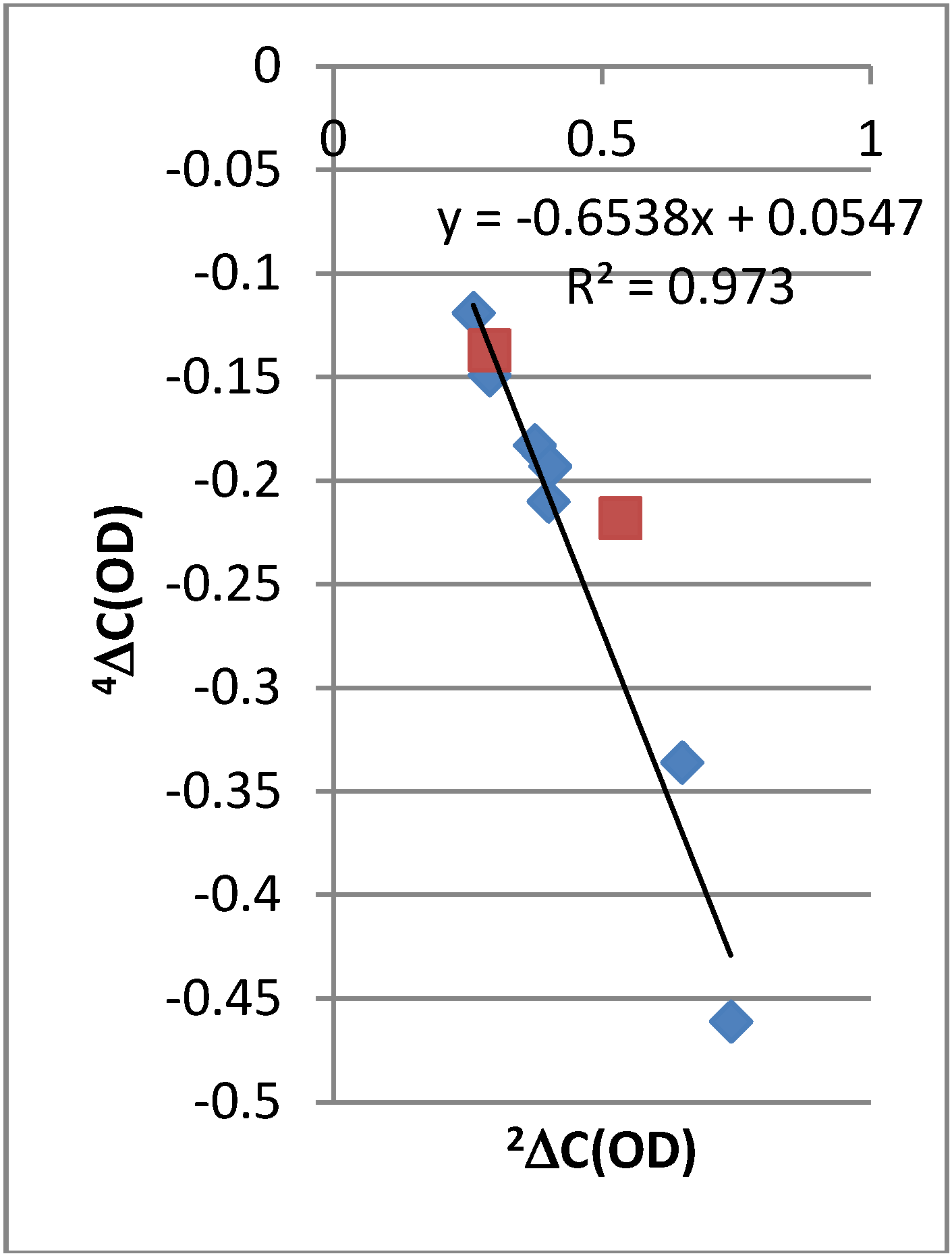
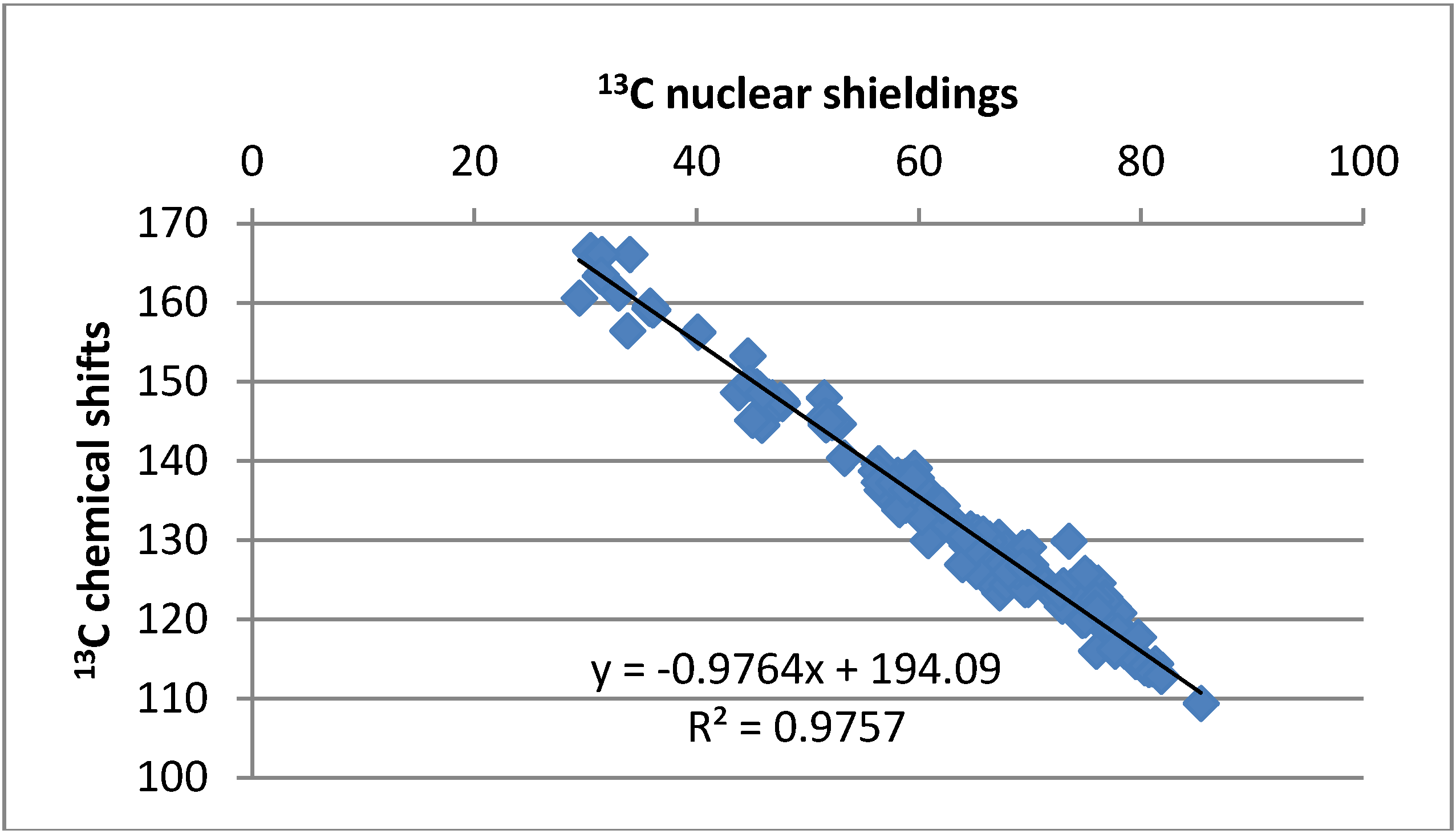
2.3. Chemical Shifts
2.3.1. Solvent Effects
| Comp. | HBQ | 7-Dodecanoyl-HBQ | 7-Formyl-HBQ | 7-Nitro-HBQ a | 7-Azo-HBQ | 9-Dode-canoyl-HBQ | 9-Azo-HBQ | 7-Nitro-9-dode-canoyl-HBQ | 7-Dodecanoyl-9-nitro-HBQ | 7,9-Dintro-HBQ b |
|---|---|---|---|---|---|---|---|---|---|---|
| H-2 | 8.80 (9.02) c | 8.80 | 8.87 | 9.13 | 8.84 | 8.86 | 8.80 | 8.92 | 8.95 | 9.11 (9.47) |
| H-3 | 7.52 (7.81) | 7.59 | 7.65 | 7.98 | 7.61 | 7.62 | 7.38 | 7.80 | 7.80 | 7.98 (8.31) |
| H-4 | 8.21 (8.61) | 8.26 | 8.35 | 8.75 | 8.31 | 8.32 | 8.28 | 8.50 | 8.48 | 8.77 (9.23) |
| H-5 | 7.51 (7.90) | 7.55 | 7.87 | 8.22 | 7.79 | 7.73 | 7.65 | 8.01 | 8.01 | 8.27 (8.58) |
| H-6 | 7.97 (7.98) | 8.95 | 9.43 | 8.55 | 9.10 | 7.80 | 7.74 | 9.05 | 8.80 | 8.92 (9.01) |
| H-7 | 7.39 (7.55) | - | - | - | - | 7.38 | d | - | - | - |
| H-8 | 7.61 (7.69) | 8.11 | 8.09 | 8.68 | 8.22 | 8.13 | d | 8.87 | 8.95 | 9.18 (9.27) |
| H-9 | 7.24 (7.21) | 7.19 | 7.30 | 7.25 | 7.29 | - | - | - | - | - |
| OH | 14.90 (14.80) | 16.11 | 16.40 | 16.68 | 15.74 | 16.39 | 15.69 | 19.11 | 18.21 | 19.45 (20.2) |
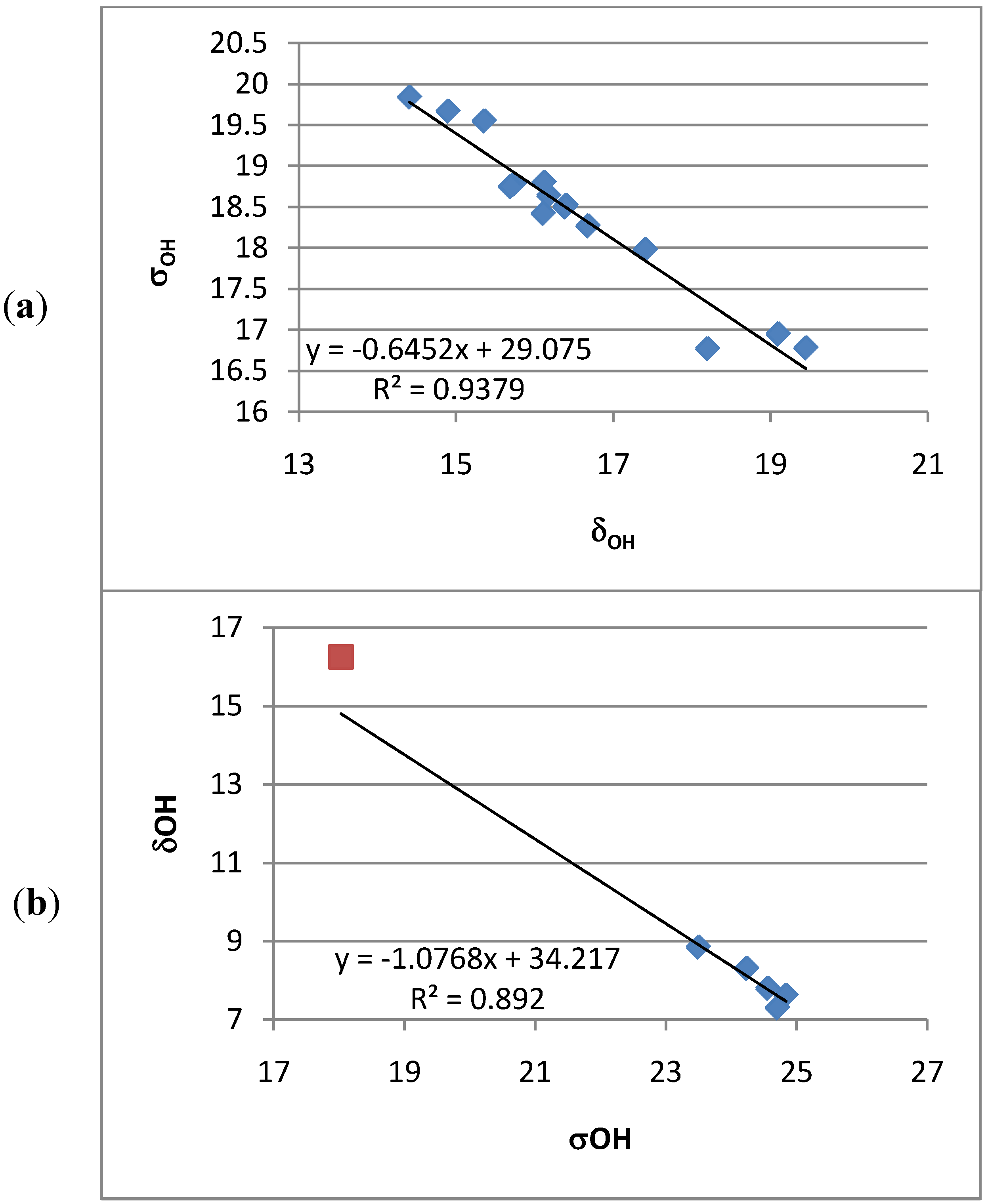
2.3.2. Ring Current Effects
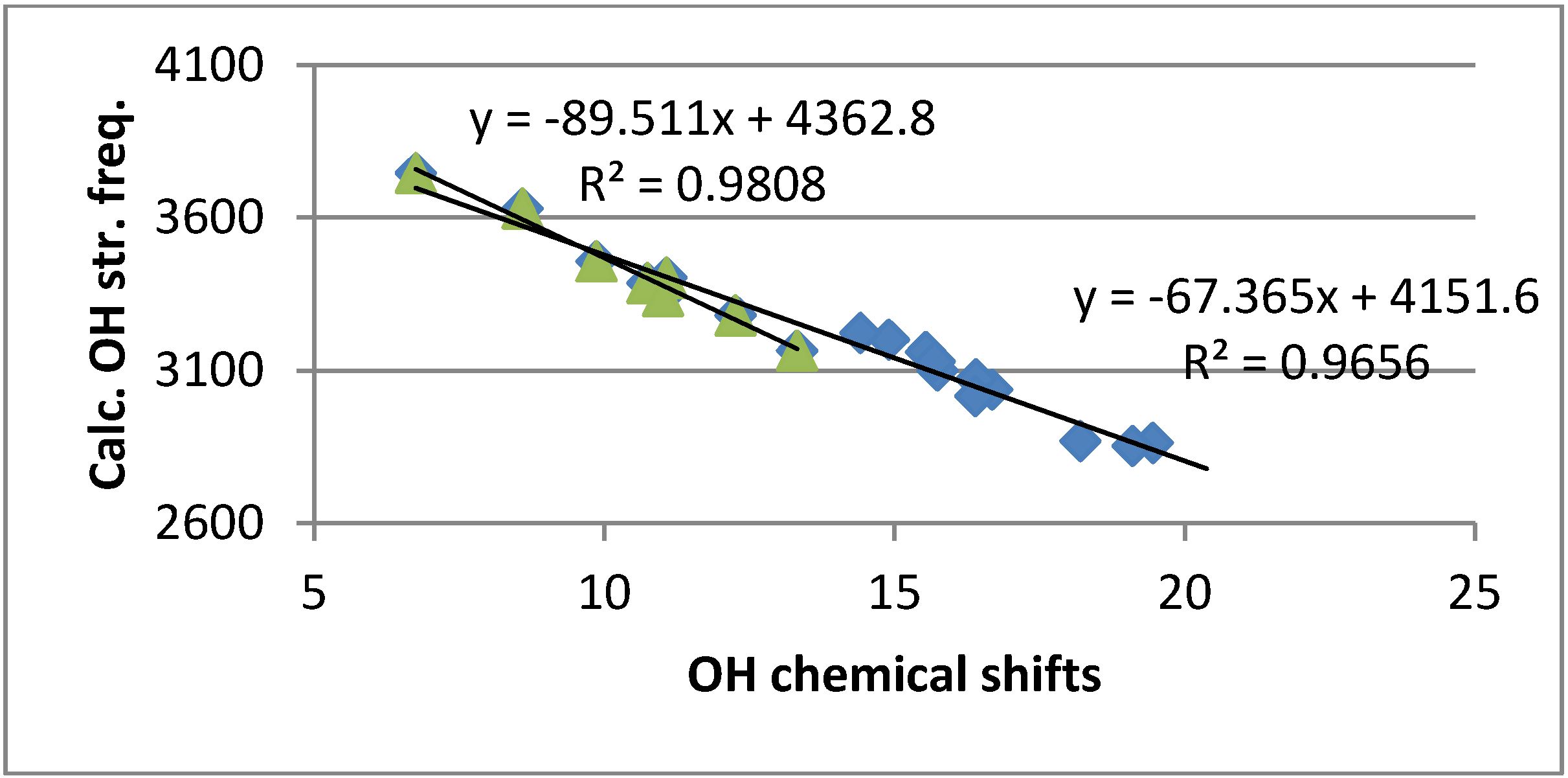
