Novel Substituted Heteroaromatic Piperazine and Piperidine Derivatives as Inhibitors of Human Enterovirus 71 and Coxsackievirus A16

Abstract
:1. Introduction
2. Results and Discussion
2.1. Computer Screening
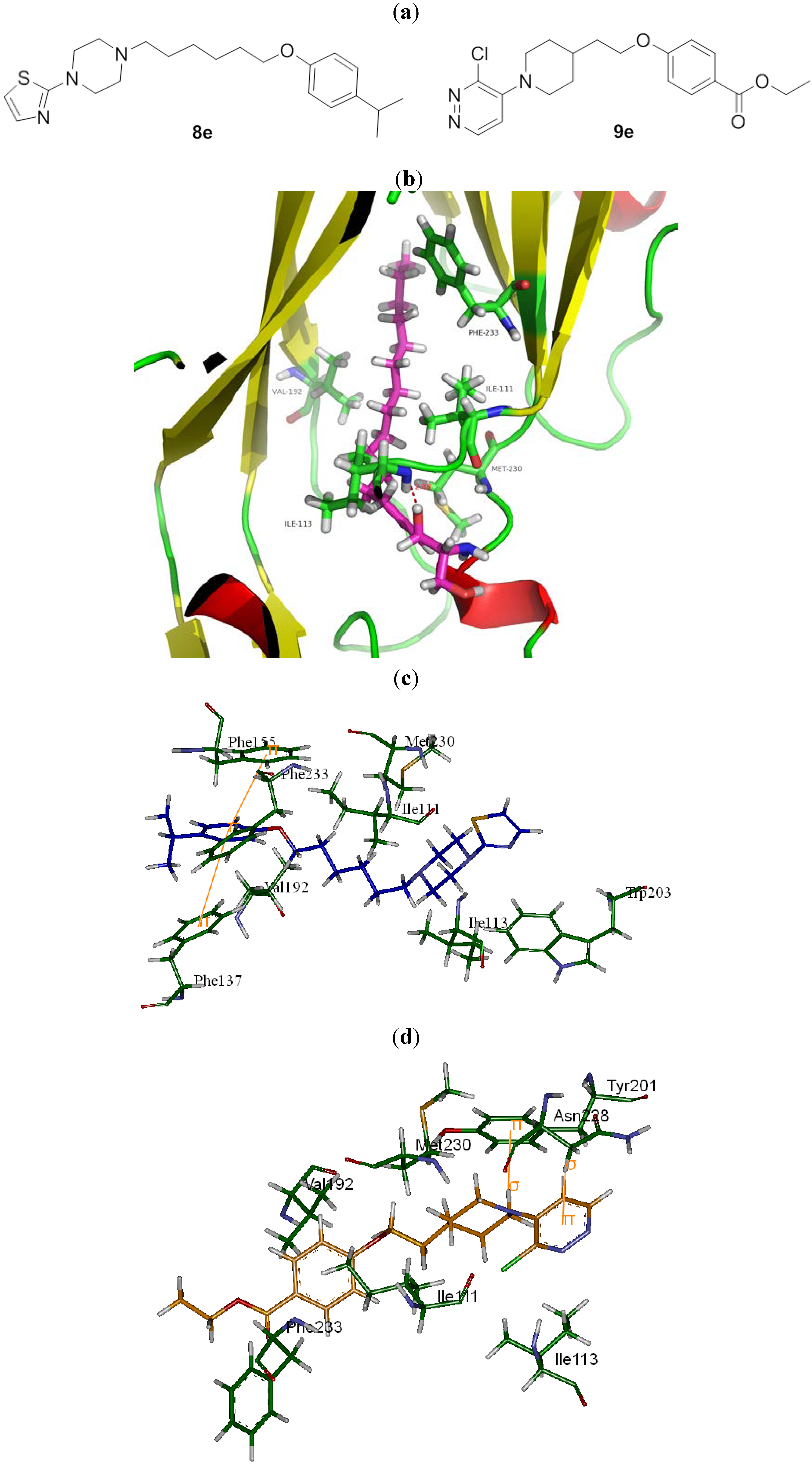

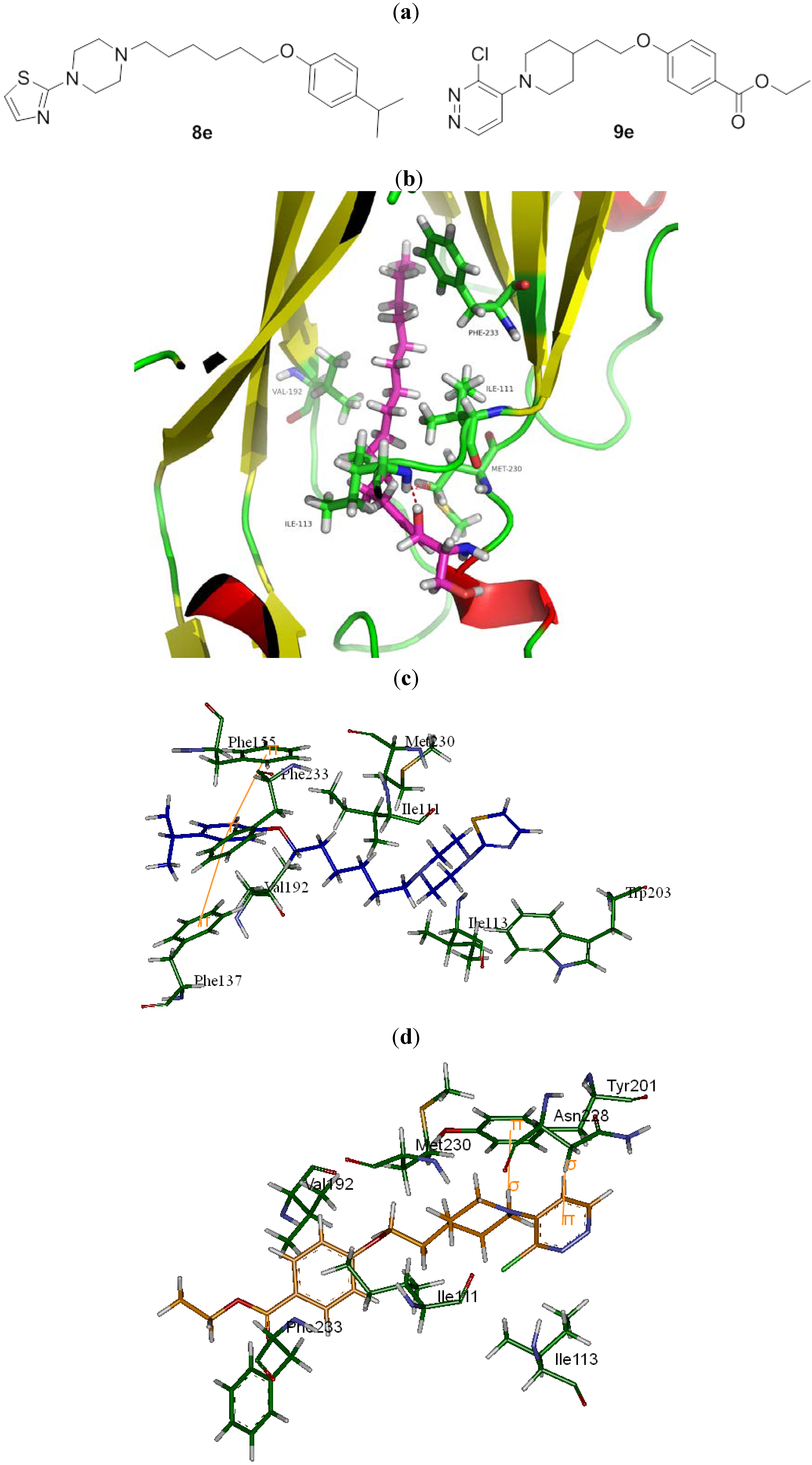{kind=link}
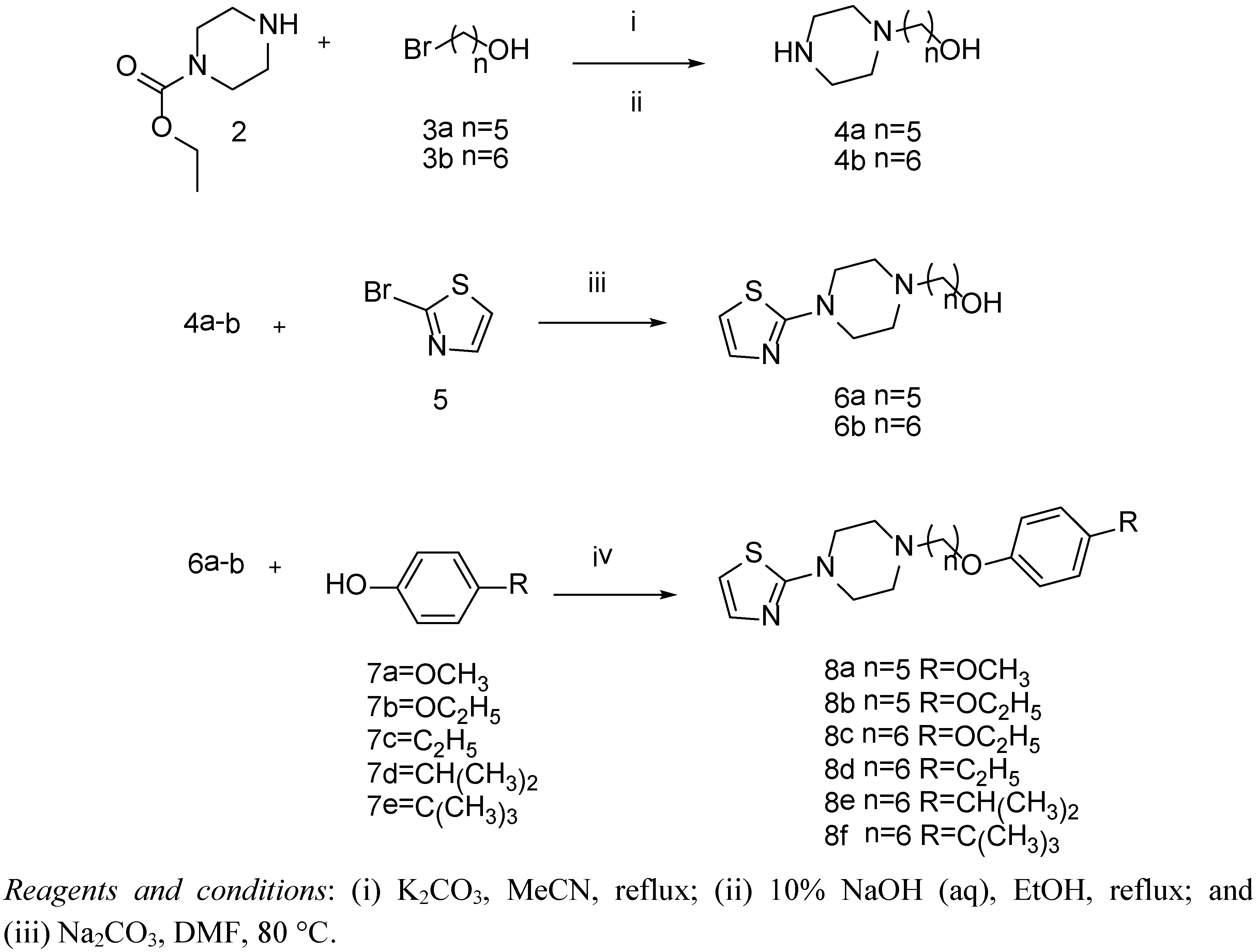{kind=link}

| Cpd | Ar | X | Y | n | R | TC50 (μM) | TC0 (μM) | EV71 | CVA16 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | SI | IC50 (μM) | SI | ||||||||
| 1 | - | - | - | - | - | 78 ± 5.3 | 7.7 ± 0.5 | 0.6 ± 0.0 | 137 | >7.7 | - |
| 9a |  | N | C | 2 | C4H9 | 19.2 ± 0.7 | 7.7 ± 0.3 | 1.2 ± 0.0 | 16.1 | 1.4 ± 0.1 | 13.8 |
| 9b | 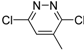 | N | C | 2 |  | 189 ± 17.3 | 29.7 ± 2.2 | 21.6 ± 1.4 | 8.7 | >29.7 | - |
| 9c | 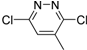 | N | C | 2 | OC2H5 | 62.9 ± 2.4 | 31.5 ± 2.1 | >31.5 | _ | 10.8 ± 0.3 | 5.8 |
| 9d |  | C | C | 2 | CO2CH3 | 193 ± 19.3 | 30.5 ± 2.0 | >31.5 | _ | 22.1 ± 1.1 | 8.7 |
| 9e | 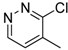 | C | C | 2 | CO2C2H5 | >513 | >513 | 1.0 ± 0.2 | 513 | 12.7 ± 0.4 | >40.3 |
| 9f | 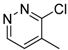 | C | N | 2 | CO2C2H5 | >512 | 128 ± 9.5 | 25.4 ± 1.7 | >20.2 | 16.0 ± 0.9 | >32.0 |