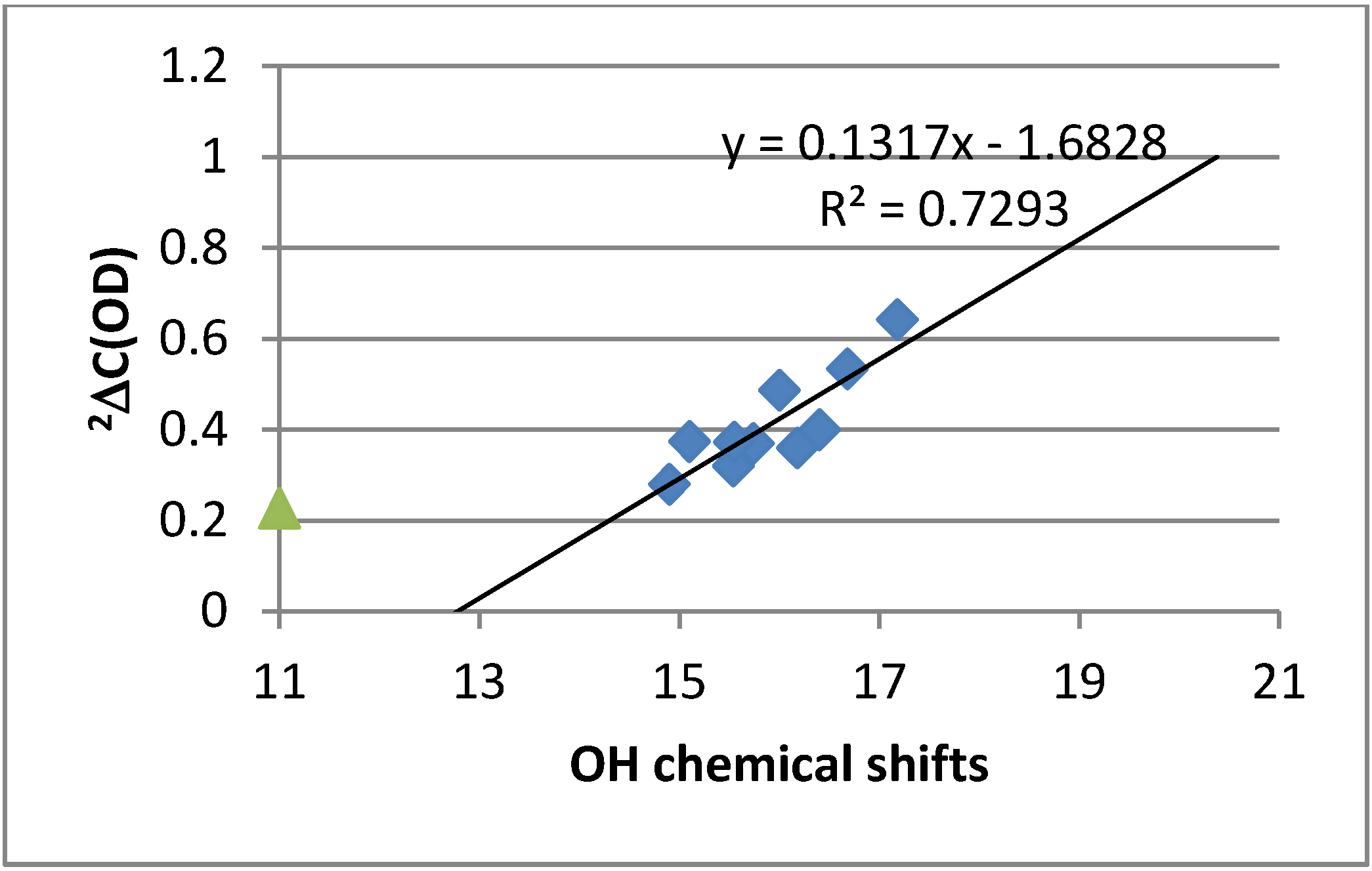
2.4. Calculations
2.4.1. Structures
2.4.2. Chemical Shifts
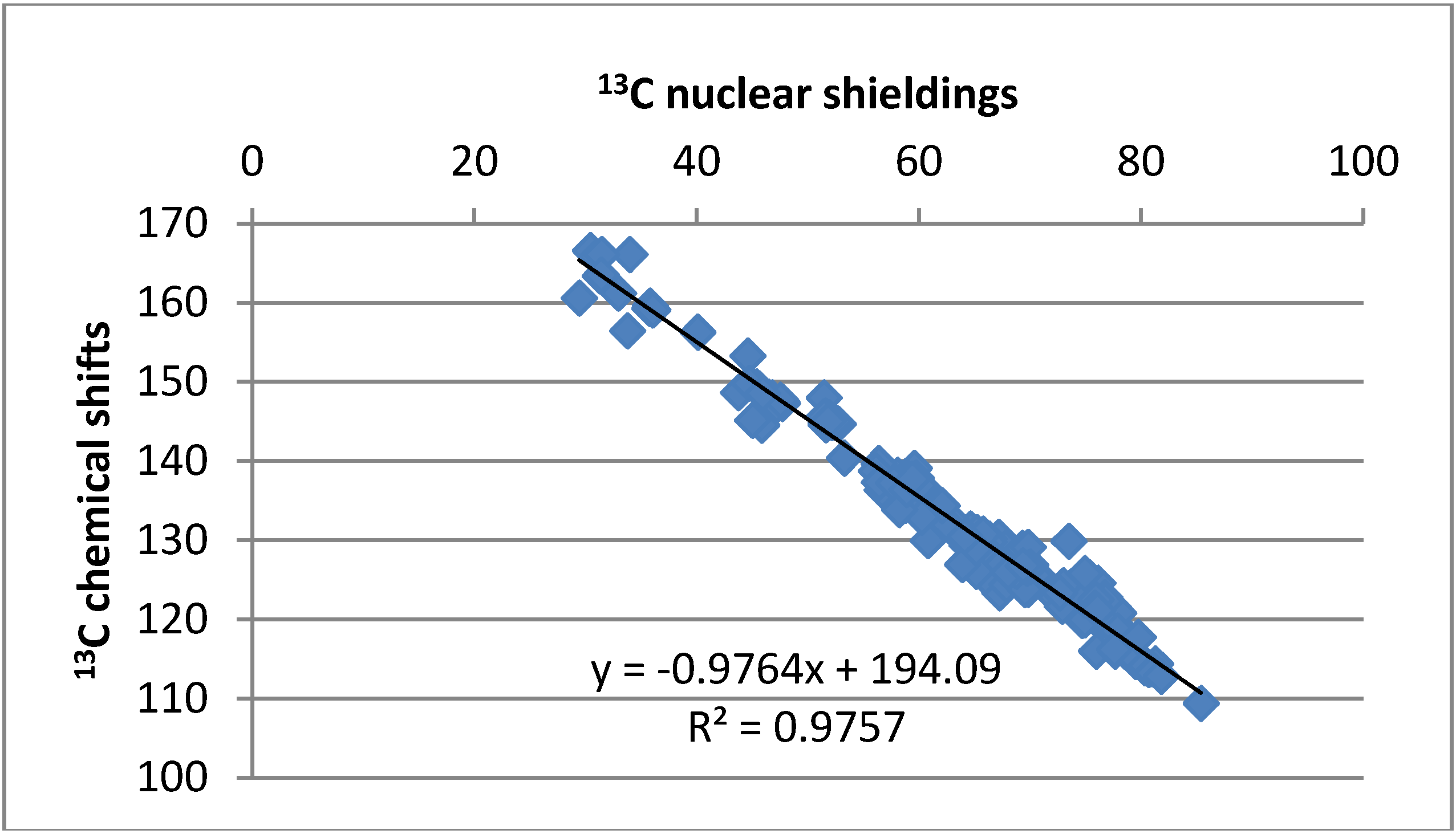
2.4.3. Calculations of Changes in Nuclear Shieldings

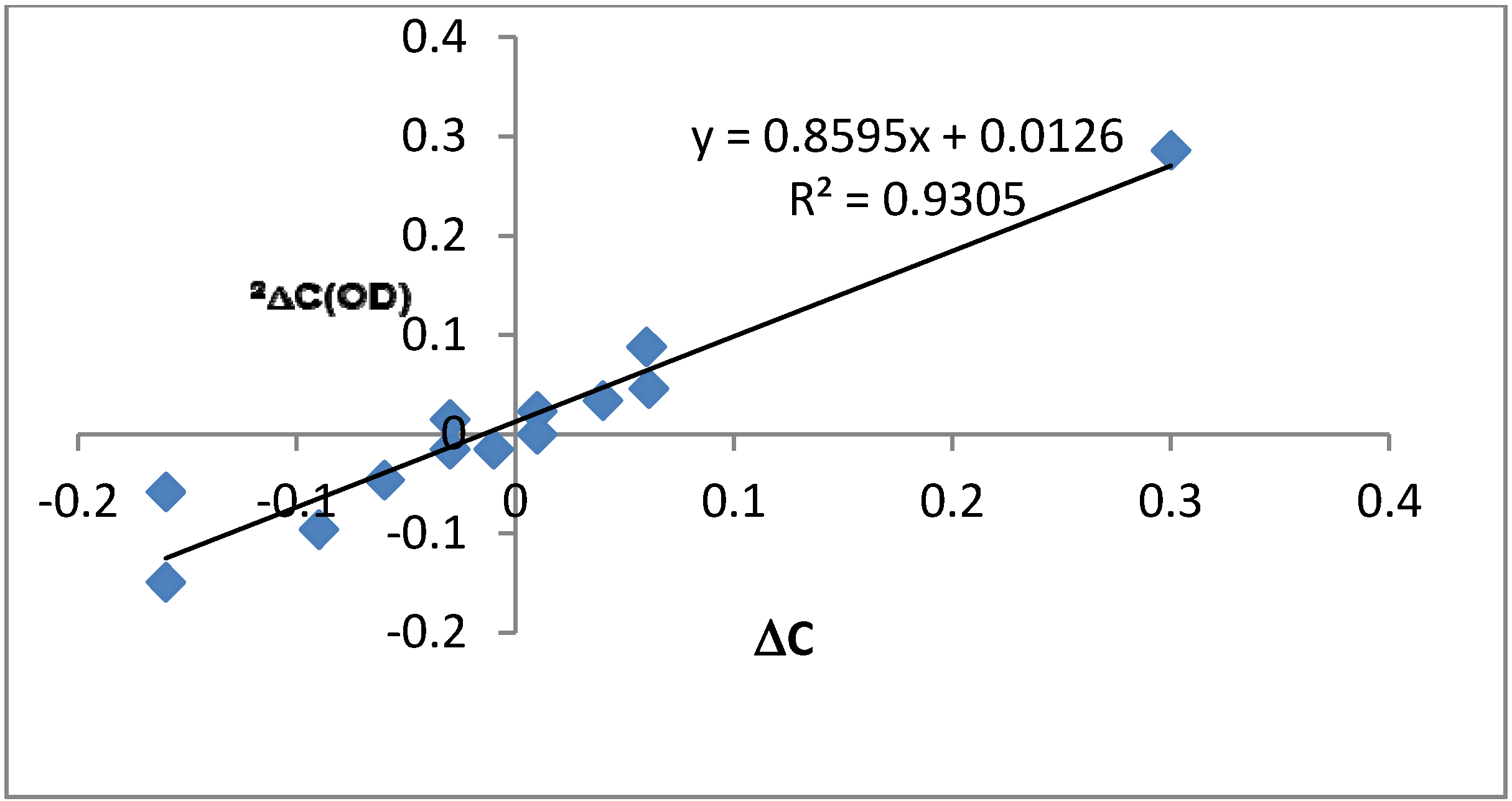
| Compounds | HBQ | 7-Dodecanoyl-HBQ | 7-Carb-aldehyde-HBQ | 7-Nitro-HBQ | 7-Nitro-9-dodecanoyl-HBQ | 9-Dodecanoyl-HBQ b | 7,9-Dinitro-HBQ (NH-form) |
|---|---|---|---|---|---|---|---|
| C-2 | −160 | −160 | −170 | −160 | −188 | 20 | 190 |
| C-3 | −30 | −30 | −20 | −10 | −31 | −10 | 130 |
| C-4 | 10 | 30 | 30 | 35 | 39 | −10 | −60 |
| C-4a | −10 | −20 | −30 | −10 | −28 | −20 | -80 |
| C-5 | −60 | −70 | −70 | −60 | −76 | 20 | 40 |
| C-6 | 60 | 90 | 70 | 110 | 102 | 0 | −90 |
| C-6a | −30 | −10 | −20 | 30 | 0 | 50 | −130 |
| C-7 | −90 | −100 | −90 | −150 | −128 | −80 | 70 |
| C-8 | 40 | 50 | 20 | 60 | 77 | −30 | −40 |
| C-9 | 60 | 30 | 40 | 30 | 41 | −110 | −140 |
| C-10 | 300 | 300 | 290 | 330 | 321 | 380 | −20 |
| C-10a | −160 | −150 | −140 | −150 | −139 | 50 | −70 |
| C-10b | 10 | −0 | −20 | −10 | −38 | 0 | 200 |
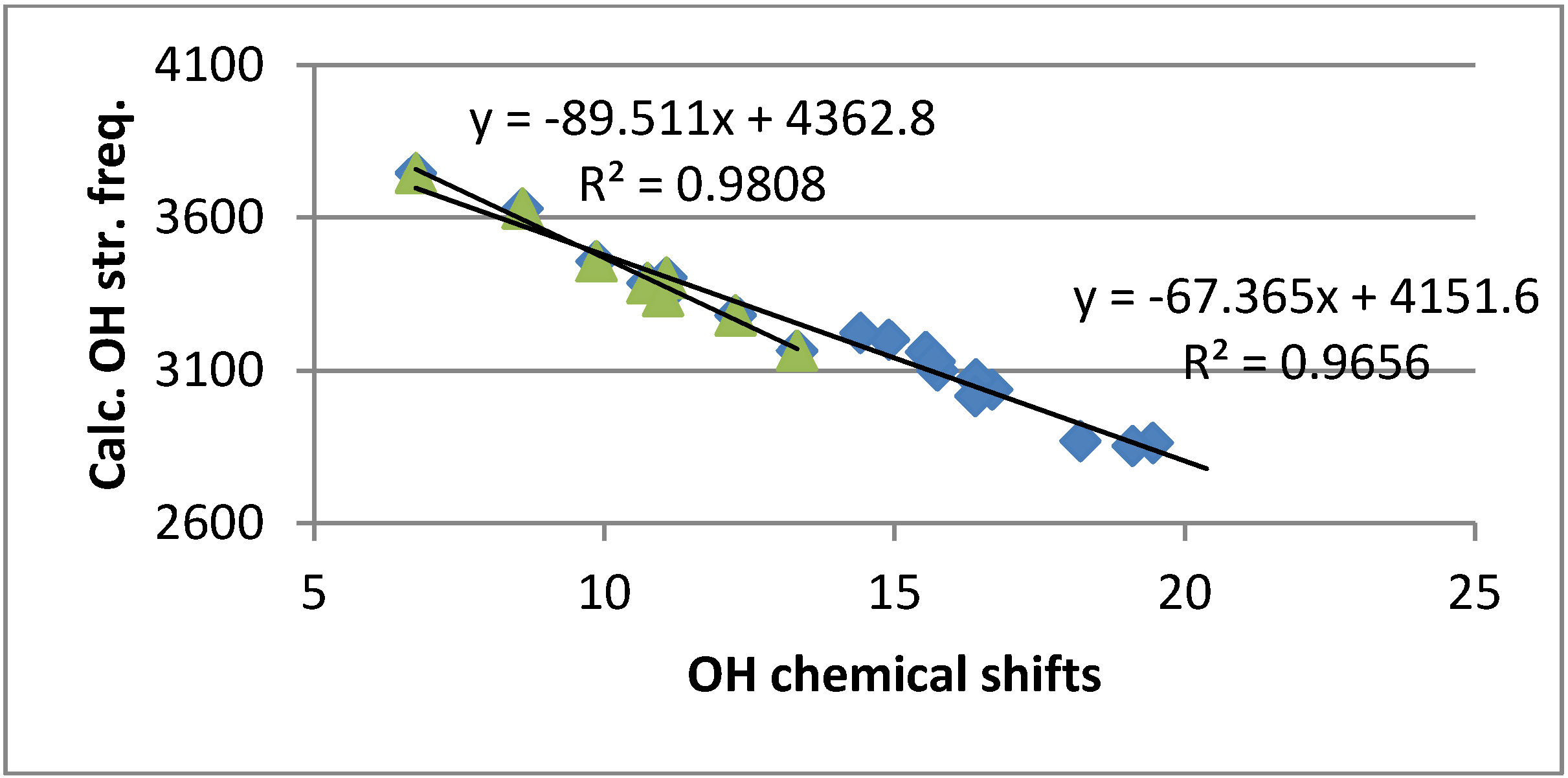
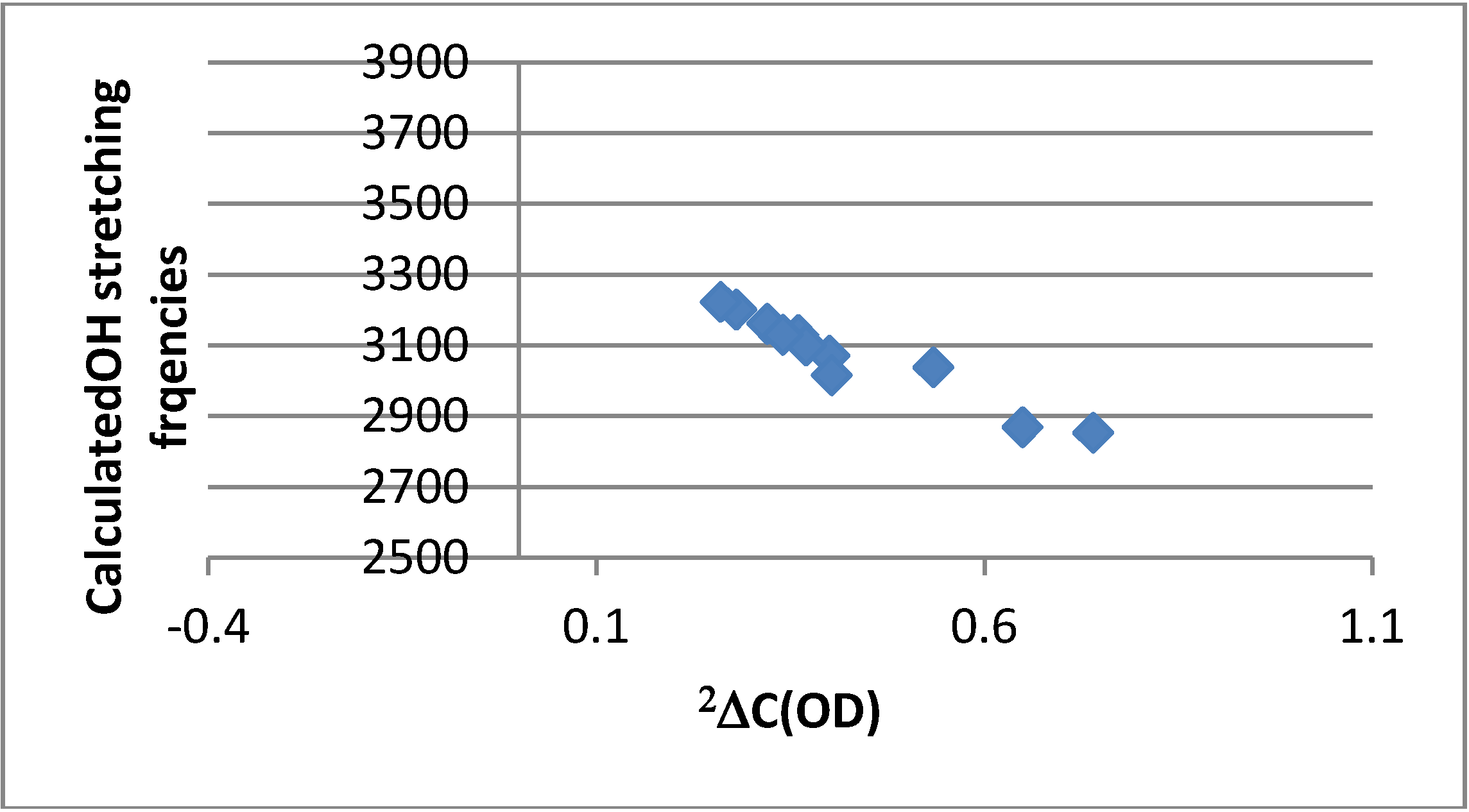
2.4.4. Calculation of Frequencies

3. Discussion
| Compounds | RO…N | ROH (B3LYP) |