| 9g | 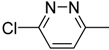 | N | C | 3 | CO2CH3 | 256 ± 22.7 | 128 ± 3.8 | 16.0 ± 0.6 | 16.0 | 50.8 ± 4.1 | 5.0 |
| 9h |  | N | C | 3 | CO2C2H5 | 359 ± 21.5 | 31 ± 4.3 | 4.9 ± 0.2 | 73.7 | 4.9 ± 0.2 | 73.7 |
| 8a |  | N | C | 5 | OCH3 | 277 ± 2.2 | 138 ± 9.6 | 100 ± 9.0 | 2.8 | 69.2 ± 5.5 | 4.0 |
| 8b |  | N | C | 5 | OC2H5 | 66.6 ± 1.0 | 33.3 ± 3.9 | 21 ± 1.2 | 3.2 | 5.3 ± 0.3 | 12.7 |
| 8c | 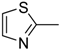 | N | C | 6 | OC2H5 | 54.1 ± 8.1 | 6.8 ± 0.1 | 4.3 ± 0.1 | 12.7 | 6.8 ± 0.4 | 8.0 |
| 8d | 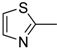 | N | C | 6 | C2H5 | 56.0 ± 3.3 | 28.0±2.5 | 4.4 ± 0.3 | 12.7 | 14.0 ± 0.5 | 4.0 |
| 8e |  | N | C | 6 | CH(CH3)2 | 43.1 ± 4.7 | 6.8 ± 0.8 | 4.3 ± 0.0 | 10.1 | 1.2 ± 0.5 | 34.8 |
| 8f | 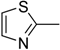 | N | C | 6 | C(CH3)3 | 15.6 ± 0.3 | 7.8 ± 0.4 | 3.9 ± 0.1 | 4.0 | 7.8 ± 0.6 | 2.0 |
2.2. Chemistry
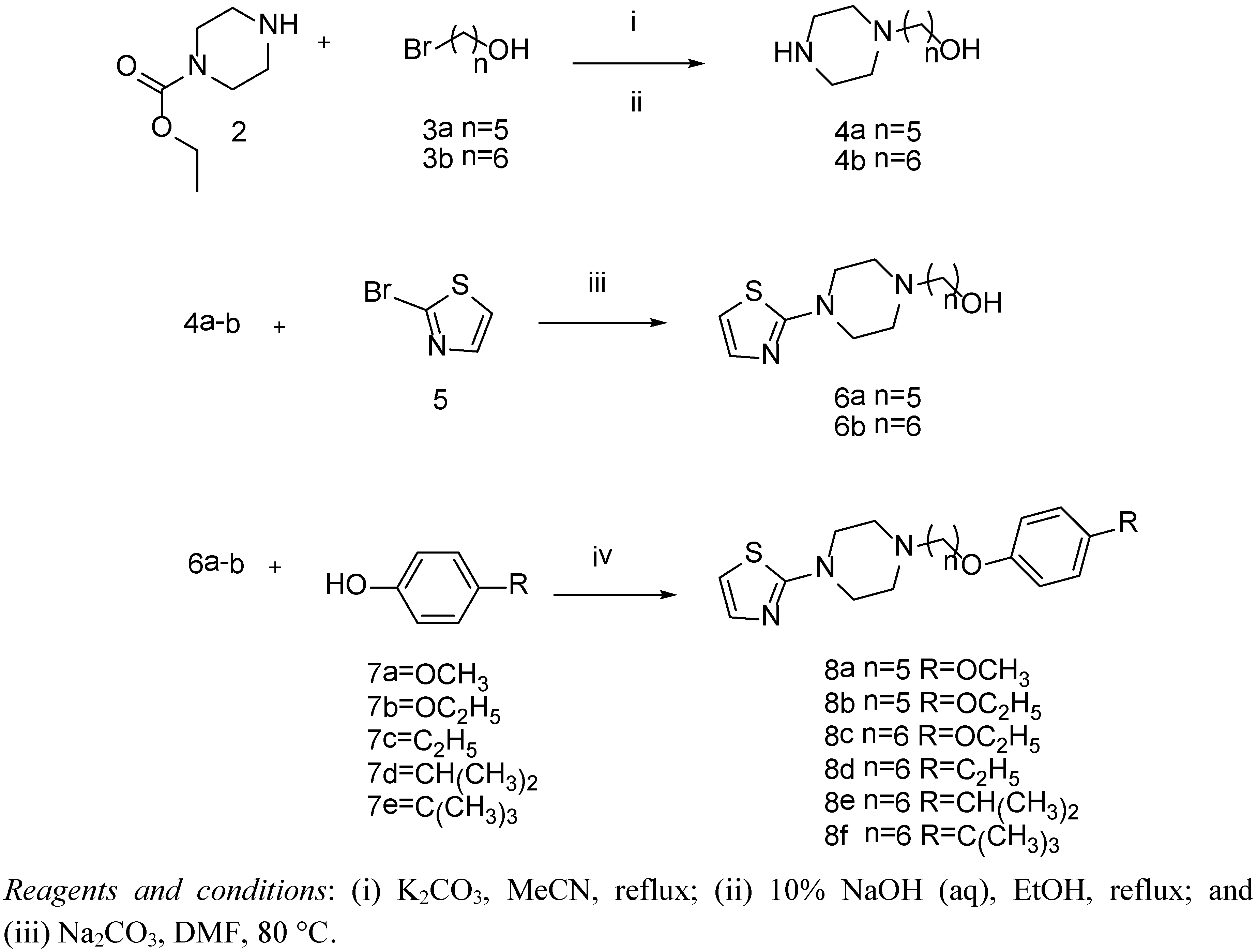
2.3. Biology
2.3.1. Results for Anti-EV71 Activity
2.3.2. Results for anti-CVA16 Activity
3. Experimental
3.1. General
3.2. Computer Screening
3.3. Chemical Synthesis
3.3.1. Synthesis of Compounds 4a,b, and 6a,b
3.3.2. Preparation of Compounds 8a–f
3.4. Biological Evaluation
3.4.1. Neutralization Test [22]
3.4.2. Cytotoxicity Assay [23]
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Racaniello, V.R. Fields Virology, 5th; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 796–839. [Google Scholar]
- Bruu, A.L. Enteroviruses: Polioviruses, Coxsackieviruses, Echoviruses and newer Enteroviruses. In A Practical Guide to Clinical Virology; John Wiley & Sons, Ltd.: Chichester, UK, 2003; pp. 44–45. [Google Scholar]
- Blomberg, J.; Lycke, E. New enterovirus type associated with epidemic of aseptic meningitis and/orhand, and mouth disease. Lancet 1974, 304, 112. [Google Scholar] [CrossRef]
- Chang, L.Y.; Lin, T.Y. Clinical features and risk factors of pulmonary oedema after enterovirus-71-related hand, foot, and mouth disease. Lancet 1999, 354, 1682–1686. [Google Scholar] [CrossRef]
- Lum, L.C.S.; Wong, K.T. Neurogenic pulmonary oedema and enterovirus 71 encephalomyelitis. Lancet 1998, 352, 1391. [Google Scholar]
- Lu, M.; He, Y.X. Pathology of enterovirus 71 infection: An autopsy study of 5 cases. Zhonghua Bing Li Xue Za Zhi 2009, 38, 81–85. [Google Scholar]
- Chern, J.H.; Lee, C.C. Synthesis and antienteroviral activity of a series of novel, oxime ether-containing pyridyl imidazolidinones. Bioorg. Med. Chem. Lett. 2004, 14, 5051–5056. [Google Scholar] [CrossRef]