|---|---|---|
| HBQ | 2.618 | 0.997 |
| 7-Dodecanoyl-HBQ | 2.574 | 1.004 |
| 7-Formyl-HBQ | 2.585 | 1.004 |
| 7-Nitro-HBQ | 2.570 | 1.005 |
| 7-Azo-HBQ | 2.597 | 1.002 |
| 9-Dodecanoyl-HBQ | 2.588 | 1.000 |
| 7-Nitro-9-dodecanoyl-HBQ | 2.533 | 1.014 |
| 7-Dodecanoyl-9-nitro-HBQ | 2.537 | 1.014 |
| 4-Morpholino-HBQ | 2.604 | 0.999 |
| 7,9-Dinitro-HBQ (NH-form) | 2.478 | 1.079 (NH) |
4. Experimental
4.1. General
4.2. Compounds
4.3. Theoretical Calculations
5. Conclusions
Acknowledgments
References
- Piechowska, J.; Gryko, D.T. Preparation of a Family of 10-hydroxybenzo[h]quinolone Analogues via a Modified Sanford Reaction and their Excited State intramolecular Proton Transfer properties. J. Org. Chem. 2011, 76, 10220–10228. [Google Scholar] [CrossRef]
- Martinez, M.L.; Cooper, W.C.; Chou, P.T. A novel excited-state intramolecular proton transfer molecule, 10-hydroxybenzo[h]quinoline. Chem. Phys. Lett. 1992, 193, 151–154. [Google Scholar] [CrossRef]
- Chou, P.-T.; Chen, Y.-C.; Yu, W.-S.; Chou, Y.-H.; Wei, C.-Y.; Cheng, Y.-M. Excited-state intramolecular proton transfer in 10-hydroxybenzo[h]quinolone. J. Phys. Chem. A 2001, 105, 1731–1740. [Google Scholar]
- Chou, P.T.; Wei, C.Y. Photophysics of 10-hydroxybenzo[h]quinolone in aqueous solution. J. Phys. Chem. 1996, 100, 17059. [Google Scholar] [CrossRef]
- Takeuchi, S.; Tahara, T. Coherent nuclear wave packet motions in ultrafast excited-state intramolecular proton transfer: Sub-30-fs resolved pump-probe absorption spectroscopy of 10-hydroxybenzo[h]quinolone in solution. J. Phys. Chem. A 2005, 109, 10199–10207. [Google Scholar] [CrossRef]
- Schriever, Ch.; Barbatti, M.; Stock, K.; Aquino, A.J.A.; Tunera, D.; Lochbrunner, S.; Riedle, E.; de Vivie-Riedle, R.; Lischka, H. The interplay of skeletal deformation and ultrafast excited-state intramolecular proton transfer: Experimental and theoretical investigation of 10-hydroxybenzo[h]quinolone. Chem. Phys. 2008, 347, 446–461. [Google Scholar] [CrossRef]
- Kim, C.H.; Joo, T. Coherent excited state intramolecular proton transfer probed by time-resolved fluorescence. Phys. Chem. Chem. Phys. 2009, 11, 10266–10269. [Google Scholar] [CrossRef]
- Higashi, M.; Saito, S. Direct simulation of excited-state intramolecular proton transfer and vibrational coherence of 10-hydroxybenzo[h]quinoline in solution. J. Phys. Chem. Lett. 2011, 2, 2366–2371. [Google Scholar] [CrossRef]