- Kuo, C.J.; Shie, J.J. Design, Synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorgan. Med. Chem. 2008, 16, 7388–7398. [Google Scholar] [CrossRef]
- Li, H.; Li, W. Research progress of enterovirus 71 vaccine. Prog. Microbiol. Immunol. 2012, 40, 89–94. [Google Scholar]
- Shia, K.S.; Li, W.T. Design, Synthesis, and Structure−Activity Relationship of Pyridyl Imidazolidinones: A Novel Class of Potent and Selective Human Enterovirus 71 Inhibitors. J. Med. Chem. 2002, 45, 1644–1655. [Google Scholar] [CrossRef]
- Rei, L.K.; Shin, R.S. Strategies to develop antivirals against enterovirus 71. J. Virol. 2013, 10, 28. [Google Scholar]
- Hung, J.H.; Yi, R.J. Traditional Chinese medicine application in HIV: An in silico study. J. Biomol. Struct. Dyn. 2012, 1–12. [Google Scholar]
- Su, S.C.; Hung, J.H. Two Birds with One Stone? Possible Dual-Targeting H1N1 Inhibitors from Traditional Chinese Medicine. PLoS Comput. Biol. 2011, 7, e1002315. [Google Scholar] [CrossRef] [Green Version]
- Rossmann, M.G.; Arnold, E. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 1985, 317, 145–153. [Google Scholar]
- Filman, D.J.; Syed, R. Structual factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO 1989, 8, 1567–1579. [Google Scholar]
- Wang, X.; Peng, W. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 2012, 19, 424–429. [Google Scholar] [CrossRef]
- Fan, S.Y.; Zheng, Z.B. Synthesis and evaluation of novel chloropyridazine derivatives as potent human rhinovirus (HRV) capsid-binding inhibitors. Bioorgan. Med. Chem. 2009, 17, 621–624. [Google Scholar] [CrossRef]
- Wang, H.; Xiao, J. Pharmacophore-based design, synthesis, and biological evaluation of novel chloro-pyridazine piperazines as human rhinovirus (HRV-3) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1057–1059. [Google Scholar] [CrossRef]
- Barnard, D.L.; Hubbard, V.D. In vitro activity of expanded-spectrum pyridazinyl oxime ethers related to pirodavir: Novel capsid-binding inhibitors with potent antipicornavirus activity. Antimicrob. Agents Chemother. 2004, 48, 1766–1772. [Google Scholar] [CrossRef]
- Ji, X.-Y.; Wang, H.-Q. Synthesis and antiviral activity of N-Phenylbenzamide derivatives, a novel class of Enterovirus 71 inhibitors. Molecules 2013, 18, 3630–3640. [Google Scholar] [CrossRef]
- Meng, E.C.; Shoiche, B.K. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Hsiung, G.D.; Fong, C. Picornaviridae. In Hsiung’s Diagnostic Virology; Hsiung, G.D., Fong, C.K.Y., Landry, M.L., Eds.; Yale University Press: New Haven, CT, USA, 1994; pp. 119–140. [Google Scholar]