- Paul, B.K.; Guchhait, N. TD. DFT investigation of the potential energy surface for excited-stte intramolecular proton transfer (ESIPT) reaction of 10-hydroxybenzo[h]quinoline: Topological (AIM) and population (NBO) analysis of the intramolecular hydrogen bonding interaction. J. Lumin. 2011, 131, 1918–1926. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Hsieh, C.-Cl.; Cheng, Y.-M.; Lai, C.-H.; Chou, P.T. Extensive spectral tuning of the proton transfer emission from 550 to 675 nm via a rational derivatization of 10-hydroxybenzo[h]quinolone. Chem.Comm. 2006, 2006, 4395–4397. [Google Scholar]
- West-Nielsen, M.; Dominiak, P.M.; Wozniak, K.; Hansen, P.E. Strong Hydrogen Bonding involving Nitro and Acetyl groups. Deuterium Isotope Effects on 13 Chemical Shifts. J. Mol. Struct. 2006, 789, 81–91. [Google Scholar] [CrossRef]
- Hansen, P.E. Deuterium Isotope Effects on 13C Nuclear Shielding of Intramolecular Hydrogenbonded Systems. Magn. Reson. Chem. 1986, 24, 903–910. [Google Scholar]
- Hansen, P.E. Isotope Effects on Chemical Shifts as Tools in Structural Studies; Roskilde University Press: Frederiksberg, Denmark, 1996. [Google Scholar]
- Reuben, J. Intramolecular Hydrogen Bonding as Reflected in the Deuterium Isotope Effects on Carbon-13 Chemical Shifts. Correlation with Hydrogen Bond Energies. J. Am. Chem. Soc. 1986, 108, 1735–1738. [Google Scholar] [CrossRef]
- Hansen, P.E.; Kolonicny, A.; Lycka, A. Deuterium Isotope Effects on 13C Nuclear Shielding of Amino and Acetamido Compounds. Tautomerism and Intramolecular Hydrogen-bonding. Magn. Reson. Chem. 1992, 30, 786–796. [Google Scholar]
- Bolvig, S.; Hansen, P.E. Isotope Effects on chemical shifts as an analytical tool in structural studies of intramolecularly hydrogenbonded compounds. Curr. Org. Chem. 2000, 4, 19–54. [Google Scholar] [CrossRef]
- Hansen, P.E.; Hansen, B.K.V.; Spanget-Larsen, J. OH stretching frequencies in systems with intramolecular hydrogen bonds. Harmonic and anharmonic analyses. Chem. Phys. 2011, 389, 107–115. [Google Scholar] [CrossRef]
- Hansen, P.E.; Spanget-Larsen, J. Prediction of OH stretching frequencies in systems with intramolecular hydrogen bonds. J. Mol. Struct. 2012, 1018, 8–13. [Google Scholar]
- Deperasínska, E.; Gryko, D.T.; Karpiuk, E.; Kozankiewicz, B.; Makarewicz, A.; Piechowska, J. 12-hydroxy-1-azaperylene-Limiting case of the ESIPT System: Enol-Keto tautomerism in A-0 and S-1 States. J. Phys. Chem. A 2012, 116, 2109–2116. [Google Scholar]