- Nakayama, M.; Suzuki, K. Inhibition of the infectivity of influenza virus by tea polyphenols. Antiviral Res. 1993, 21, 289–299. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 8a–f, and 9a–h are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, X.; Wang, H.; Li, Y.; Cao, R.; Zhong, W.; Zheng, Z.; Wang, G.; Xiao, J.; Li, S. Novel Substituted Heteroaromatic Piperazine and Piperidine Derivatives as Inhibitors of Human Enterovirus 71 and Coxsackievirus A16. Molecules 2013, 18, 5059-5071. https://doi.org/10.3390/molecules18055059
Zhang X, Wang H, Li Y, Cao R, Zhong W, Zheng Z, Wang G, Xiao J, Li S. Novel Substituted Heteroaromatic Piperazine and Piperidine Derivatives as Inhibitors of Human Enterovirus 71 and Coxsackievirus A16. Molecules. 2013; 18(5):5059-5071. https://doi.org/10.3390/molecules18055059
Chicago/Turabian StyleZhang, Xian, Hongliang Wang, Yuhuan Li, Ruiyuan Cao, Wu Zhong, Zhibing Zheng, Gang Wang, Junhai Xiao, and Song Li. 2013. "Novel Substituted Heteroaromatic Piperazine and Piperidine Derivatives as Inhibitors of Human Enterovirus 71 and Coxsackievirus A16" Molecules 18, no. 5: 5059-5071. https://doi.org/10.3390/molecules18055059