- Deperasińska, I.; Gryko, D.T.; Karpiuk, E.; Kozankiewicz, B.; Makarewicz, A.; Piechowska, J. Low Temperature Spectra of the Analogues of 10-Hydroxybenzo[h]quinoline as an Indication of Barrierless ESIPT. J. Phys. Chem. A 2012, 116, 12049–12055. [Google Scholar] [CrossRef]
- Gryko, D.T.; Piechowska, J.; Gałęzowski, M. A strong emitting fluorescence based on 1-azaperylene scaffold. J. Org. Chem. 2010, 75, 1297–1300. [Google Scholar] [CrossRef]
- Piechowska, J.; Huttunen, K.; Gryko, D.T.; Wróbel, Z.; Lemmetyinen, H.; Tkachenko, N.V. Excited state intramolecular proton transfer in electron-rich and electron-poor derivatives of 10-Hydroxybenzo[h]quinoline. J. Phys. Chen. A 2012, 116, 9614–9620. [Google Scholar] [CrossRef]
- Haig, C.W.; Mallion, R.B. Ring Current theories in nuclear magnetic resonance. Prog. NMR Spectrosc. 1980, 13, 303–344. [Google Scholar]
- Jameson, C.J. Isotopes in the Physical and Biomedical Sciences Isotopic Application in NMR Studies; Elsevier: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Lycka, A.; Hansen, P.E. Deuterium Isotope Effects on 13C and 15N Nuclear Shielding of o-Hydroxyazo Dyes. Org. Magn. Reson. 1984, 22, 569–571. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. I. A gage-invariant LCAO (linear combination of atomic orbitals) methods for NMR chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the Gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Kubicki, M.; Borowiak, T.; Antkowiak, W.Z. 10-Hydroxybenzo[h]quinoline. Acta Cryst. 1995, C51, 1173–1175. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density Functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Savetti correlation-energy formula into a functional of the electron-density. Phys. Rev. 1988, B37, 785–789. [Google Scholar]
- Sample Availability: Samples of the all the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hansen, P.E.; Kamounah, F.S.; Gryko, D.T. Deuterium Isotope Effects on 13C-NMR Chemical Shifts of 10-Hydroxybenzo[h]quinolines. Molecules 2013, 18, 4544-4560. https://doi.org/10.3390/molecules18044544
Hansen PE, Kamounah FS, Gryko DT. Deuterium Isotope Effects on 13C-NMR Chemical Shifts of 10-Hydroxybenzo[h]quinolines. Molecules. 2013; 18(4):4544-4560. https://doi.org/10.3390/molecules18044544
Chicago/Turabian StyleHansen, Poul Erik, Fadhil S. Kamounah, and Daniel T. Gryko. 2013. "Deuterium Isotope Effects on 13C-NMR Chemical Shifts of 10-Hydroxybenzo[h]quinolines" Molecules 18, no. 4: 4544-4560. https://doi.org/10.3390/molecules18044544